
Dva případy atypického teratoidního rhabdoidního tumoru CNS a přehled literatury
Two Cases of CNS Atypical Theratoid Rhabdoid Tumor and Review of Literature
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
H. Zítek 1; D. Sumerauer 2; J. Zámečník 3; P. Vachata 1; M. Sameš 1; A. Puchmajerová 4
Působiště autorů:
Neurochirugická klinika UJEP a Masarykova nemocnice v Ústí nad Labem, o. z, Krajská zdravotní, a. s., Ústí nad Labem
1; Klinika dětské hematologie a onkologie 2. LF UK a FN Motol, Praha
2; Ústav patologie a molekulární medicíny, 2. LF UK a FN Motol, Praha
3; Ústav biologie a lékařské genetiky, 2. LF UK a FN Motol, Praha
4
Vyšlo v časopise:
Cesk Slov Neurol N 2017; 80/113(5): 605-607
Kategorie:
Dopis redakci
doi:
https://doi.org/10.14735/amcsnn2017605
Souhrn
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Vážená redakce,
atypický teratoidní rhabdoidní tumor (AT/RT) je vzácný maligní (WHO grade IV) embryonální nádor centrální nervové soustavy (CNS) dětského věku [1]. Jde o nádor agresivní, který se projevuje progresivní symptomatikou a vede zpravidla k infaustnímu konci během několika měsíců. Práce z posledních let však přinášejí optimističtější závěry týkající se celkového přežití těchto pacientů, a to díky intenzivní multimodální terapii (MT) [1–3].
Prezentujeme dvě kazuistiky, které mají mimo jiné upozornit odbornou neurologickou a neurochirurgickou společnost, aby zařadila tuto jednotku do diferenciálně-diagnostického uvažování v rámci neuroonkologie dětských pacientů.
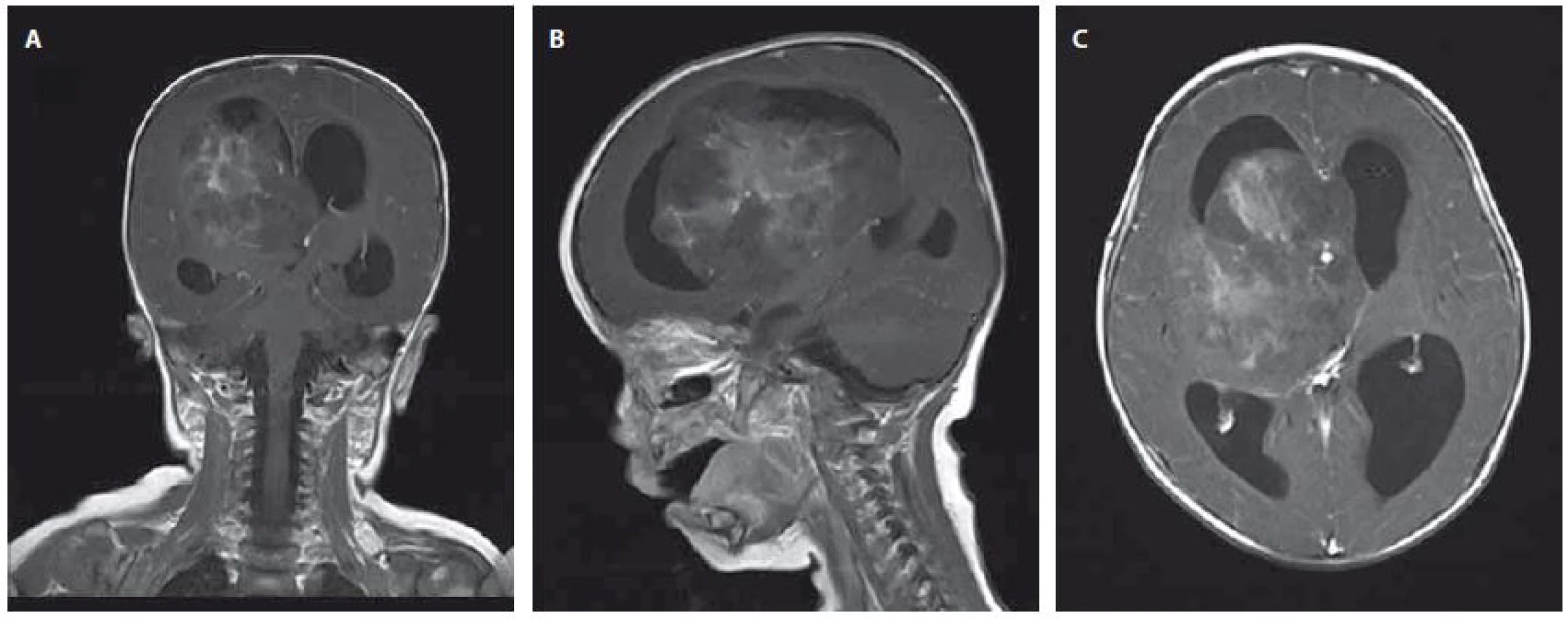
Devítiměsíční kojenec byl přijat na JIP dětské kliniky pro 3 dny trvající levostrannou hemiparézu, plačtivost a neklid. Při příjmu byl dále zjištěn obvod hlavičky nad 97. percentilem a regres v psychomotorickém vývoji. Na MR byla nalezena velká expanze zasahující do III. a pravé postranní komory se známkami akutního hydrocefalu a přesunem středočárových struktur. Po podání kontrastní látky se tumor nehomogenně sytí (obr. 1A–C). Během 24 hod klinický stav dále progredoval. Pacient prodělal parciální epileptický záchvat a na CT byl popsán obraz centrální tentoriální herniace s bilaterální mydriázou. Pacient byl zaintubován a byl indikován urgentní operační výkon z vitální indikace. Byla provedena subtotální resekce a bazální stomie III. komory. Na kontrolním CT však byl popsán obraz difúzního edému mozku s infaustní prognózou. Desátý den od prvních příznaků byla definitivně stanovena smrt mozku a pacient své nemoci podlehl. Výsledek histologie potvrdil klasifikaci tumoru jako AT/ RT s imunohistochemickou negativitou INI-1 vč. pozitivní mutační analýzy genu SMARCB1/INI1.
Druhý pacient, starý 23 měsíců, byl hospitalizován pro půl roku trvající neprospívání a opakované zvracení. Přivolaný dětský neurolog diagnostikoval centrální hypotonický syndrom, pravostrannou periferní parézu n. VII, horizontální nystagmus a parézu n. VI vpravo. Je provedena MR mozku s nálezem nehomogenně sytícího se ložiska v zadní jámě lební vpravo infiltrující pravou mozečkovou hemisféru a přilehlý kmen s vícečetnými metastázami. Komorový systém je supratentoriálně rozšířen se známkami incipientní dekompenzace hydrocefalu (obr. 2A).
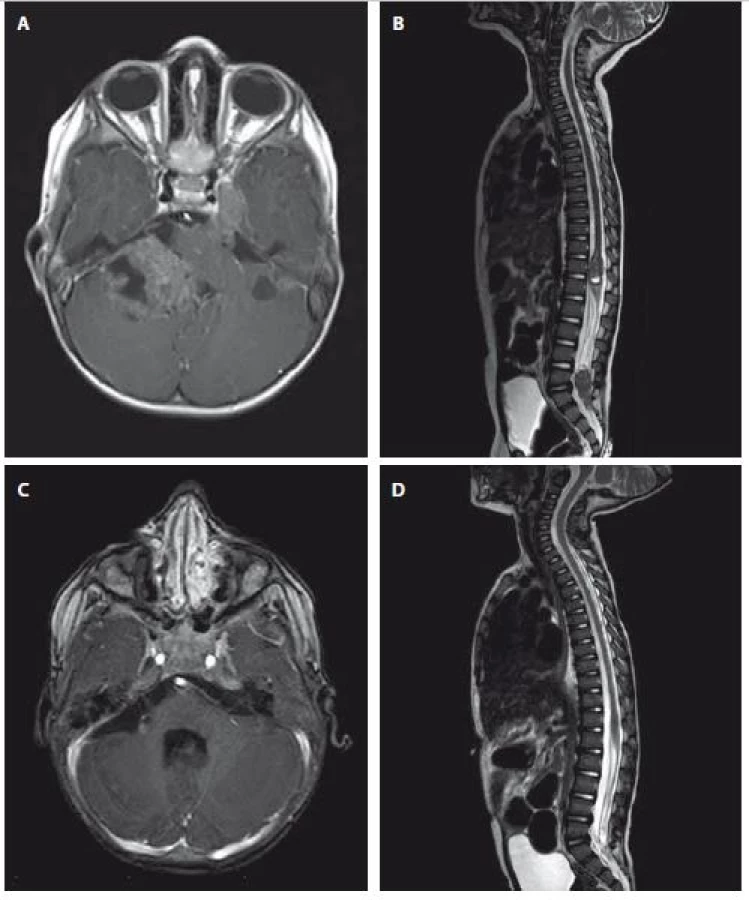
Nejprve byla provedena parciální resekce tumoru z retrosigmoidálního přístupu vpravo. Morfologický obraz a imunohistochemické vyšetření (negativita INI1) prokázaná mutace v genu SMARCB1/ INI1 vedly k diagnóze AT/RT. Při následných stagingových vyšetřeních byla objevena další metastatická ložiska v oblasti páteřního kanálu v oblasti Th11–L1 a L4–S2 a dále 4 % atypických nádorových elementů v mozkomíšním moku odebraném lumbální punkcí (obr. 2B).
Vzhledem k závažnému stavu dítěte byla bezprostředně zahájena chemoterapie kombinující systémové a intratékální podání cytostatik. Následné kontrolní MR mozku a míchy prokázalo postupnou významnou regresi nádoru ve všech lokalizacích (obr. 2C, D). Následně chlapec v parciální remisi a za zlepšujícího se klinického stavu podstoupil vysokodávkovanou chemoterapii s Stem Cell Rescue (SCR). Vzhledem k vysokému riziku recidivy onemocnění pokračoval chlapec v léčbě režimem kombinujícím nízcedávkovanou p.o. chemoterapii a intratékalní aplikaci cytosinarabinosidu. Radioterapie s ohledem na věk dítěte a rozsah ozařovacího pole nebyla v primární léčbě indikována.
Po přechodné stabilizaci onemocnění však došlo 13 měsíců od primární operace k progresi klinického obrazu. Na MR byla prokázána recidiva tumoru v mostomozečkovém koutu vpravo částečně infiltrující mozkový kmen. Vzhledem ke klinickému obrazu a infaustní prognóze při recidivě bylo po domluvě s rodiči pacienta rozhodnuto o ukončení onkologické terapie. Pacient v následujících dnech svému onemocnění podlehl.
AT/ RT tvoří méně než 5 % dětských tumorů CNS, ale až 20 % tumorů CNS u dětí do 3 let věku [1]. Diagnostika tohoto onemocnění se kromě klinického obrazu opírá především o radiodiagnostiku, histologický, molekulární a imunohistochemický rozbor bioptické tkáně a cytologický rozbor mozkomíšního moku.
AT/ RT postihuje typicky nejmenší děti, jejichž stav se často rychle zhoršuje [4]. Progresivní neurotopická symptomatika byla přítomna v obou námi prezentovaných případech.
AT/ RT se vyskytuje ve všech částech CNS bez výrazné predilekce [1,5–7]. Primární postižení míchy se vyskytuje v 4 % [4,6]. Na CT i MR zobrazení má tato léze heterogenní obraz. Četnost metastáz, po nichž by se mělo aktivně pátrat, se dle dostupné literatury pohybuje kolem 30 % [6,7]. Vícečetné metastatické postižení bylo diagnostikováno i v jedné z našich kazuistik.
Histologický obraz bioptické tkáně může být velmi proměnlivý. Patognomické pro tento tumor jsou rhabdoidní buňky, zastoupení těchto elementů je však ve vzorcích velmi variabilní, což dále ztěžuje diagnostiku [3]. Nedílnou součástí diagnostiky je také imunohistochemické vyšetření, které prakticky ve všech případech, vč. našich dvou kazuistik, prokáže absenci nukleární exprese INI-1 [4,7]. Tento marker je nejužitečnější v rámci diferenciální diagnostiky maligních tumorů CNS a byl pozitivní v obou námi popisovaných případech. Molekulární vyšetření dále prokáže mutaci či deleci v genu SMARCB1/ INI-1/ hSNF5 na chromozomu 22q1 [7].
Prvním terapeutickým krokem v rámci navrhované intenzivní MT bývá chirurgická resekce tumoru. V literatuře nejsou jednoznačné závěry, zda rozsah a radikalita operačního zákroku zlepšuje prognózu. Nicméně ve větších souborech pacientů není rozdíl v době přežití u pacientů léčených chemoterapií dle radikality operace signifikantní [2].
Rozhodující adjuvantní terapeutickou modalitou se ukazuje být chemoterapie. Výsledky recentně publikovaných studií se přiklánějí k vysokodávkované chemoterapii (HDC) nebo také kombinaci systémové a intratékální chemoterapie (IT). HDC byla vzhledem ke své toxicitě doplněna o autologní transplantaci hematopoetických kmenových buněk (SCR). Tato kombinace bývá spojena s lepší prognózou, ale počet zdokumentovaných případů zůstává malý [2]. U IT se předpokládá, že má vliv na vzdálený relaps onemocnění [6,8]. Náš druhý pacient taktéž podstoupil HDC doplněnou o SCR. V kombinaci s primární resekcí došlo k dočasné stabilizaci onemocnění s parciální regresí ložisek na MR.
Na rozdíl od chemoterapie je užití radioterapie (RT) kontroverznější. Toto onemocnění postihuje nejmladší pacienty, u kterých jsou dobře popsány nežádoucí účinky užití RT v podobě narušení vývoje CNS. V poslední době však autoři poukazují na souvislost mezi dobou přežití a užití RT v první linii adjuvantní terapie [9,10]. Vzhledem k riziku při použití RT u malých dětí mělo by vždy jít o vysoce fokální ozáření a difúzní ozáření ponechat pro starší pacienty [10].
AT/RT patří mezi vzácná a smrtící onemocnění. Na základě podobnosti s ostatními dětskými maligními nádory CNS se lze domnívat, že jde o poddiagnostikovanou jednotku. Odlišení od ostatním podobných jednotek je ale zásadní vzhledem k odlišnostem v prognóze a terapii tohoto tumoru. V poslední době se množí práce, které publikují optimističtější výsledky v léčbě, a to především zásluhou intenzivní MT.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Hynek Zítek
Neurochirugická klinika UJEP a Masarykova nemocnice, o.z, Krajská zdravotní a.s. Ústí nad Labem
Sociální péče 3316/12A
401 13 Ústí nad Labem
email: hynek.zitek@kzcr.eu
Přijato k recenzi: 4. 7. 2017
Přijato do tisku: 3. 8. 2017
Na webu www.csnn.eu najdete nezkrácenou verzi tohoto článku.
Zdroje
1. Buscariollo DL, Park HS, Roberts KB, et al. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 2012;118(17):4212– 19. doi: 10.1002/ cncr.27373.
2. Schrey D, Carceller Lechón F, Malietzis G, et al. Multimodal therapy in children and adolescents with newly diagnosed atypical teratoid rhabdoid tumor: individual pooled data analysis and review of the literature. J Neurooncol 2016;126(1):81– 90. doi: 10.1007/ s11060-015-1904-0.
3. Slavc I, Chocholous M, Leiss U, et al. Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992– 2012. Cancer Med 2014;3(1):91– 100. doi: 10.1002/ cam4.161.
4. DiPatri AJ, Sredni ST, Grahovac G, et al. Atypical teratoid rhabdoid tumors of the posterior fossa in children. Childs Nerv Syst 2015;31(10):1717– 28. doi: 10.1007/ s00381-015-2844-x.
5. Ostrom QT, Chen Y, M de Blank P, et al. The de-scriptive epidemiology of atypical teratoid/ rhabdoid tumors in the United States, 2001-2010. Neuro Oncol 2014;16(10):1392– 9. doi: 10.1093/ neuonc/ nou090.
6. Lafay-Cousin L, Hawkins C, Carret AS, et al. Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 2012;48(3):353– 9. doi: 10.1016/ j.ejca.2011.09.005.
7. Dufour C, Beaugrand A, Le Deley MC, et al. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 2012;118(15):3812– 21. doi: 10.1002/ cncr.26684.
8. Chi SN, Zimmerman MA, Yao X, et al. Intensive multimodality treatment for children with newly diagnos-ed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 2009;27(3):385– 9. doi: 10.1200/ JCO.2008.18.7724.
9. von Hoff K, Hinkes B, Dannenmann-Stern E, et al. Frequency, risk-factors and survival of children with atypical teratoid rhabdoid tumors (AT/ RT) of the CNS diag-nosed between 1988 and 2004, and registered to the German HIT database. Pediatr Blood Cancer 2011 Dec 1;57(6):978– 85. doi: 10.1002/ pbc.23236.
10. Pai Panandiker AS, Merchant TE, Beltran C, et al. Sequencing of local therapy affects the pattern of treatment failure and survival in children with atypical teratoid rhabdoid tumors of the central nervous system. Int J Radiat Oncol Biol Phys 2012;82(5):1756– 63. doi: 10.1016/ j.ijrobp.2011.02.059.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2017 Číslo 5
Nejčtenější v tomto čísle
- Esenciální tremor – vzniká nový nozologický koncept?
- Leberova hereditární neuropatie optiku
- Statiny indukovaná nekrotizující autoimunitní myopatie
- Terapia pokročilých štádií Parkinsonovej choroby invazívnymi postupmi