
Neuromyelitis optica a poruchy jejího širšího spektra – retrospektivní analýza klinických a paraklinických nálezů
Neuromyelitis Optica Spectrum Disorders – Retrospective Analysis of Clinical and Paraclinical Findings
Aim:
The aim of this study was to describe the clinical picture of patients with NMO spectrum disorders (NMO SD) in the Czech Republic, including comparison of patients with disease onset before and after 50 years of age in the context of longitudinally extensive transverse myelitis (LETM) and optic nerve involvement. Another objective was to evaluate cerebrospinal fluid samples and aquaporin-4 antibody (AQP4-IgG) seropositivity.
Methods:
A systematic retrospective analysis of clinical and paraclinical findings in 32 patients with NMO SD was performed. The retinal nerve fiber layer (RNFL) thickness in each eye was measured with optical coherence tomography. Severity of neurological deficit was expressed by the EDSS score.
Results:
NMO SD are more frequent in women (8 : 1). Median age of disease onset was 36 years. Median disease duration was 10.5 years. Median EDSS score was 4.5 (range 1.0–8.5) and median disease duration 10.5 years (range 0.5–24). AQP4-IgG were found in the serum of 31 out of the 32 patients and their titer positively correlated with the EDSS score. The RNFL thickness of the right eye (median 63 µm) and the left eye (median 58 µm) was below the lower limit of normal (97 µm) in 57% and 64% of patients, respectively. The RNFL thickness correlated with the disease duration. Myelitis as the first manifestation of the disease was more frequent in patients older than 50 years (p < 0.05). Compared to remission, patients with a relapse had increased protein levels and element count in the cerebrospinal fluid (p < 0.001 for both parameters).
Conclusion:
LETM is typically the first clinical manifestation of NMO patients older than 50 years and is associated with severe neurological deficit at the early stages of the disease.
Key words:
neuromyelitis optica – antibodies against aquaporin-4 – longitudinally extensive transverse myelitis
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
P. Nytrová 1; P. Kleinová 1; J. Preiningerová Lízrová 1; V. Král 2; M. Hinďoš 3; M. Vaněčková 4; E. Havrdová 1; D. Horáková 1
![]()
Působiště autorů:
Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN v Praze
1; Centrum imunologie a mikrobiologie, Zdravotní ústav se sídlem v Ústí nad Labem
2; Laboratoř klinické imunologie a alergologie, Ústav lékařské biochemie a laboratorní diagnostiky, 1. LF UK a VFN v Praze
3; Oddělení magnetické rezonance, Radiodiagnostická klinika 1. LF UK a VFN v Praze
4
Vyšlo v časopise:
Cesk Slov Neurol N 2015; 78/111(1): 72-77
Kategorie:
Krátké sdělení
doi:
https://doi.org/10.14735/amcsnn201572
Souhrn
Cíl:
Cílem této práce je popis klinického obrazu pacientů s neuromyelitis optica a poruch jejího širšího spektra (NMO Spectrum Disorders; NMO SD) v České republice, včetně srovnání pacientů se začátkem onemocnění před 50. rokem života a po něm v kontextu výskytu longitudinálně extenzivní myelitis (LETM) a poškození zrakového nervu. Dalším cílem je hodnocení likvorových nálezů a séropozitivity protilátek proti akvaporinu‑ 4 (AQP4- IgG).
Soubor a metodika:
Byla provedena systematická retrospektivní analýza klinických a paraklinických nálezů 32 pacientů s NMO SD. Tloušťka nervových vláken sítnice (RNFL) každého oka byla měřena metodou optické koherentní tomografie. Tíže neurologického deficitu byla vyjádřena pomocí EDSS skóre.
Výsledky:
NMO SD častěji postihují ženy (8 : 1). Medián věku počátku onemocnění je 36 let. Medián délky trvání onemocnění je 10,5 roku. Medián EDSS skóre je 4,5 (rozmezí 1,0– 8,5) a délky onemocnění 10,5 let (rozmezí 0,5– 24). U 31 z 32 pacientů nacházíme AQP4- IgG v séru a jejich titr pozitivně koreluje s EDSS skóre. RNFL oka pravého (medián 63 µm) a oka levého (medián 58 µm) je sníženo pod dolní hranici normy (97 µm) u 57 %, resp. 64 % pacientů. RNFL koreluje s délkou choroby. Myelitida jako první manifestace onemocnění je častější u pacientů starších 50 let (p < 0,05). Ve srovnání s remisí nacházíme v likvoru během relapsů vyšší koncentraci bílkoviny a počet elementů (p < 0,001; pro oba parametry).
Závěr:
LETM je typicky první klinickou manifestací NMO u pacientů starších 50 let a je spojena s těžkým neurologickým deficitem již v úvodu onemocnění.
Klíčová slova:
neuromyelitis optica – protilátky proti akvaporinu-4 – longitudinálně extenzivní transverzální myelitis
Úvod
Neuromyelitis optica (NMO), též známá jako Devicova choroba, je autoimunitní onemocnění postihující převážně zrakový nerv a míchu a méně často mozkový kmen či jiné části centrálního nervového systému [1,2]. Autoři Lennon et al popsali sérový marker této choroby, kterým jsou protilátky proti akvaporinu‑ 4 (AQP4- IgG, též NMO‑ IgG) [3,4]. AQP4 je vodní kanál exprimovaný astrocyty v centrálním nervovém systému. Navázání AQP4- IgG na cílový antigen spouští imunitní kaskádu vedoucí k devastujícím zánětlivým změnám postižených struktur a invaliditě pacienta. Tyto protilátky nacházíme v séru až u 80 % pacientů a jsou také součástí revidovaných Wingerchukových kritérií pro NMO [5].
Klinický průběh je ve většině případů relaps remitentní, nicméně můžeme se také setkat s průběhem monofázickým, kdy myelitida a optická neuritida (ON) probíhají současně nebo těsně po sobě následují [2]. Ataky onemocnění mají těžký nebo středně těžký průběh a velmi často dochází pouze k částečné úpravě. Invalidita pacienta je zejm. následkem relapsů onemocnění. Jejich adekvátní terapie spolu s trvalou imunosupresivní léčbou snižuje jejich tíži i výskyt a zlepšuje prognózu pacientů.
Díky objevu AQP4- IgG byly ke „klasické“ NMO přiřazeny tzv. limitované formy, jako jsou izolované nebo rekurentní ON, longitudinálně extenzivní transverzální myelitidy (LETM) a méně časté poruchy endokrinní při lézi hypothalamu anebo kmenové encefalitidy s pozitivitou těchto protilátek. V roce 2007 byla Wingerchukem definována skupina poruch širšího spektra NMO (NMO Spectrum Disorders; NMO SD), do níž tedy kromě NMO řadíme pacienty s pozitivitou AQP4- IgG, kteří prodělali pouze ON nebo myelitidu [6].
V současnosti nemáme žádný prediktivní marker aktivity choroby. Nicméně dle práce Collonguese et al jsou negativními prediktivními faktory průběhu onemocnění věk počátku nad 50 let a vyšší počet atak za rok. V kohortě pacientů s mediánem sledování 4,6 let došlo k úmrtí z důvodu myelitidy nebo oportunní infekce u více jak 10 % pacientů [7].
Zájmem naší studie byla monocentrická retrospektivní analýza klinických a paraklinických nálezů pacientů s NMO SD, zvláště srovnání průběhu choroby u pacientů s počátkem před 50. rokem života a po něm. Analýza zahrnuje také hodnocení základních likvorových parametrů a tloušťky nervových vláken sítnice (Retinal Nerve Fibre Layer; RNFL) pro oko pravé i levé u těchto pacientů.
Materiál a metodika
Pacienti
Klinická data, nálezy na magnetické rezonanci, RNFL a likvorové nálezy byly hodnoceny u pacientů s NMO SD definovaných podle Wingerchuka [1,8]. Pacienti byli sledováni v Centru pro demyelinizační onemocnění 1. LF UK a VFN v Praze. Neurologický deficit byl vyjádřen pomocí škály Expanded Disability Status Scale (EDSS) [9]. Nálezy na magnetické rezonanci byly posouzeny radiologem s dlouholetou zkušeností v oblasti demyelinizačních onemocnění centrálního nervového systému.
Data pacientů byla vygenerována z databáze iMED (www.imed.org) a hodnocena retrospektivně. V době analýzy obsahovala databáze retrospektivně a prospektivně zadaná data 32 pacientů, z čehož revidovaná Wingerchukova kritéria pro NMO splňovalo 25 pacientů. Dalších sedm AQP4- IgG pozitivních subjektů bylo sledováno pro recidivující ON (n = 2), izolovanou (n = 2) nebo recidivující LETM (n = 2) a kmenovou encefalitidu (n = 1). Dvacet devět pacientů bylo původu indoevropského, dva asijského a jeden hispánského. Osm pacientů se začátkem onemocnění po 50. roce života jsme zařadili do skupiny NMO SD s pozdním začátkem.
Osmnáct subjektů bylo sledováno a léčeno pro diagnózu roztroušené sklerózy před rokem 2008, kdy bylo zavedeno rutinní testování AQP4- IgG v České republice.
Všichni pacienti podepsali informovaný souhlas se sledováním. Informovaný souhlas byl schválen Etickou komisí VFN v Praze.
Pomocná laboratorní vyšetření
Stanovení protilátek proti AQP4
AQP4- IgG v séru byly stanoveny u všech pacientů metodou nepřímé imunofluorescence na komerčně dostupných AQP4 transfekovaných buňkách izoformou M1 (Euroimmun, Lübeck, Německo). V séru byly stanoveny titry protilátek, které odpovídaly pozitivitě při nejvyšším ředění séra (pozitivní titr 1 : 10 a více). Pro statistické analýzy byly použity hodnoty z prvního dostupného vyšetření každého pacienta.
Vyšetření mozkomíšního moku
U 27 pacientů byla provedena lumbální punkce, u některých opakovaně. Celkem proběhlo 55 vyšetření mozkomíšního moku získaného během relapsů (n = 31) i remisí (n = 24). Byla hodnocena koncentrace celkové bílkoviny, albuminu, albuminový kvocient, počet elementů a přítomnost oligoklonálních pásů (OCB). Nález OCB v likvoru byl rozdělen do tří skupin: likvor bez průkazu OCB (vzorec I), OCB v likvoru, které nejsou přítomny v séru nebo průkaz OCB v obou kompartmentech a některé jen v likvoru (vzorec II + III) a průkaz identických OCB v likvoru i séru (vzorec IV). Počet elementů a celková bílkovina byly hodnoceny ve vztahu k aktivitě onemocnění (relaps/ remise) v okamžiku lumbální punkce.
Vyšetření optickou koherentní tomografií
Míra poškození zrakového nervu byla kvantifikována pomocí optické koherentní tomografie (Heidelberg Spectralis OCT) a vyjádřena parametrem celkové tloušťky retinálních nervových vláken sítnice (RNFL). Tloušťku retinálních nervových vláken jsme hodnotili na kružnici o průměru 3,4 mm v okolí hlavy optického nervu a tloušťku sítnice (ohraničenou vnitřní limitující membránou a retinálním pigmentovým epitelem) v oblasti makuly. Vyšetření bylo provedeno u 28 pacientů s minimálním odstupem tří měsíců od poslední ON.
Statistická analýza
Data byla analyzována pomocí programu SoftSTAT. Statistická analýza byla považována za průzkumnou analýzu dat a nebylo použito korekce pro mnohočetné testování. Mannův‑ Whitneyho U test byl použit pro stanovení signifikantní významnosti pro skupinové srovnání. Korelační analýza byla provedena pomocí Spearmanova testu.
Výsledky
Demografická data a počátek onemocnění
Poměr žen a mužů ve sledované skupině NMO SD byl 8 : 1. Medián počátku onemocnění byl 36 let (rozmezí 19– 70 let). Z toho 8 z 32 (25 %) nemocných bylo starších 50 let. ON byla prvním příznakem onemocnění u 13 z 32 (40,5 %), zatímco myelitis u 12 z 32 (37,5 %) nemocných. U pěti pacientů (15,5 %) byly první manifestací kmenové příznaky, zejm. singultus s nauzeou nebo vomitem. Dva pacienti (6,5 %) souběžně rozvinuli ON a myelitidu. Myelitida jako první příznak choroby byla signifikantně častější u skupiny NMO SD s pozdním začátkem (p < 0,05). Výsledky jsou shrnuty v tab. 1.

Léčba pacientů
Většina pacientů byla léčena různými léčebnými režimy také proto, že správná diagnóza byla stanovena až po zavedení testování AQP4- IgG do rutinní praxe. Čtyři pacientky byly léčeny interferonem beta. Léčba byla ukončena pro neefektivitu (těžký relaps) nebo v jednom případě pro těžký flu‑like syndrom. Jedna pacientka byla léčena glatiramer acetátem bez efektu na frekvenci relapsů. Část pacientů původně s diagnózou roztroušené sklerózy byla léčena nízkodávkovanými kortikoidy, někdy v kombinaci se subterapeutickou dávkou azathioprinu. Přehled léčby v okamžiku analýzy souboru je uveden v tab. 2.
Průběh choroby
Relaps remitentní průběh byl zaznamenán u 29/ 32 pacientů (91 %). U tří žen s doposud jedinou atakou onemocnění byl medián délky trvání choroby 13 měsíců. Medián doby dosažení druhého relapsu onemocnění byl necelých 11 měsíců (s rozmezím 2– 60 měsíců). Všichni pacienti s pozdním začátkem onemocnění (po 50. roce života) rozvinuli v průběhu onemocnění LETM. Myelitidy u těchto pacientů vedly vždy k poměrně závažným reziduálním postižením, a to i v důsledku jediné ataky. U této skupiny byl tři měsíce po atace medián EDSS skóre 5,5 (rozmezí 4,0– 7,0). U pacientů s počátkem nemoci před 50. rokem života dosahovala tíže postižení během míšní ataky od lehčích senzitivních příznaků až po těžkou myelitidu s kvadruplegií a nutností umělé plicní ventilace pro neurogenně podmíněné respirační selhání.
Nebyl signifikantní rozdíl v tíži neurologického deficitu u skupiny pacientů s délkou choroby více nebo méně než pět let. Medián EDSS skóre u pacientů s více než pětiletým průběhem choroby byl 4,5 (1,5– 8,5) a u pacientů s průběhem kratším pěti let byl 4,0 (1,0– 6,5). Medián délky choroby u 11 pacientů s invaliditou odpovídající EDSS ≥ 5,0 (interval chůze bez opory a odpočinku minimálně 200 m) byl 5,3 let.
Koincidence s jiným autoimunitním onemocněním je u NMO častá. Dvacet jedna pacientů (66 %) bylo sledováno pro jednu a více autoimunitních komorbidit (tab. 3), které předcházely rozvoji neurologické symptomatiky o několik měsíců až let. Dvě pacientky v předchorobí prodělaly thymektomii pro diagnózu myasthenia gravis.
Longitudinálně extenzivní transverzální myelitis
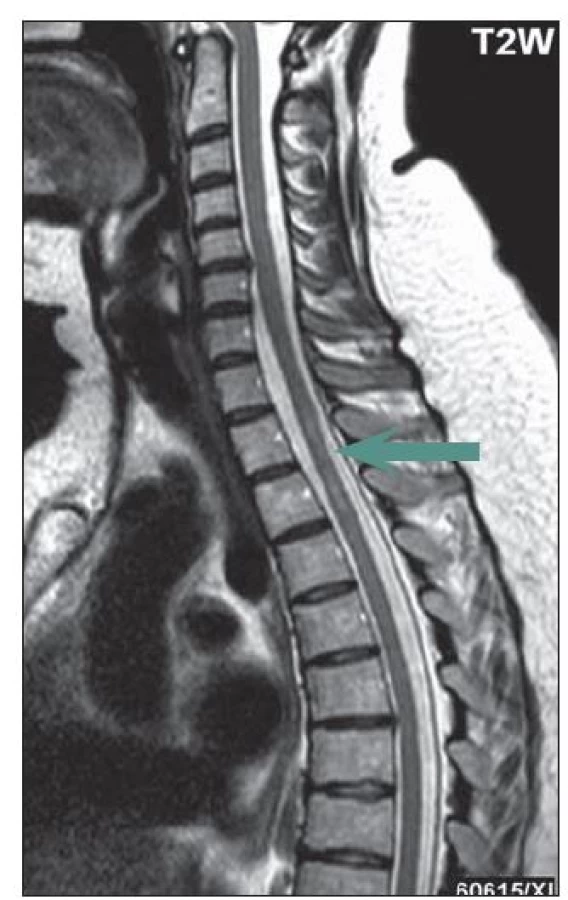
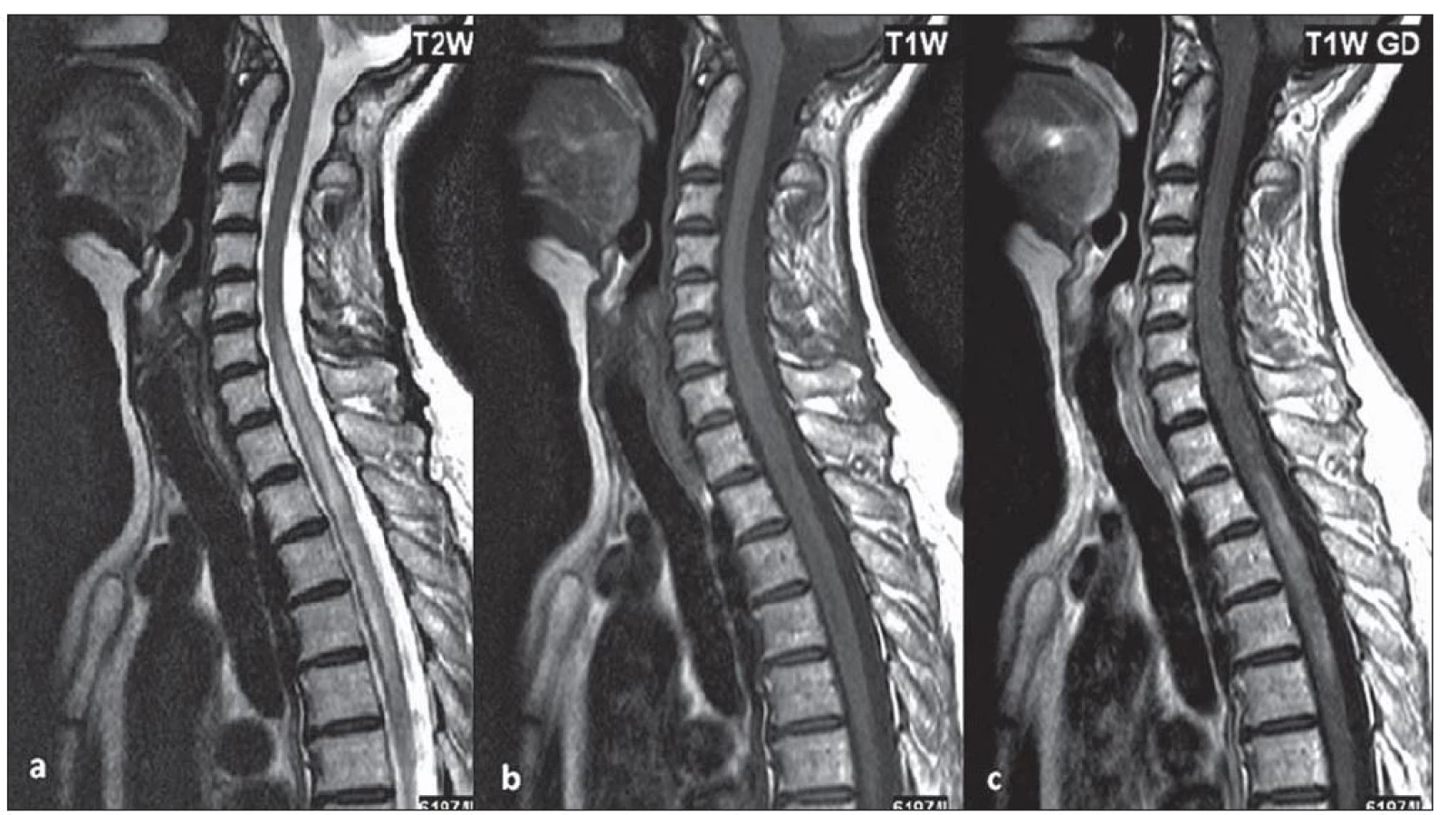
U části pacientů s LETM může dojít na MR míchy k významné regresi nálezu v důsledku léčby, a to zejm. ve skupině pacientů s počátkem onemocnění před 50. rokem života. U některých pacientů bylo na MR míchy zachyceno pouze rozšíření centrálního kanálu nebo myelitida nepřesahující tři obratlové segmenty, častěji u nemocných, kteří byli na imunosupresivní léčbě pro předcházející ON (obr. 1). Typicky se setkáváme s gadolinium enhancujícími lézemi, pokud je magnetická rezonance provedena během relapsu myelitidy (obr. 2a– c).
Likvorové nálezy
Celková bílkovina v likvoru u pacientů s NMO byla v relapsu signifikantně vyšší než v remisi (p < 0,001; rozmezí 210– 2 430 mg/ l), graf 1. Taktéž koncentrace albuminu a albuminový kvocient byly vyšší v okamžiku relapsů (p = 0,002; rozmezí 203– 1 540 mg/ l, resp. p < 0,001; rozmezí 3,90– 40,63). Počet elementů v likvoru byl také signifikantně vyšší v okamžiku relapsu v porovnání s remisí (p < 0,001; rozmezí 1– 182 elementů/ 1µl), graf 2.


OCB nebyly nalezeny ve 21 (40 %) vyšetřených vzorcích likvoru (vzorec I). Přítomnost OCB izolovaně v likvoru nebo s částečně shodným zastoupením v séru (vzorec II + III) byla zjištěna ve 20 případech (38 %) a shodná přítomnost OCB v séru a likvoru (vzorec IV) ve 12 vzorcích (22 %). U dvou vyšetření nebyl výsledek OCB dostupný. V rámci kvalitativního cytologického hodnocení byla u některých pacientů zaznamenána přítomnost neutrofilních a eozinofilních granulocytů.
Tloušťka nervových vláken sítnice
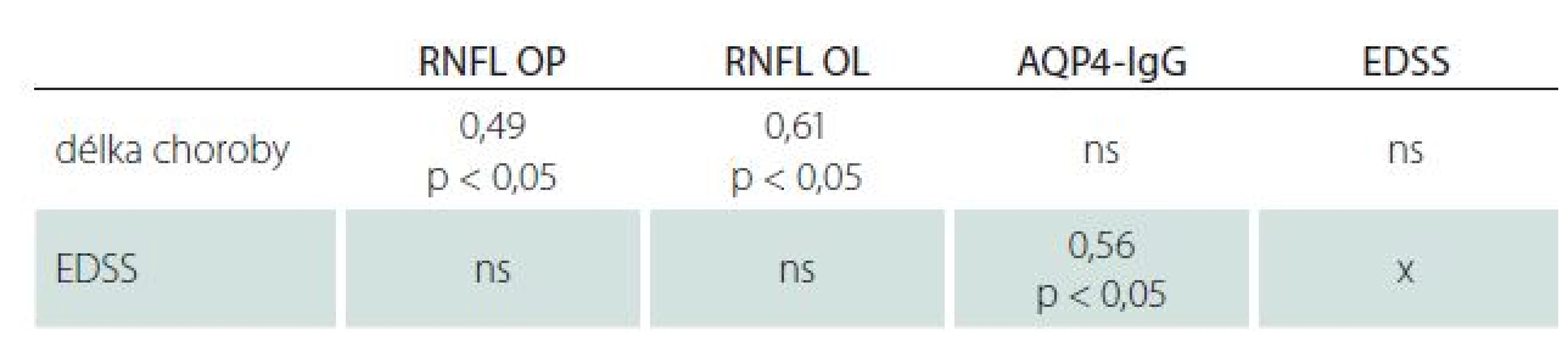
Celkové RNFL bylo sníženo pod dolní hranici normy (97 µm) pro oko pravé (OP) u 57 % a pro oko levé (OL) u 64 % pacientů. Medián RNFL OP byl 63 µm (35– 113 µm) a 58 µm pro OL (31– 110 µm). Pro OP ani OL nebyl zjištěn statisticky významný rozdíl RNFL u pacientů s počátkem choroby před 50. rokem života a po něm. Pokles RNFL pro OP i OL koreloval s délkou trvání choroby (tab. 4).
Protilátky proti akvaporinu‑ 4
U 31 pacientů (97 %) byly v séru prokázány protilátky proti AQP4 (medián titru 80, rozmezí 10– 2 560). Titr protilátek koreloval s tíží neurologického deficitu vyjádřeného pomocí EDSS skóre (graf 3). Neprokázali jsme závislost na délce choroby. Nebyla prokázána korelace mezi poklesem RNFL a titrem sérových protilátek. U čtyř pacientek došlo k vymizení protilátek, které nebyly detekovatelné ve více než dvou dalších odběrech v minimálním odstupu dvou měsíců.

Diskuze
V naší práci shrnujeme základní klinická data a paraklinické nálezy pacientů s NMO a poruch jejího širšího spektra sledovaných v Centru pro demyelinizační onemocnění 1. LF UK a VFN v Praze.
Jedná se o onemocnění převážně postihující ženy s první klinickou manifestací prakticky v kterékoliv dekádě života. U pacientů s pozdním počátkem onemocnění je typická LETM s výrazným reziduálním nálezem. Naše zjištění podporuje práce Kitleyho et al, která poukazuje na věk počátku onemocnění jako prediktivní faktor typu invalidity u pacientů ve Velké Británii a Japonsku. Konkrétně u pacientů s počátkem NMO do 30. roku života je typické výrazné postižení vizu, naopak u pacientů s pozdním začátkem je typická invalidita motorická a vyšší riziko úmrtí [10]. Collongues et al se ve své práci přímo soustřeďují na prognózu pacientů s NMO s počátkem onemocnění ve věku před 50. rokem života a po něm. U skupiny s pozdním začátkem je relativně mírné postižení vizu a naopak vysoké riziko motorické invalidity a úmrtí (v důsledku postižení CNS nebo oportunní infekce) [7]. Vysvětlení může souviset s věkem vázanou remodelací cévní stěny, integrity endotelové bariéry a poklesu její funkce [11]. Víme, že průnik AQP4- IgG endotelovou bariérou a navázání na výběžky astrocytů je stěžejní v patogenezi NMO [4]. Přesné molekulární mechanizmy však známy nejsou. Výskyt myelitidy nedosahující na T2 vážených obrazech tří obratlových segmentů může mít souvislost s imunosupresivní léčbou pro předcházející optickou neuritidu nebo další autoimunitní onemocnění v předchorobí [12] nebo načasováním vyšetření mimo relaps [13]. U akutních myelitid vídáme na MR míchy gadolinium enhancement, který se na terapii kortikoidy a plazmaferézami redukuje. Také může při úspěšné terapii dojít k regresi ložiska na T2 vážených sekvencích, nicméně často provázené atrofií zejm. v oblasti hrudní míchy.
V likvoru je obraz porušené hematoencefalické bariéry u NMO SD typicky vyjádřen zvýšenou koncentrací bílkoviny a albuminu během relapsů, a to zvláště u ataky myelitidy. U těžkých myelitid může koncentrace bílkoviny nebo albuminu dosahovat vysokých hodnot (kolem 2 000 mg/ ml, respektive 1 500 mg/ ml) a současně s přítomností neutrofilních granulocytů v likvoru mohou napodobovat likvorový nález infekční myelitidy. Byla provedena analýza všech 55 likvorových nálezů (tzn. u několika pacientů s více než jedním vyšetřením). Oligoklonální pásy byly přítomny v 38 % vzorků. U některých pacientů byla jejich pozitivita přechodná a naopak u některých přetrvávala i v době dlouhodobé remise. Podíváme‑li se pouze na přítomnost OCB u jednotlivých pacientů, u 11 z 27 (41 %) byly minimálně jednou OCB v likvoru nalezeny. Intratékální syntéza protilátek pravděpodobně navazuje na tkáňovou destrukci různého stupně, nicméně ve většině případů je přechodná a vázaná na relaps [14]. Předchozí práce uvádějí pozitivitu OCB asi u 35 % pacientů včetně možnosti vymizení pásů v průběhu onemocnění [2,15]. Přechodná produkce OCB může souviset s porušením hematoencefalické bariéry a indukcí imunitní odpovědi „odkrytím nových antigenů“ a následnou úklidovou reakcí, nicméně pravděpodobně nedochází u většiny pacientů k rozvoji sekundárních germinálních center s přítomností dlouhožijících plazmatických buněk. Absence OCB v likvoru jsou pro NMO typické, ale naopak jejich přítomnost diagnózu NMO nevylučuje.
Na histologických řezech lézí u NMO můžeme vidět různý stupeň ztráty astrocytů, demyelinizace a tkáňové nekrózy [6]. Právě tkáňová nekróza podmiňuje trvalý neurologický deficit. Titr AQP4- IgG pozitivně koreluje s výší EDSS skóre, které ovšem nekoreluje s délkou choroby. Tato skutečnost je pravděpodobně dána závažnou invaliditou již po první atace LETM u skupiny pacientů s pozdním začátkem choroby. U dvou pacientek léčených anti‑CD20 terapií (rituximabem) došlo k poklesu AQP4- IgG pod detekční limit vyšetřovací metody. U třetí pacientky došlo k vymizení protilátek po prodělané heterologní transplantaci kostní dřeně a u čtvrté spontánně bez léčby.
Klasický průběh současně nebo těsně po sobě probíhající myelitidy a optické neuritidy dle původního Devicova konceptu vídáme u minority pacientů. Naopak se často můžeme setkat s klinicky limitovanými formami a jejich zařazení do skupiny NMO SD je stěžejní nejen pro správnou léčbu relapsu, ale i volbu chronické imunosupresivní léčby odlišné od terapie roztroušené sklerózy, která prevencí atak zabrání rozvoji těžkého neurologického deficitu nebo jeho dalšímu zhoršení.
Závěr
Naše výsledky podporují dosud publikované práce o vlivu věku na manifestaci onemocnění a tíži motorického deficitu u NMO pacientů s pozdním začátkem, které je v této skupině závažnější. Rutinní testování AQP4- IgG významně zlepšilo diagnostiku tohoto onemocnění.
Poděkování
Podpořeno Grantovou agenturou grantem Univerzity Karlovy (GAUK 132010), grantem IGA MZ ČR NT13239- 4, PRVOUK‑ P26/ LF1/ 4 a výzkumným záměrem RVO‑ VFN64165.
MUDr. Petra Nytrová
Neurologická klinika a Centrum klinických neurověd
1. LF UK a VFN v Praze
Kateřinská 30
120 00 Praha 2
e-mail: Petra.Nytrova@lf1.cuni.cz
Přijato k recenzi: 19. 8. 2014
Přijato do tisku: 23. 10. 2014
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Zdroje
1. Wingerchuk DM. Diagnosis and treatment of neuromyelitis optica. Neurologist 2007; 13(1): 2– 11.
2. Wingerchuk DM, Hogancamp WF, O‘Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic‘s syndrome). Neurology 1999; 53(5): 1107– 1114.
3. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364(9451): 2106– 2112.
4. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SROV. IgG marker of optic‑ spinal multiple sclerosis binds to the aquaporin‑4 water channel. J Exp Med 2005; 202(4): 473– 477.
5. Waters P, Jarius S, Littleton E, Leite MI, Jacob S, Gray B et al. Aquaporin‑4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol 2008; 65(7): 913– 919. doi: 10.1001/ archneur.65.7.913.
6. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM et al. A role for humoral mechanisms in the pathogenesis of Devic‘s neuromyelitis optica. Brain 2002; 125(7): 1450– 1461.
7. Collongues N, Marignier R, Jacob A, Leite M, Siva A,Paul F et al. Characterization of neuromyelitis optica and neuromyelitis optica spectrum disorder patients with a late onset. Mult Scler 2013 [online]. Available from URL: http:/ / msj.sagepub.com/ content/ early/ 2013/ 12/ 09/ 1352458513515085.full.pdf+html.
8. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66(10): 1485– 1489.
9. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33(11): 1444– 1452.
10. Kitley J, Leite MI, Nakashima I, Waters P, McNeillis B, Brown R et al. Prognostic factors and disease course in aquaporin‑4 antibody‑ positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012; 135(6): 1834– 1849. doi: 10.1093/ brain/ aws109.
11. Blau CW, Cowley TR, O‘Sullivan J, Grehan B, Browne TC, Kelly L et al. The age‑related deficit in LTP is associated with changes in perfusion and blood‑ brain barrier permeability. Neurobiol Aging 2012; 33(5): e1023– e1035. doi: 10.1016/ j.neurobiolaging.2011.09.035.
12. Zavada J, Nytrova P, Wandinger KP, Jarius S, Svobodova R, Probst C et al. Seroprevalence and specificity of NMO‑ IgG (anti‑aquaporin 4 antibodies) in patients with neuropsychiatric systemic lupus erythematosus. Rheumatol Int 2013; 33(1): 259– 263. doi: 10.1007/ s00296‑ 011‑ 2176‑ 4.
13. Vaněčková M, Horáková D, Havrdová E, Nytrová P, Němcová J, Seidl Z. Retrospektivní studie nálezů na magnetické rezonanci míchy a mozku u pacientů s diagnózou neuromyelitis optica. Cesk Slov Neurol N 2010; 73/ 106(2): 164– 168.
14. Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R et al. Cerebrospinal fluid findings in aquaporin‑4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 2011; 306(1– 2): 82– 90. doi: 10.1016/ j.jns.2011.03.038.
15. Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol 2010; 17(8): 1019– 1032. doi: 10.1111/ j.1468‑ 1331.2010.03066.x.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2015 Číslo 1
Nejčtenější v tomto čísle
- Protokol diagnostiky a léčby hyponatremie a hypernatremie v neurointenzivní péči
- Mini‑Mental State Examination – česká normativní studie
- Autoimunitní encefalitidy
- Asymptomatická spondylogenní komprese krční míchy