
Neuromyelitis optica
Neuromyelitis Optica
Neuromyelitis optica (NMO), also known as Devic‘s disease, is a devastating autoimmune disorder of the central nervous system and is clinically characterized by inflammation of the optic nerve, spinal cord, and, less frequently, brainstem. The discovery of antibodies against aquaporin‑4 (NMO‑ IgG/ AQP4- IgG) has definitively distinguished this disease from multiple sclerosis. AQP4- IgG testing is crucial in differential diagnosis that also includes magnetic resonance imaging of the brain and spinal cord. Typically, spinal cord lesions exceeding the length of three vertebral segments are found. Changes in magnetic resonance imaging of the brain do not exclude the diagnosis of NMO; by contrast, they are relatively common and are fairly typical. Optical coherence tomography provides important information, and quantifies the involvement of the retina and optic nerve. Neurologic deficit in this disease is a result of disease relapse, and it is therefore crucial to establish the diagnosis early and initiate an appropriate treatment that can change the unfavorable prognosis. The main therapeutic strategies include aggressive treatment of relapses (with high‑dose corticosteroids or plasmapheresis), followed by chronic therapy (first choice drugs include azathioprine and rituximab).
Key words:
neuromyelitis optica – autoantibodies against aquaporin-4 – neuromyelitis optica spectrum disorders
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
P. Nytrová; D. Horáková
![]()
Působiště autorů:
Neurologická klinika a Centrum klinických neurověd 1. LF UK a VFN v Praze
Vyšlo v časopise:
Cesk Slov Neurol N 2015; 78/111(2): 130-137
Kategorie:
Minimonografie
doi:
https://doi.org/10.14735/amcsnn2015130
Souhrn
Neuromyelitis optica (NMO), také známá jako Devicova choroba, je devastující autoimunitní onemocnění centrálního nervového systému klinicky charakterizované záněty zrakového nervu, míchy a méně často mozkového kmene. Objev protilátek proti akvaporinu‑ 4 (NMO‑ IgG/ AQP4- IgG) definitivně odlišil tuto chorobu od roztroušené sklerózy. Testování AQP4- IgG je klíčové v diferenciálně diagnostickém procesu, ve kterém se dále neobejdeme bez magnetické rezonance mozku a míchy. Typickým nálezem jsou míšní ložiska přesahující délku tří míšních segmentů. Změny na magnetické rezonanci mozku diagnózu NMO nevylučují, jsou naopak relativně časté a mají poměrně typický charakter. Důležité informace přináší i optická koherentní tomografie, která kvantifikuje postižení sítnice a zrakového nervu. Neurologický deficit u této choroby je následkem relapsů onemocnění, a proto je zcela zásadní časné stanovení diagnózy a nasazení adekvátní terapie, která může změnit nepříznivou prognózu pacienta. Mezi hlavní léčebné strategie patří agresivní léčba relapsů (vysokodávkované steroidy či plazmaferéza) následované zahájením chronické terapie (mezi léky první volby dnes řadíme azathioprin a rituximab).
Klíčová slova:
neuromyelitis optica – protilátky proti akvaporinu-4 – onemocnění ze širšího spektra neuromyelitis optica
Úvod
Neuromyelitis optica (NMO), známá také jako Devicova choroba, je autoimunitní astrocytopatie klinicky typicky charakterizovaná záněty zrakového nervu, míchy a méně často mozkového kmene [1,2]. Dlouhá desetiletí byla považována za agresivní podtyp roztroušené sklerózy, což zásadně změnil objev V. Lennonové et al v roce 2004. Ti popsali sérový marker tzv. Neuromyelitis Optica Imunoglobulin G (NMO‑ IgG/ AQP4‑ IgG) u pacientů splňujících původní Wingerchukova kritéria [3]. Cílovým antigenem těchto protilátek je akvaporin‑4 (AQP4) [4], který je exprimován zejm. astrocyty a patří do rodiny transmembránových vodních kanálů, které se významně podílejí na vodní homeostáze a jsou součástí hematoencefalické bariéry [5,6]. AQP4 zprostředkovává transport vody přes plazmatickou membránu, který je především regulován hustotou těchto proteinů v membráně a osmotickým gradientem. Selektivní destrukce a dysfunkce astrocytů vede dále k demyelinizaci a ztrátě neuronů. NMO se definitivně vyčlenila jako samostatná nozologická jednotka a dnes ji řadíme mezi protilátkově zprostředkovaná autoimunitní onemocnění nebo autoimunitní ionforopatie centrálního nervového systému.
Epidemiologie
Před objevem protilátek proti AQP4 byla NMO považována za onemocnění s extrémně vzácným výskytem u indoevropské populace v našich geografických šířkách. Údaje o incidenci a prevalenci jsou limitované a vycházejí zvláště z národních registrů. Nejvyšší prevalence onemocnění byla zaznamenána v Dánsku – 4,4 na 100 000 obyvatel. Odhaduje se, že toto onemocnění tvoří asi 1,5 % získaných demyelinizačních onemocnění CNS u této populace. Naopak u asijské populace je výskyt mnohem vyšší a tvoří asi 20– 40 % onemocnění v této skupině. Častěji onemocní ženy (poměr se pohybuje od 2,8– 9 : 1), medián věku začátku onemocnění je okolo 39 let [7,8]. První manifestace nemoci může být prakticky v kterémkoliv věku, včetně první a sedmé dekády života. Přesná prevalence v České republice není známa, ale odhadujeme ji na 1 : 100 000 obyvatel.
Patogeneze
Studium histopatologických nálezů a zvířecích modelů významně přispělo k poznání patogeneze NMO. Klíčovým mechanizmem je navázání protilátek na cílový antigen, kterým je astrocyty exprimovaný AQP4. Jedná se o protilátky podtřídy IgG1, které po navázání na svůj cílový antigen mohou aktivovat komplement, což vede k formování membránu atakujícího komplexu s lýzou buněk [9,10]. Štěpné produkty komplementu a další prozánětlivé cytokiny působí chemotakticky pro leukocyty, zejm. neutrofily a eozinofily [11], které se spolupodílejí na destrukci nervové tkáně uvolněnými proteázami a volnými radikály. Zvýšené sérové koncentrace C3a složky komplementu korelují s neurologickým deficitem a aktivitou choroby [12]. Formování AQP4 imunokomplexů s destrukcí buněk je provázeno ztrátou pozitivity barvení na AQP4 s vaskulocentrickou depozicí imunoglobulinů a produktů aktivace komplementu v patologických ložiscích [11,13,14]. Tato histopatologická charakteristika odpovídá lézím u myší indukovaných podáním lidských protilátek proti AQP4 spolu s lidským komplementem intracerebrálně. Akutní NMO léze je charakterizována splývající a/ nebo fokální perivaskulární demyelinizací, prominentní infiltrací makrofágů, ztrátou axonů, nekrózami šedé i bílé hmoty míchy, provázené ztrátou astrocytů [10,11].
Základním rozdílem v patogenezi NMO v porovnání s roztroušenou sklerózou (RS) je primární postižení astrocytů po navázání protilátek v oblasti hematoencefalické bariéry (HEB), systémová aktivace komplementu a infiltrace granulocytů způsobující primární postižení. Demyelinizace a ztráty neuronů jsou pak sekundárním projevem indukovaného zánětu. U RS dochází k průniku mononukleárních buněk přes relativně intaktní HEB, za kterou probíhá zánětlivá reakce v oblasti bílé i šedé hmoty, dochází také k formování sekundárních germinálních center v oblasti mening s přítomností plazmatických buněk produkujících různé protilátky.
Klinický obraz
Neuromyelitis optica byla popsána klinicky již na konci 19. století jako monofázické zánětlivé onemocnění postihující míchu a zrakové nervy souběžně nebo v těsné návaznosti. Tento monofázický průběh je vzácnější a vyskytuje se asi u 10 % případů. Nejčastěji je průběh relaps remitentní. Délka remise je variabilní od několika týdnů po několik let. Díky objevu protilátek proti AQP4 se rozšířily klinické příznaky tohoto onemocnění, které nebyly dříve dobře známy. Zejména se jedná o příznaky při postižení mozkového kmene, hypothalamu nebo sluchu [1,8].
Optická neuritida (ON) se projevuje bolestí za okem, výpadky zorného pole, změnou barvocitu nebo snížením zrakové ostrosti. Tíže postižení zrakového nervu, popřípadě jeho bilaterální postižení, by nás mělo vést k úvaze o zánětlivém postižení v rámci onemocnění NMO. Dále je typický poměrně rychlý rozvoj s výrazným otokem na papile, těžký průběh bez úplné úpravy, popřípadě amauróza, kterou vyvine asi 22 % pacientů po první atace [1]. Někdy zánět zrakového nervu nemusí být doprovázen bolestí při pohybu očním bulbem. U některých pacientů může dojít k rozvoji amaurózy během několika hodin. Zde pak může být mylně stanovena diagnóza přední ischemické neuropatie optiku i vzhledem k věku, protože počátek onemocnění v 5. dekádě života a později je poměrně častý.
Kmenové příznaky jsou vyjádřeny asi u čtvrtiny pacientů, často v úvodu onemocnění. Typicky se jedná o zvracení s nauzeou provázené neztišitelným singultem, často v souvislosti s lézí v oblasti area postrema [15]. Není vzácná ani ztráta sluchu, diplopie, neuralgie trigeminu a nystagmus [16]. V důsledku postižení dechového centra v oblasti medulla oblongata může dojít k neurogenně podmíněnému respiračnímu selhání, s kterým se setkáváme zejm. u pacientů se souběžnou rozsáhlou myelitidou oblasti krční a hrudní míchy. Postižení hypothalamu se může projevit polyurií, nadměrnou denní spavostí, hypotermií, hypotenzí a obezitou [17,18]. Příznaky bývají doprovázeny T2 hyperintenzitami na magnetické rezonanci (MR) mozku v oblasti hypothalamu.
Myelitida probíhá často pod obrazem para‑ či kvadruparézy nebo plegie s hranicí čití a sfinkterovými poruchami. Inkompletní míšní léze diagnózu NMO nevylučuje, naopak setkáváme se s ní u pacientů, kteří užívají imunosupresivní léčbu pro prodělanou ON nebo mají jiné autoimunitní onemocnění (např. systémový lupus erythematodes) [19]. Radikulární bolest, paroxyzmální tonické velmi bolestivé svalové spazmy trvající 20– 45 sekund a decharge electrique (Lhermittův příznak) se vyskytují u 1/ 3 relaps remitentních pacientů, ale nenacházíme je u pacientů s monofázickým průběhem NMO [8,20]. Myelitida typická pro NMO dosahuje nebo přesahuje na T2 obrazech tři obratlové segmenty, tzn. longitudinálně extenzivní transverzální myelitida (LETM). Klinická úprava těžkého neurologického deficitu závisí na zahájení adekvátní terapie, ale často zůstává výrazné reziduum. V kohortě pacientů s mediánem sledování 75 měsíců s počátkem onemocnění nad 46. rokem života bylo více než 40 % odkázáno na invalidní vozík. S vyšším věkem počátku onemocnění roste riziko trvalého motorického deficitu [21].
Pomocné vyšetřovací metody
Magnetická rezonance představuje u NMO klíčové zobrazovací vyšetření, a to zejm. při stanovení diagnózy a v diferenciálně diagnostickém procesu. Bez ohledu na počáteční příznaky by měla být vždy provedena MR mozku i míchy, doporučeno je i podání kontrastní látky [22].
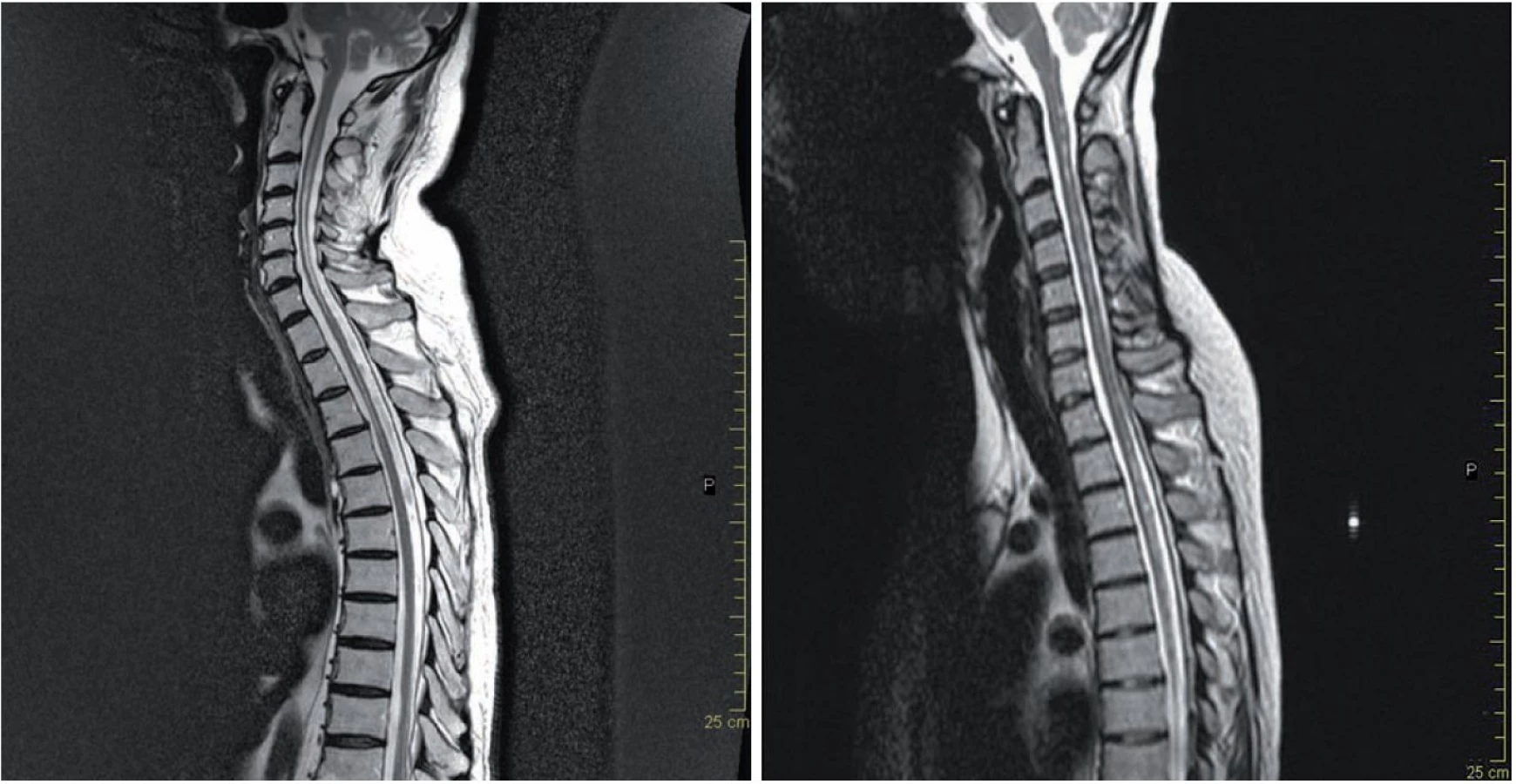
Typickým nálezem podporujícím diagnózu NMO jsou LETM, u kterých tato míšní ložiska dosahují v průměru délky 4,5– 8,7 obratlových segmentů, postihují centrální šeď i oblast bílé hmoty, typicky jsou lokalizována v krční a hrudní míše (obr. 1a). Přibližně 30– 70 % těchto lézí se v akutní fázi barví kontrastní látkou, enhancement může přetrvávat i několik týdnů. Po léčbě kortikoidy nebo s odstupem času může dojít k vymizení nebo fragmentaci původních míšních T2 lézí. Postupně dochází u většiny pacientů k významné míšní atrofii (obr. 1b).
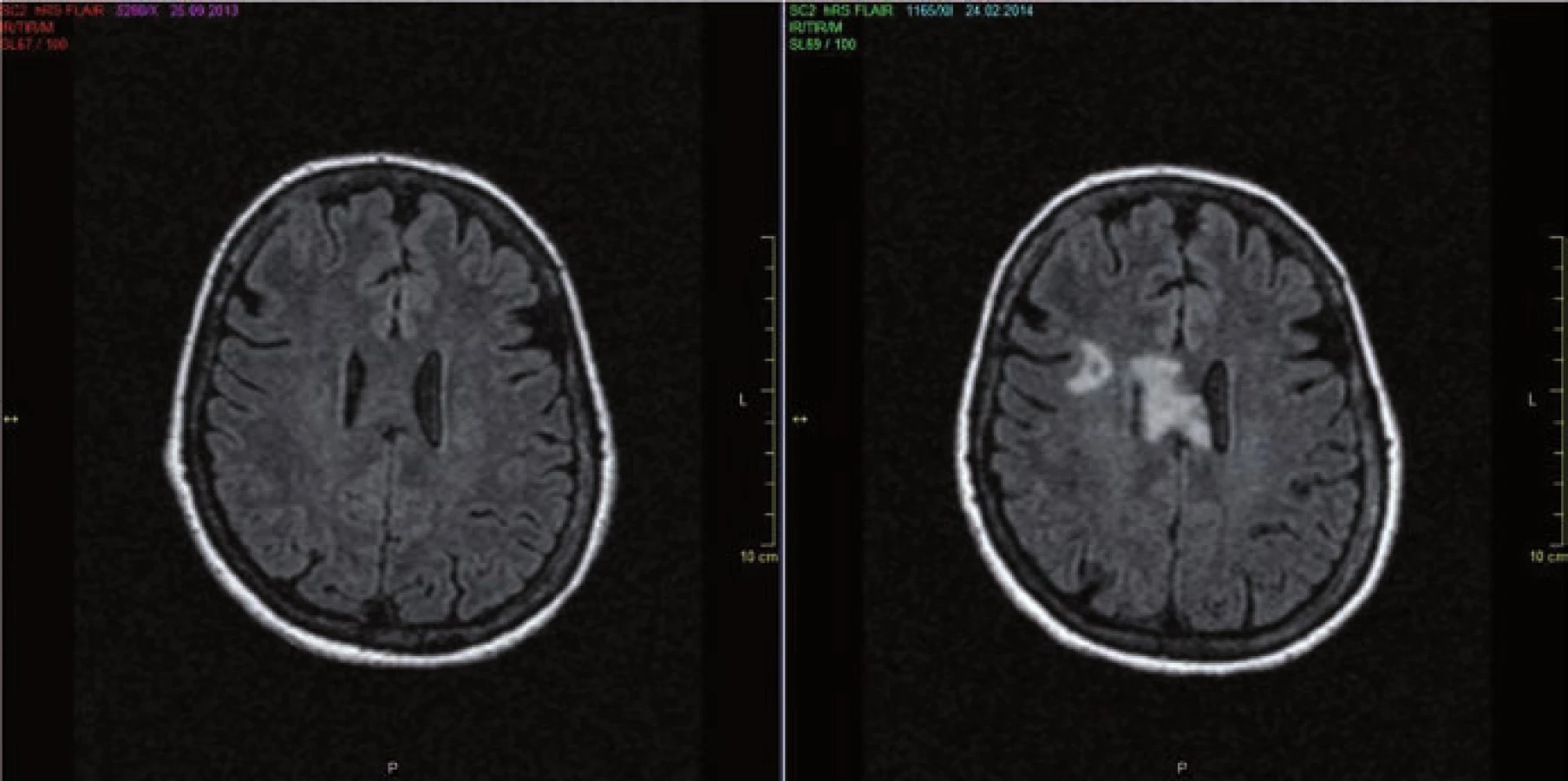
Původní představa, že pacienti s NMO mají na MR mozku negativní nález, je dnes již vyvrácena. Z různých studií vyplývá, že asi 50 % NMO pacientů má MR nález již na počátku onemocnění a s přibývající délkou se toto číslo zvětšuje (obr. 2a,b). Nález je ale nespecifický, může sice napodobovat RS, nicméně atypická lokalizace, často ve spojitosti s atypickými klinickými projevy, by měla upozornit na možnost NMO [23]. Léze bývají v místech vysoké exprese AQP4, tj.:
- v periependymální oblasti Sylviova akveduktu, kolem III. a IV. komory, někdy zasahující i do oblasti hypothalamu;
- v oblasti prodloužené míchy často navazující na ložisko míšní, typicky s postižením area postrema (obr. 3a,b);
- lemující periependymálně postranní komory (na rozdíl od RS, kde vídáme ložiska ovoidní, kolmá na postranní komory, označované jako tzv. Dawsonovy prsty), v případě NMO vídáme ložiska rozsáhlejší, často vyplňující celé corpus callosum a přecházející přes střední čáru (obr. 2b);
- postižení pyramidové dráhy v oblasti zadního raménka capsula interna a v oblasti mozkových penduklů;
- vzácněji mohou být i rozsáhlé, edematózní léze v oblasti bílé hmoty supratentoriálně [23,24].

Zajímavé nálezy ukazuje 7 tesla MR, kde u NMO na rozdíl od RS chybí kortikální postižení a léze v oblasti bílé hmoty nemají typicky centrálně uloženou venulu [25]. Tyto MR charakteristiky by mohly v budoucnu pomoci v diagnostickém procesu. Po podání kontrastní látky je někdy popisován „cloud like enhancement“, pro NMO ale jinak není specifický vzorec.
Stanovení protilátek proti AQP4 (AQP4- IgG nebo též NMO‑ IgG) v séru pacienta je základní vyšetření v rámci stanovení diagnózy NMO nebo poruch jejího širšího spektra. Zlatým standardem je stanovení protilátek na buňkách transfekovaných AQP4 (tzv. Cell Based Assay; CBA) pomocí metody nepřímé imunofluorescence nebo průtokové cytometrie. Specificita je prakticky 100 %, senzitivita CBA je vyšší než 80 % [26]. Z toho vyplývá i důležitý fakt, že negativita protilátek proti AQP4 nevylučuje diagnózu NMO. U většiny pacientů přetrvává sérová pozitivita při opakovaných stanoveních. U některých pacientů může v důsledku léčby dojít k snížení koncentrace protilátek pod detekční limit nebo jsou přítomny pouze v okamžiku relapsů. Indikace k vyšetření AQP4- IgG vychází z kombinace klinických příznaků uvedených výše, které nás nutí přemýšlet o diagnóze NMO. Zcela jistě lze ještě uvést některé specifické případy, kdy indikujeme vyšetření protilátek proti AQP4. Jedná se o každou ON u pacienta asijské populace, u pacientek s atakou ON nebo myelitidy vzniklé v těhotenství, u pacientů s myasthenia gravis, systémovým lupus erythematodes a Sjögrenovým syndromem.
Vyšetření likvoru je důležité v diferenciálně diagnostickém procesu. Nálezy v mozkomíšním moku závisí na okamžiku provedení lumbální punkce ve vztahu k aktivitě choroby. V okamžiku relapsu nacházíme zvýšenou koncentraci bílkoviny odpovídající poruše hematoencefalické bariéry, pleiocytózu typicky s přítomností neutrofilních a eozinofilních granulocytů včetně mononukleárních buněk v různém poměru. U těžkých myelitid je možné nalézt i shluky ependymových buněk. V okamžiku remise nacházíme prakticky normální koncentraci bílkovin a počet buněk nebo mírnou oligocytózu [27]. Asi u 30 % pacientů mohou být pozitivní oligoklonální pásy v likvoru, popřípadě se můžeme setkat i obrazem shodného výskytu oligoklonálních pásů v séru i likvoru. Právě negativita OCB (Oligoclonal Bands) v likvoru u pacientů s podezřením na RS by měla vést také k úvaze o diagnóze NMO.
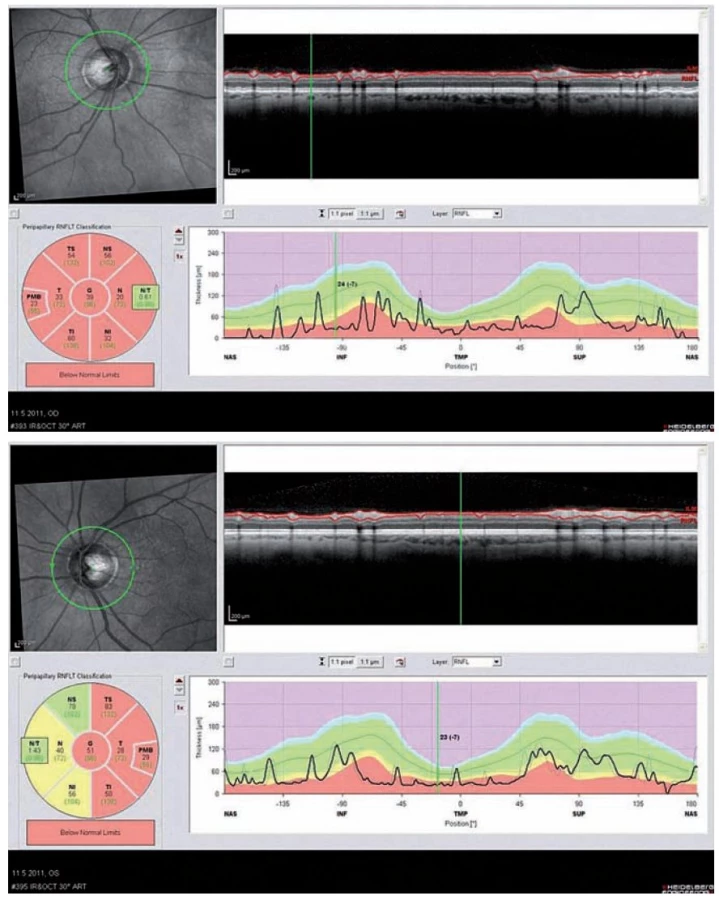
Optická koherentní tomografie (OCT) je nekontaktní neinvazivní zobrazovací metoda, která zobrazuje příčné řezy sítnicí a její abnormality. Toto vyšetření není součástí diagnostických kritérií, ale může nám velmi napomoci v diferenciální diagnostice a odkrýt určitý vzorec postižení zrakového nervu po prodělané ON, který je pro pacienty s NMO příznačný. Jedním ze základních měřených parametrů je tloušťka vrstvy nervových vláken peripapilárně, tzv. RNFL (Retinal Nerve Fiber Layer thickness). U těchto pacientů dochází po odeznění ON k poklesu hodnoty RNFL ve většině nebo všech kvadrantech (obr. 4a,b). V akutních fázích vidíme často i edém papily. U pacientů s RS vídáme častěji pokles RNFL pouze v temporálních kvadrantech. Vizuální evokované potenciály vykazují abnormality charakteru prodloužení vlny P100 u 40 % a redukci amplitudy nebo chybění potenciálu u 25 % nemocných [28].
Stanovení diagnózy NMO a onemocnění jejího širšího spektra (NMO SD)
V rámci stanovení diagnózy vycházíme z klinického obrazu a výsledků pomocných vyšetřovacích metod. V praxi používáme revidovaná Wingerchukova kritéria z roku 2006, mezi jejichž hlavní kritéria patří ON a myelitida. Tři vedlejší kritéria se vztahují k nálezu na MR míchy, mozku a stanovení AQP4- IgG v séru (tab. 1) [29]. V případě naplnění obou hlavních a dvou ze tří vedlejších kritérií je možné stanovit diagnózu NMO, a to i v případě, že by AQP4- IgG byly negativní, při naplnění ostatních kritérií. O této skupině mluvíme jako tzv. AQP4- IgG séronegativní NMO, kde se také častěji setkáváme s monofázickým průběhem [30]. U relaps remitentní formy je časový interval mezi první a dalšími atakami různý. Vzhledem k různému časovému intervalu mezi atakami ON a myelitidy nemusí pacienti se sérovou pozitivitou AQP4- IgG poměrně dlouho naplnit revidovaná Wingerchukova diagnostická kritéria pro NMO. Pacienty s anamnézou izolované nebo rekurentní optické neuritidy bez myelitidy a naopak pouze s myelitidou a pozitivitou AQP4- IgG řadíme do skupiny onemocnění širšího spektra NMO poruch (Neuromyelitis Spectrum Disorders; NMO SD). Někteří autoři používají pro tyto případy jiný termín, tzv. limitované formy NMO [8]. Tito pacienti jsou léčeni dle stejných schémat jako nemocní s definitivní diagnózou. U pacientů s LETM nacházíme pozitivitu AQP4- IgG až v 75 % případů [31]. Pozitivita těchto protilátek predikovala rozvoj ON nebo recidivy míšní ataky do jednoho roku u 55 % pacientů. U rekurentních izolovaných ON nacházíme tyto protilátky u čtvrtiny nemocných [32].

U AQP4-IgG séronegativních pacientů s NMO je diferenciální diagnóza širší a je nutné vyloučit jiná autoimunitní onemocnění, infekce, tumory, metabolické příčiny a cévní malformace. Část AQP4-IgG séronegativních pacientů s NMO může mít pozitivní protilátky proti myelinovému oligodendrocytárnímu proteinu (MOG‑ IgG) nebo alfa podjednotce glycinového receptoru [33,34]. Pacienti se sérovou pozitivitou MOG‑ IgG mají ve srovnání s pacienty s protilátkami proti AQP4 lepší prognózu s dobrou odpovědí na terapii kortikoidy [33].
Lze také očekávat, že dojde k úpravě Wingerchukových kritérií, a to zejm. ve smyslu zařazení kmenových příznaků mezi hlavní diagnostická kritéria.
Léčba
U NMO zatím nemáme léky, které by nemoc zcela vyléčily. Za posledních 10 let ale došlo k významnému posunu v poznání patogenetických mechanizmů a tím i možnostem cílené léčby, která nemoc u velké části pacientů dobře stabilizuje. Vzhledem k tomu, že u přirozeného průběhu nemoci hrozí již časně těžká invalidita, která je většinou následkem proběhlého relapsu, tak je maximum úsilí směrováno právě na agresivní léčbu akutního zhoršení a následně na dlouhodobou/ trvalou prevenci dalších atak. Nedílnou součástí péče je i léčba symptomatická (sfinkterové dysfunkce, neuropatická bolest, spasticita atd.) a léčba v souvislosti s terapií navozeného sekundárního imunodeficitu a prevence infekcí [35,36].
Vzhledem k tomu, že se jedná o relativně vzácné onemocnění s vysokým rizikem časné invalidity, je provádění randomizovaných, placebem kontrolovaných studií velmi obtížné. Většina doporučení je tedy založena na zkušenosti expertů, retrospektivním hodnocení efektů léčby (off‑ label indikace) a open‑ label studií, popřípadě pozorování z běžné klinické praxe. Jednotlivá doporučení mezi léky první a druhé linie se mohou dle různých pracovních skupin mírně lišit. Všechny níže uvedené léky jsou v současnosti podávány v off‑ label indikaci.
Důležitým poznatkem posledních let jsou data, která ukazují, že klasické imunomodulační léky užívané u RS, jako jsou interferony beta, fingolimod či natalizumab, v terapii Devicovy choroby selhávají či dokonce průběh zhoršují [37– 41].
Léčba relapsu onemocnění je zahajována vysokodávkovanými kortikoidy – metylprednisolon v dávce 5 g (1 g/ den) při respektování známých nežádoucích účinků této léčby. Pokud efekt není dostatečný, tak je doporučeno bez prodlení zahájit sérii plazmaferéz (obvykle v počtu 5– 7 výkonů obden). U pacientů s těžkým relapsem a známou diagnózou, kdy byl již v minulosti dobrý efekt plazmaferézy, je doporučeno zahájit jako první přímo sérii plazmaferéz, možno použít i střídání plazmaferéz a 1 g metylprednisolonu obden. U plazmaferéz doporučujeme provádění výkonů pouze na pracovištích se zkušenostmi s těmito výkony. V případě nedostupnosti můžeme jako alternativu doporučit pulzy cyklofosfamidu (800 mg/ m2, 1× za měsíc, celkem 3×). V případě intolerance předchozích režimů je další variantou podání intravenózních imunoglobulinů (IVIG).
Úspěšná chronická léčba vede k redukci frekvence relapsů a jejich tíže. V případě definitivní diagnózy NMO nebo u limitovaných forem s pozitivitou AQP4 protilátek je doporučeno ihned zahájení chronické terapie s cílem prevence dalších devastujících atak. Toto také platí pro pacienty AQP4-IgG séronegativní v případě těžšího relapsu s neúplnou úpravou. U těchto pacientů je nicméně šance na monofázický průběh choroby, proto současné doporučení zahrnuje po několika letech stabilizace zvážit snížení či přerušení chronické terapie. U pacientů s izolovanými nebo rekurentními atakami LETM nebo ON a pozitivitou AQP4- IgG je také doporučeno nasazení imunosupresivní terapie. V těchto případech by imunosupresivní léčba měla být ponechána alespoň pět let [42].
Mezi léky první volby dnes patří azathioprin a rituximab buď v monoterapii či v kombinační terapii s kortikoidy (tab. 2) [35]. U všech těchto léků je nutné sledování krevního obrazu, jaterních testů a dalších vyšetření krve či moči v rámci monitorování nežádoucích účinků. Azathioprin má doporučenou denní dávku 2,5– 3 mg/ kg tělesné hmotnosti/ den. V úvodu je doporučeno kombinovat s vyšší dávkou orálních steroidů (např. prednisonem v dávce až 1 mg/ kg/ den s velmi pomalým snižováním dávky), dlouhodobě je pak možné kombinovat s nižší dávkou prednisonu (5– 15 mg/ den) [42]. Před nasazením azathioprinu je třeba vyšetřit aktivitu enzymu TPMT (thiopurinmetyltransferáza), v případě nedostatečné aktivity je doporučeno azathioprin nepodávat pro vysoké riziko dřeňového útlumu. Rituximab je chimérická myší/ lidská anti‑CD20 monoklonální protilátka. Při jeho podání dochází k depleci pre‑B a B buněk. V praxi používáme terapeutické schéma 1 000 mg i.v. v odstupu 14 dní. Ve skupině 30 pacientů léčených rituximabem po dobu pěti let dosáhlo 93 % pacientů stabilizace EDSS skóre nebo došlo k mírnému zlepšení neurologického nálezu. Podání další terapeutické dávky (stejné terapeutické schéma) je doporučeno v okamžiku před znovuobjevením se CD19 pozitivních buněk (vyšetření průtokovou cytometrií) [43,44]. Tento interval je u pacientů individuální, ale nejčastěji se pohybuje mezi šesti až osmi měsíci. Z našich zkušeností je u některých pacientů monoterapie rituximabem nedostačující, proto kombinujeme s nízkou dávkou kortikoidů.
![Doporučená léčba pro NMO/NMOSD.
Upraveno dle doporučení Wingerchuka, EFNS 2010 a NMEOS 2014 [33,40,43].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/e9b6314979e461252fb9f3f86effd303.png)
U pacientů s relativně mírnějším průběhem je doporučen pro dlouhodobou terapii jako první volba azathioprin. Rituximab je určen pro pacienty s NMO nebo NMO SD se závažným průběhem choroby (těžký relaps nebo relaps na stávající imunosupresivní léčbě) nebo první ataka onemocnění s těžkým reziduálním nálezem.
V případě intolerance, nežádoucích účinků či nedostatečné efektivity výše uvedené léčby jsou doporučovány další léky v rámci druhé linie, jako je např. mykofenolát mofetil, IVIG, methotrexát atd. Mykofenolát mofetil je vhodný zejm. u pacientů, u kterých nelze podat azathioprin (intolerance nebo nedostatečná aktivita enzymu TMPT). Doporučené dávkování je v úvodu 500 mg 2× denně, po týdnu zvýšit na 1 000 mg 2× denně. Jinou možností je podání IVIG v dávce 0,4g/ kg/ den po dobu pěti dnů v rámci relapsu s následnou udržovací dávkou 1g/ kg/ den ve dvou po sobě následujících dnech každý měsíc. Další možností je mitoxantron a cyklofosfamid v pulzním podání s respektováním kumulativní dávky nebo perorální podání metotrexátu [35,42,45]. Všechny léčebné režimy vyžadují dodržování bezpečnostních doporučení týkajících se např. kontroly krevního obrazu, jaterních funkcí a dalších parametrů.
Vzhledem k zásadnímu posunu ve znalostech o patogenezi choroby je dnes snaha o cílenou léčbu, která by lépe postihla tyto mechanizmy. Ve fázi klinických studií nebo experimentální léčby je dnes několik nadějných preparátů. Příkladem jsou eculizumab, který blokuje aktivaci komplementové kaskády vazbou na C5 složku komplementu a tocilizumab, což je monoklonální protilátka proti receptoru pro interleukin‑6 [46,47]. Výsledky vysokodávkované imunoablace s podporou autologních hematopoetických buněk (AHSCT) jsou shrnuty v reportu EBMT (European Group for Blood and Marrow Transplantation) a jsou zatím nejednoznačné [48].
Těhotenství
Těhotenství a období po porodu je u pacientek s NMO/ NMOSD velmi rizikové. Jakkoliv je celosvětově v literatuře popsáno několik desítek případů, tak výsledná data hovoří o rizikovosti zejm. v 1. a 3. trimestru a období po porodu. V odstupu jednoho roku po porodu došlo ke zhoršení EDSS skóre z hodnoty 1,33 ± 1, 6 před graviditou na 3,01 ± 1,83, včetně jednoho případu úmrtí v důsledku těžké ataky po porodu. Některé pacientky byly po dobu gravidity léčeny prednisolonem, metotrexátem nebo azathioprinem [49].
Závěr
Přestože se NMO a poruchy jejího širšího spektra vyskytují v naší klinické praxi poměrně vzácně, nesmíme na tuto možnost v diferenciální diagnostice zapomínat. Stěžejní je zvláště odlišení NMO a RS vzhledem k odlišné terapii. Diagnostika se výrazně zjednodušila díky vyšetření AQP4- IgG, které je dnes dobře dostupné i v České republice. Vzhledem k riziku časné těžké invalidity je důležité určení diagnózy v okamžiku první manifestace onemocnění, protože pouze agresivní léčba akutního zhoršení a následná dlouhodobá imunosupresivní léčba může změnit negativní prognózu pacienta.
Podpořeno výzkumným záměrem projektem PRVOUK‑ P26/ LF1/ 4. Děkujeme za zapůjčení obrazové dokumentace z magnetické rezonance prof. MU Dr. Manuele Vaněčkové, Ph.D. (Oddělení MR, Radiodiagnostická klinika 1. LF UK a VFN v Praze).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 1. 10. 2014
Přijato do tisku: 13. 1. 2015
doc. MUDr. Dana Horáková, Ph.D.
Neurologická klinika a Centrum klinických neurověd
1. LF UK a VFN v Praze
Kateřinská 30
128 08 Praha 2
e-mail: dana.horakova@vfn.cz
Recenzenti
MUDr. Jiří Piťha, CSc.
doc. MUDr. Pavel Štourač, Ph.D.
doc. MUDr. Radomír Taláb, CSc.
MUDr. Petra Nytrová

Petra Nytrová promovala na 1. lékařské fakultě Univerzity Karlovy v Praze v roce 2007. Po ukončení studií nastoupila jako lékařka na Neurologickou kliniku 1. LF UK a VFN v Praze, kde také zahájila postgraduální studium zaměřující se na demyelinizační onemocnění centrálního nervového systému. S podporou grantového projektu Univerzity Karlovy začala výzkum v oblasti markerů aktivity choroby pacientů s neuromyelitis optica. Vědecké zkušenosti v oblasti detekce neuronálních protilátek získala během tříměsíční stáže na Institute for Experimental Immunology v Lübecku a dále během sedmiměsíční stáže na Univerzitě v Oxfordu, Nuffi eld Department of Clinical Neuroscience, za podpory Newsom-Davis Visiting Fellow udělené časopisem Brain. V současnosti jsou jejími hlavními výzkumnými zájmy kromě imunitních aspektů neuromyelitis optica také otázky imunogenicity biologické léčby řešené v rámci evropského vědeckého projektu ABIRISK (Anti-Biopharmaceutical Immunization: prediction and analysis of clinical relevance to minimize the RISK).
Zdroje
1. Wingerchuk DM, Hogancamp WF, O‘Brien PC, Weinshenker BG. The clinical courseof neuromyelitis optica (Devic‘s syndrome). Neurology 1999; 53(5): 1107– 1114.
2. de Seze J, Stojkovic T, Ferriby D, Gauvrit JY, Montagne C,Mounier‑ Vehier F et al. Devic‘s neuromyelitis optica: clinical, laboratory, MRI and outcome profile. J Neurol Sci 2002; 197(1– 2): 57– 61.
3. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364(9451): 2106– 2112.
4. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR.IgG marker of optic‑ spinal multiple sclerosis binds to the aquaporin‑4 water channel. J Exp Med 2005; 202(4): 473– 477.
5. Hasegawa H, Ma T, Skach W, Matthay MA, Verkman AS. Molecular cloning of a mercurial‑ insensitive water channel expressed in selected water‑ transporting tissues. J Biol Chem 1994; 269(8): 5497– 5500.
6. Rash JE, Yasumura T, Hudson CS, Agre P, Nielsen S. Direct immunogold labeling of aquaporin‑4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci U S A 1998; 95(20): 11981– 11986.
7. Marrie RA, Gryba C. The incidence and prevalence of neuromyelitis optica: a systematic review. Int J MS Care 2013; 15(3): 113– 118. doi: 10.7224/ 1537‑ 2073.2012‑ 048.
8. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007; 6(9): 805– 815.
9. Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP,Kryzer TJ et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007; 69(24): 2221– 2231.
10. Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra‑ cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010; 133(2): 349– 361. doi: 10.1093/ brain/ awp309.
11. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM et al. A role for humoral mechanisms in the pathogenesis of Devic‘s neuromyelitis optica. Brain 2002; 125(7): 1450– 1461.
12. Nytrova P, Potlukova E, Kemlink D, Woodhall M, Horakova D, Waters P et al. Complement activation in patients with neuromyelitis optica. J Neuroimmunol 2014; 274(1– 2): 185– 191. doi: 10.1016/ j.jneuroim.2014.07.001.
13. Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld Ret al. Mechanisms of disease: aquaporin‑4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol 2008; 4(4): 202– 214. doi: 10.1038/ ncpneuro0764.
14. Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W et al. Pattern‑ specific loss of aquaporin‑4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007; 130(5): 1194– 1205.
15. Iorio R, Lucchinetti CF, Lennon VA, Farrugia G, Pasricha PJ, Weinshenker BG et al. Intractable nausea and vomiting from autoantibodies against a brain water channel. Clin Gastroenterol Hepatol 2013; 11(3): 240– 245. doi: 10.1016/ j.cgh.2012.11.021.
16. Chan KH, Tse CT, Chung CP, Lee RL, Kwan JS, Ho PW et al. Brain involvement in neuromyelitis optica spectrum disorders. Arch Neurol 2011; 68(11): 1432– 1439. doi: 10.1001/ archneurol.2011.249.
17. Samart K, Phanthumchinda K. Neuromyelitis optica with hypothalamic involvement: a case report. J Med Assoc Thai 2010; 93(4): 505– 509.
18. Viegas S, Weir A, Esiri M, Kuker W, Waters P, Leite MI et al. Symptomatic, radiological and pathological involvement of the hypothalamus in neuromyelitis optica. J Neurol Neurosurg Psychiatry 2009; 80(6): 679– 682. doi: 10.1136/ jnnp.2008.157693.
19. Zavada J, Nytrova P, Wandinger KP, Jarius S, Svobodova R, Probst C et al. Seroprevalence and specificity of NMO‑ IgG (anti‑aquaporin 4 antibodies) in patients with neuropsychiatric systemic lupus erythematosus. Rheumatol Int 2013; 33(1): 259– 263. doi: 10.1007/ s00296‑ 011‑ 2176‑ 4.
20. Wingerchuk DM, Weinshenker BG. Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology 2003; 60(5): 848– 853.
21. Kitley J, Leite MI, Nakashima I, Waters P, McNeillis B, Brown R et al. Prognostic factors and disease course in aquaporin‑4 antibody‑ positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012; 135(6): 1834– 1849. doi: 10.1093/ brain/ aws109.
22. Tackley G, Kuker W, Palace J. Magnetic resonance imaging in neuromyelitis optica. Mult Scler 2014. pii:1352458514531087. [Epub ahead of print].
23. Kim W, Park MS, Lee SH, Kim SH, Jung IJ, Takahashi Tet al. Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin‑4 autoimmunity. Mult Scler 2010; 16(10): 1229– 1236. doi: 10.1177/ 1352458510376640.
24. Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol 2006; 63(3): 390– 396.
25. Sinnecker T, Dorr J, Pfueller CF, Harms L, Ruprecht K,Jarius S et al. Distinct lesion morphology at 7– T MRI differentiates neuromyelitis optica from multiple sclerosis. Neurology 2012; 79(7): 708– 714. doi: 10.1212/ WNL.0b013e3182648bc8.
26. Jarius S, Wildemann B. Aquaporin‑4 antibodies (NMO‑ IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol 2013; 23(6): 661– 683. doi: 10.1111/ bpa.12084.
27. Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M,Bergamaschi R et al. Cerebrospinal fluid findings in aquaporin‑4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 2011; 306(1– 2): 82– 90. doi: 10.1016/ j.jns.2011.03.038.
28. Ringelstein M, Kleiter I, Ayzenberg I, Borisow N, Paul F,Ruprecht K et al. Visual evoked potentials in neuromyelitis optica and its spectrum disorders. Mult Scler 2014; 20(5): 617– 620. doi: 10.1177/ 1352458513503053.
29. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66(10): 1485– 1489.
30. Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 2012; 9: 14. doi: 10.1186/ 1742‑ 2094‑ 9‑ 14.
31. Jiao Y, Fryer JP, Lennon VA, McKeon A, Jenkins SM, Smith CY et al. Aquaporin 4 IgG serostatus and outcome in recurrent longitudinally extensive transverse myelitis. JAMA Neurol 2014; 71(1): 48– 54. doi: 10.1001/ jamaneurol.2013.5055.
32. Matiello M, Lennon VA, Jacob A, Pittock SJ, Lucchinetti CF, Wingerchuk DM et al. NMO‑ IgG predicts the outcome of recurrent optic neuritis. Neurology 2008; 70(23): 2197– 2200. doi: 10.1212/ 01.wnl.0000303817.82134.da.
33. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J et al. Myelin‑oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012; 79(12): 1273– 1277. doi: 10.1212/ WNL.0b013e31826aac4e.
34. Woodhall M, Coban A, Waters P, Ekizoglu E, Kurtuncu M,Shugaiv E et al. Glycine receptor and myelin oligodendrocyte glycoprotein antibodies in Turkish patients with neuromyelitis optica. J Neurol Sci 2013; 335(1– 2): 221– 223. doi: 10.1016/ j.jns.2013.08.034.
35. Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 2014; 261(1): 1– 16. doi: 10.1007/ s00415‑ 013‑ 7169‑ 7.
36. Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state‑ of‑ the‑ art and emerging therapies. Nat Rev Neurol 2014; 10(9): 493– 506. doi: 10.1038/ nrneurol.2014.141.
37. Shimizu J, Hatanaka Y, Hasegawa M, Iwata A, Sugimoto I, Date H et al. IFNbeta‑1b may severely exacerbate Japanese optic‑ spinal MS in neuromyelitis optica spectrum. Neurology 2010; 75(16): 1423– 1427. doi: 10.1212/ WNL.0b013e3181f8832e.
38. Papeix C, Vidal JS, de Seze J, Pierrot‑ Deseilligny C, Tourbah A, Stankoff B et al. Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler 2007; 13(2): 256– 259.
39. Kleiter I, Hellwig K, Berthele A, Kumpfel T, Linker RA, Hartung HP et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 2012; 69(2): 239– 245. doi: 10.1001/ archneurol.2011.216.
40. Barnett MH, Prineas JW, Buckland ME, Parratt JD, Pollard JD. Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult Scler 2012; 18(1): 108– 112. doi: 10.1177/ 1352458511421185.
41. Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler 2012; 18(1): 113– 115. doi: 10.1177/ 1352458511431973.
42. Wingerchuk DM, Weinshenker BG. Neuromyelitis optica. Curr Treat Options Neurol 2008; 10(1): 55– 66.
43. Pellkofer HL, Krumbholz M, Berthele A, Hemmer B,Gerdes LA, Havla J et al. Long‑term follow‑up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 2011; 76(15): 1310– 1315. doi: 10.1212/ WNL.0b013e3182152881.
44. Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5‑year follow‑up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol 2013; 70(9): 1110– 1117.
45. Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol 2010; 17(8): 1019– 1032. doi: 10.1111/ j.1468‑ 1331.2010.03066.x.
46. Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF et al. Eculizumab in AQP4- IgG‑ positive relapsing neuromyelitis optica spectrum disorders: an open‑ label pilot study. Lancet Neurol 2013; 12(6): 554– 562. doi: 10.1016/ S1474‑ 4422(13)70076‑ 0.
47. Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M et al. Efficacy of the anti‑IL‑6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology 2014; 82(15): 1302– 1306. doi: 10.1212/ WNL.0000000000000317.
48. Greco R, Bondanza A, Oliveira MC, Badoglio M, Burman J, Piehl F et al. Autologous hematopoietic stem cell transplantation in neuromyelitis optica: a registry study of the EBMT Autoimmune Diseases Working Party. Mult Scler 2014; 21(2): 189– 197. doi: 10.1177/ 1352458514541978.
49. Fragoso YD, Adoni T, Bichuetti DB, Brooks JB, Ferreira ML, Oliveira EM et al. Neuromyelitis optica and pregnancy. J Neurol 2013; 260(10): 2614– 2619. doi: 10.1007/s00415‑ 013‑ 7031‑ y.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2015 Číslo 2
Nejčtenější v tomto čísle
- Agresivní hemangiom obratle
- Neuromyelitis optica
- Kongenitální centrální hypoventilační syndrom (Ondinina kletba)
- Radiologické hodnocení lumbální spinální stenózy a jeho klinická korelace