
Nemalinová myopatie asociovaná s monoklonální gamapatií – kazuistika
Nemaline Myopathy Associated with Monoclonal Gammopathy – a Case Report
Nemaline (rod) myopathy is an uncommon muscle disease with a wide spectrum of phenotypes. Since 1966 until 2008, 71 patients with sporadic late‑ onset nemaline myopathy have been described. Among them, 12 had monoclonal gammopathy. This case report describes a 64‑year‑ old man with IgG kappa monoclonal gammopathy of undetermined significance, who developed progressive muscle weakness and symmetric hypotrophy, mainly of the proximal limb muscles a year after diagnosis. Muscle biopsy and electron microscopy revealed the presence of numerous typical rod‑ shaped inclusions in muscle fibers and the diagnosis of nemaline myopathy was established. The patient did not respond to therapy with corticosteroids, application of IVIG had also been considered. The case report is supplemented with basic information related to sporadic late‑ onset nemaline myopathy.
Key words:
nemaline myopathy – monoclonal gammopathy – muscle biopsy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
M. Forgáč 1; T. Uher 1; J. Zámečník 2
Působiště autorů:
Neurologická klinika 1. LF UK a VFN v Praze
1; Ústav patologie a molekulární medicíny 2. LF UK a FN v Motole, Praha
2
Vyšlo v časopise:
Cesk Slov Neurol N 2014; 77/110(2): 247-250
Kategorie:
Kazuistika
Práce byla podpořena granty VZ MŠM 0021620849, PRVOUK‑ P26/ LF/ 4 a projektem (Ministerstva zdravotnictví ČR) koncepčního rozvoje výzkumné organizace 00064203 (FN Motol).
Souhrn
Nemalinová (rod) myopatie je vzácné svalové onemocnění se širokým spektrem fenotypů. Od roku 1966 do roku 2008 bylo popsáno 71 případů sporadické formy nemalinové myopatie se začátkem v dospělosti (sporadic late onset nemaline myopathy), z toho u 12 pacientů byla myopatie asociována s monoklonální gamapatií. Popisujeme kazuistiku 64letého muže s monoklonální gamapatií IgG kappa neurčeného významu. Jeden rok po stanovení této diagnózy se u nemocného rozvinula progredující svalová slabost a symetrické hypotrofie převážně proximálního pletencového svalstva končetin. Diagnóza nemalinové myopatie byla stanovena svalovou biopsií, kde dominoval nález typických tyčinkovitých inkluzí na úrovni elektronového mikroskopu. Nemocný nereagoval na terapii kortikosteroidy, zvažovala se aplikace IVIG. Kazuistika je doplněna základními informacemi, které se týkají sporadické formy nemalinové myopatie se začátkem v dospělosti.
Klíčová slova:
nemalinová myopatie – monoklonální gamapatie – svalová biopsie
Úvod
Nemalinová (též tyčinková) myopatie byla poprvé popsána v roce 1963 jako neprogredující myopatie dětského věku [1,2].
Název byl odvozen od vzhledu tmavě červenomodrých struktur připomínajících vlákno příze (řecky nema to) patrných ve tkáňovém řezu barveném Gömöriho trichromem [3]. Je to velmi heterogenní skupina myopatií (klinicky i etiopatogeneticky) charakterizovaná přítomností tyčinkovitých inkluzí ve svalových vláknech.
Tradičně se rozeznávají tři formy nemalinové myopatie:
- forma kongenitální s rychlým začátkem a fatálním průběhem,
- mírná forma s pomalu progredující svalovou slabostí,
- forma se začátkem v dospělosti [4,5], známá jako Sporadic Late Onset Nemaline Myopathy (SLONM) [6].
Časné formy mají většinou genetický základ (identifikováno bylo už sedm kauzálních genů) [7]. U SLONM je však situace jiná, předpokládá se degenerativní či autoimunitní etiologie.
Ve svalové biopsii je typický nález atrofie svalových vláken, často s centrálně uloženými jádry, ve kterých jsou v barvení Gömöriho trichromem přítomny vláknité struktury připomínající vlákna příze. Elektronmikroskopicky odpovídají tyto inkluze tyčinkovitým tělískům doutníkovitého tvaru (tyčinky = rods), proto název nemalinová myopatie není přesný.
Tyčinkovité inkluze vycházejí ze Z linie sarkomery a obsahují a‑ aktinin, tropomyozin a desmin. Vzácně mohou být přítomny i známky zánětu [3,8– 10].
Histopatologické nálezy u kongenitálních forem nemalinové myopatie a forem se začátkem v dospělosti jsou prakticky shodné [11].
V roce 1966 W. K. Engel a Resnick na jedné straně a A. G. Engel na druhé nezávisle na sobě informovali o subakutně probíhající myopatii se začátkem v dospělosti – Sporadic Late Onset Nemaline Myopathy – SLONM (s nálezem tyčinek – „rods“ v atrofovaných vláknech) [12,13].
Od roku 1966 do roku 2008 bylo popsáno 71 případů sporadické formy nemalinové myopatie se začátkem v dospělosti, z toho u 12 pacientů byla myopatie asociována s monoklonální gamapatií [6].
SLONM je vzácná idiopatická myopatie. Klinicky je charakterizována subakutní svalovou slabostí, která postihuje proximální i distální končetinové svaly a axiální svalstvo (typicky oslabení extenzorů šíje). Může vést k respiračnímu selhání a bývá provázena i kardiomyopatií [3,14]. Raritně může být prvním příznakem SLONM i progresivní zevní oftalmoplegie [15].
Začátek onemocnění je obvykle po 40. roku života [16]. I když většina nemalinových myopatií se začátkem v dospělosti byly sporadické formy, bez familiárního výskytu, u jednoho nemocného byla verifikována ACTA1 mutace [17].
Kreatinkináza v séru je nejčastěji normální, ojediněle lehce zvýšená, elektromyografie může ukazovat myogenní změny a denervace (fibrilace) [18].
V roce 1975 W. K. Engel a Oberc publikují první případ pacienta postiženého nemalinovou myopatií se začátkem v dospělosti, která byla asociována s monoklonální gamapatií [8]. Od té doby bylo publikováno několik dalších případů nemalinové myopatie asociované s monoklonální gamapatií [6].
V největší dlouhodobě sledované skupině (1975– 2003) 14 nemocných se SLONM z Mayo Clinic mělo sedm nemocných nemalinovou myopatii asociovanou s monoklonální gamapatií. U této skupiny 14 nemocných nebyly pozorovány známky svědčící pro mnohočetný myelom, ani systémovou amyloidózu. Monoklonální gamapatie byla prezentována jak s řetězci IgG kappa, tak i lambda [16].


Kazuistika
Čtyřiašedesátiletý nemocný s uvedenou diagnózou neměl žádné pozoruhodnosti v osobní a rodinné anamnéze. Pro dlouhodobé bolesti bederní páteře byla u nemocného v rámci širší diferenciální diagnostiky vedle dalších vyšetření provedena i imunoelektroforéza bílkovin séra, kde byla zjištěna přítomnost paraproteinu IgG kappa se sérovou koncentrací tohoto monoklonálního imunoglobulínu 2,2 g/ l. V moči paraprotein detekován nebyl.
Následné provedení punkce kostní dřeně sice prokázalo 1% výskyt buněk s fenotypem plazmocytů, neprokázalo ale změny, které by svědčily pro diagnózu myelomu.
Doplněná zobrazovací vyšetření (RTG a MR) celé páteře nezobrazila žádná osteolytická ložiska.
Na základě provedených vyšetření byl nález paraproteinu v séru uzavřen jako monoklonální gamapatie IgG kappa neurčeného významu (Monoclonal Gammopathy of Undetermined Significance, MGUS).
Přibližně rok po stanovení diagnózy MGUS se u nemocného objevila progredující svalová slabost stehenního a postupně i pletencového svalstva horních končetin.
Během následujících 10 měsíců došlo k oslabení flexe v kyčli a extenze v koleni oboustranně na 50 % maximální svalové síly. Abdukce v rameni a flexe v lokti se oboustranně oslabila na 75 % maximální svalové síly.
Rozvinuly se svalové atrofie akcentované v oblasti pažních pletenců a kvadricepsů, bez výraznějších myalgií.
Nemocný neměl postiženo axiální svalstvo, byl bez dechových a polykacích obtíží. Opakovaně v tříměsíčních intervalech absolvoval spirometrické vyšetření.
Usilovná vitální kapacita plic se při každém vyšetření pohybovala nad 80 %. Nedošlo k rozvoji chronické respirační insuficience jako komplikace základního (svalového) onemocnění.
Základní laboratorní skríning, včetně hladiny kreatinkinázy v séru, testů na HIV infekci a likvorologického vyšetření byl v normě.
Provedené EMG nevykazovalo abnormity motorických a senzitivních neurogramů.
Při vyšetření koncentrickou jehlovou elektrodou byly přítomny ojedinělé denervace (fibrilace, pozitivní ostré vlny), myogenní změny MUPs (potenciály se sníženou amplitudou a zkráceným trváním) a předčasný nábor motorických jednotek.
Na horních končetinách byla myogenní léze s maximem v musculus deltoideus bilaterálně a biceps brachii vlevo, na dolních končetinách byly změny nejvíc vyjádřeny v musculus vastus medialis a lateralis oboustranně.
Na základě EMG nálezů a klinického stavu byla indikována svalová biopsie z musculus biceps brachii vlevo.
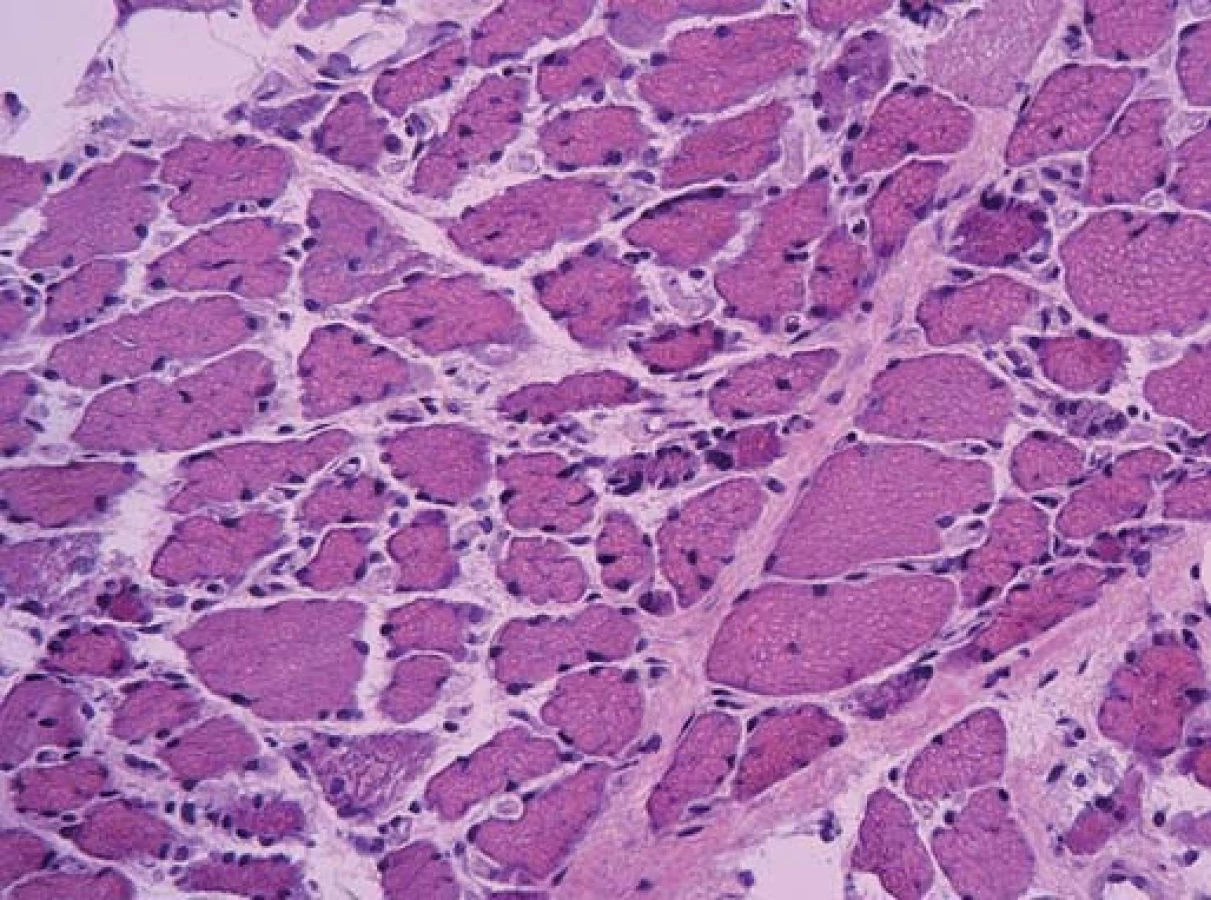
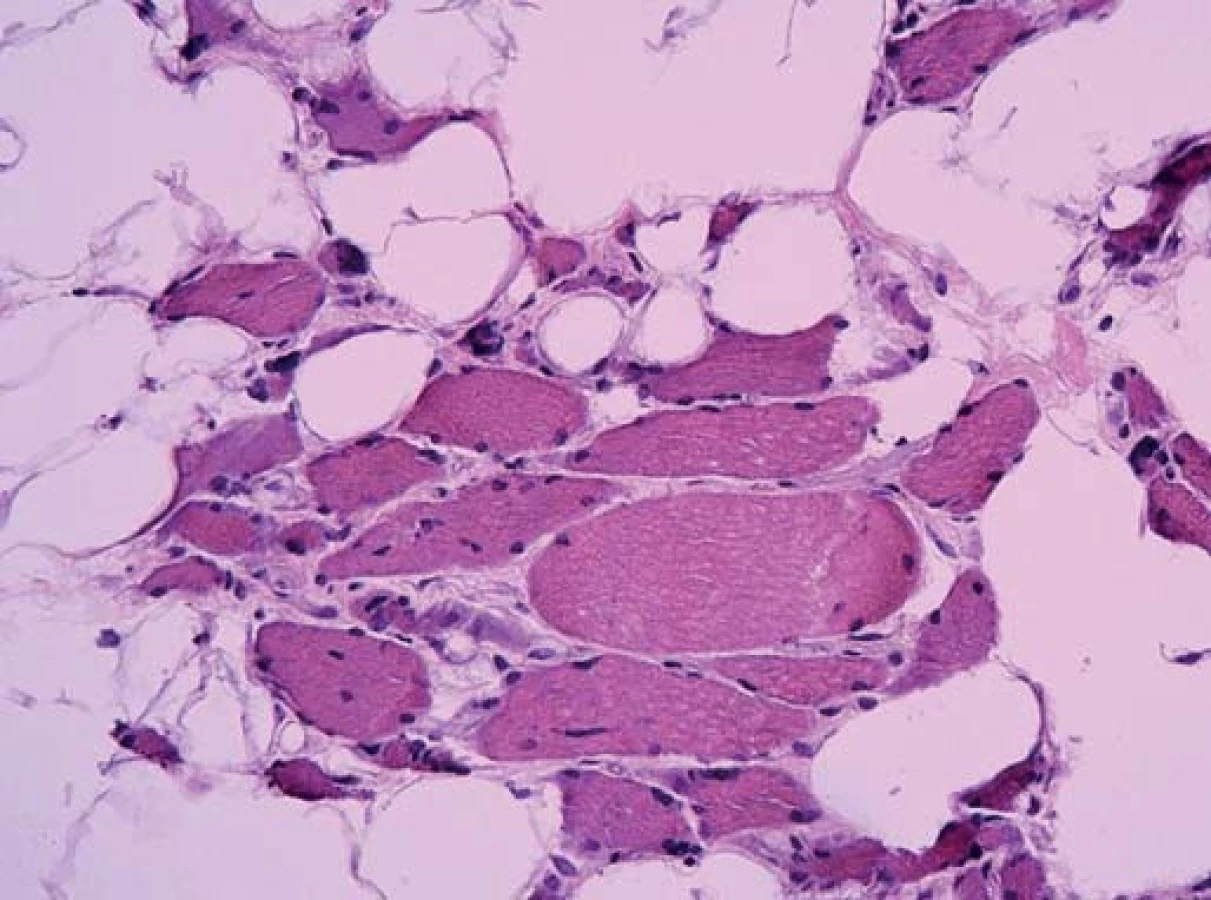
Ta ukázala myopatické změny s velikostním kolísáním svalových vláken od atrofie po normu, bez zánětlivé infiltrace. Atrofická vlákna se vyskytovala jak kulatá (myopatická), tak i angulární (denervovaná). Častá byla přítomnost internalizovaných jader. Intersticium svalové tkáně bylo rozšířené zmnožením vazivově tukové tkáně. Typizace svalových vláken ozřejmila predominanci vláken I. typu. Panel enzymově histochemických vyšetření ani imunohistologie sarkolemálních proteinů nedetekovala jinou poruchu.
Diagnózu přinesla elektronová mikroskopie. Už v polotenkých řezech barvených toluidinovou modří byla zřetelná přítomnost drobných tmavých tyčinkovitých inkluzí ve většině vláken. V elektronovém mikroskopu odpovídaly tyto inkluze osmiofilním tyčinkovitým tělískům, která se nacházela v sarkoplazmě svalových vláken, většinou náhodně distribuovaná, často však s akumulací subsarkolemálně. Tyčinkovité inkluze byly uloženy většinou v podélné ose vlákna, jen některé kolmo. Ultrastrukturálně měly tyčinkovité inkluze protáhlý doutníkový tvar, na kolmých řezech s polygonálním ohraničením.
Inkluze byly výrazně elektrondenzní a byly uloženy často v oblasti Z‑ disků sarkomer. V postižených vláknech byla dále přítomna disrupce sarkomer s jejich porušeným uspořádáním.
Byla tak stanovena diagnóza nemalinové (tyčinkovité) myopatie.
Následně byla zahájena imunosupresivní terapie. Nejprve proběhla aplikace 2,5 g metylprednizolonu i.v., s následným přechodem na perorální glukokortikoidy v sestupných dávkách (z 60 mg prednizonu denně byly dávky snižovány o 5mg/ 14 dní). Vzhledem k tomu, že terapie kortikosteroidy nebyla účinná, byla v plánu aplikace intravenózních imunoglobulinů. Po ukončení terapie kortikosteroidy pacient podstoupil dlouhodobou rehabilitaci. K plánované aplikaci intravenózních imunoglobulinů již nedošlo.
Pacient zemřel 25 měsíců od objevení se prvních symptomů svalového postižení. Příčinou smrti byla těžká hnisavá bronchopneumonie spojená s respirační insuficiencí.
Diskuze
Nemalinové myopatie tvoří vzácnou skupinu svalových onemocnění, která zahrnuje jak kongenitální, tak i sporadické dospělé formy. Ke sporadickým dospělým formám je často přidružen nález monoklonální gamapatie bez průkazu mnohočetného myelomu.
Hlavní příznaky typické pro SLONM jsou subakutně progredující svalová slabost, normální nebo jen lehce zvýšená hladina kreatinkinázy, myopatický obraz v EMG a event. paraproteinemie. Diagnózu potvrdí vyšetření svalové biopsie z klinicky postiženého svalu.
Příčina frekventovaného společného výskytu SLONM s monoklonální gamapatií a úloha paraproteinu v patogenezi onemocnění zatím není jasná. SLONM se vyskytla i v kombinaci s HIV infekcí [16].
SLONM je progresivní myopatie nejasné etiologie. Je obvykle rezistentní na farmakologickou léčbu (pokud není asociována s HIV infekcí, kde je evidentní pozitivní reakce na imunoterapii) [14].
V případě, že je nemalinová myopatie s pozdním začátkem asociována s monoklonální gamapatií neurčeného významu (MGUS), má nejistou prognózu.
Nedávno publikovaný případ dvou pacientů, kteří byli úspěšně léčeni vysokou dávkou melfalanu a transplantací kmenových buněk, je jistou nadějí pro nemocné postižené tímto onemocněním [6,19].
Klinický obraz našeho nemocného je v mnoha znacích podobný dříve publikovaným případům nemalinové myopatie v kombinaci s monoklonální gamapatií (progredující svalová slabost, která nereagovala na léčbu kortikosteroidy, symetrické hypotrofie převážně proximálního pletencového svalstva končetin, normální hodnota kreatinkinázy v séru, myogenní změny v EMG a nález početných tyčinkovitých inkluzí ve svalové biopsii). I když si nemůžeme být zcela jisti, že se jednalo o získanou formu nemalinové myopatie (nebylo provedeno u nás nedostupné neurogenetické vyšetření), negativní rodinná anamnéza – sporadický výskyt, pozdní začátek onemocnění a rychlá progrese svědčí spíše pro získanou než pro geneticky podmíněnou formu nemalinové myopatie.
Závěrem je potřeba zdůraznit, že nález tyčinkovitých inkluzí v elektromikroskopickém obraze není pro nemalinovou myopatii zcela patognomický. Vzácně se může vyskytovat i u jiných forem myopatií – byl popsán u dermatomyozitidy [20], polymyozitidy [21], u akutní alkoholické myopatie [22] či hypotyreoidizmu [23]. Dokonce může doprovázet neurogenní poruchy [24].
Proto správné stanovení diagnózy vyžaduje explicitní klinicko‑patologickou korelaci. Klinické, elektromyografické i histopatologické vyšetření u našeho případu všechny dříve popsané možnosti sekundárních nemalinových inkluzí vyloučilo.
Použité zkratky
ACTA1 gen kódující bílkovinu a- aktin
EMG elektromyografie
HIV virus lidské imunitní nedostatečnosti
IgG imunoglobulin G
IVIG intravenózní lidský imunoglobulin
MGUS monoklonální gamapatie neurčeného významu
MR magnetická rezonance
MUP akční potenciál motorické jednotky
RTG rentgen
SLONM Sporadic Late Onset Nemaline Myopathy
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 8. 3. 2013
Přijato do tisku: 26. 11. 2013
MUDr. Martin Forgáč
Neurologická klinika
1. LF UK a VFN
Kateřinská 30
120 00 Praha 2
e-mail: martin.forgac@vfn.cz
Zdroje
1. Shy GM, Engel WK, Somers JE, Wanko T. Nemaline myopathy: a new congenital myopathy. Brain 1963; 86: 793– 810.
2. Conen PE, Murphy EG, Donohue WL. Light and electron microscopic studies of „myogranules“ in a child with hypotonia and muscle weakness. Can Med Assoc J 1963; 89: 983– 986.
3. Bednařík J, Voháňka S, Lukáš Z, Kadaňka Z, Vytopil M,Mechl M et al. Nemoci kosterního svalstva. Praha: Triton 2001: 182– 183.
4. Fardeau M. Congenital myopathies. In: Mastaglia FL,Walton J (eds). Skeletal Muscle Pathology. 2nd ed. Edinburgh: Churchill Livingstone 1992: 237– 281.
5. Banker BQ. The congenital myopathies. In: Engel AG,Banker BQ (eds). Myology. New York: McGraw‑ Hill 1986: 1527– 1581.
6. Benveniste O, Laforet P, Dubourg O, Solly S, Musset L, Choquet S et al. Stem cell transplantation in a patient with late‑ onset nemaline myopathy and gammopathy. Neurology 2008; 71(7): 531– 532. doi: 10.1212/ 01.wnl.0000310813.79325.32.
7. Wallgren‑ Pettersson C, Sewry CA, Nowak KJ, Laing NG. Nemaline myopathies. Semin Pediatr Neurol 2011; 18(4): 230– 238. doi: 10.1016/ j.spen.2011.10.004.
8. Engel WK, Oberc MA. Abundant nuclear rods in adult‑ onset rod disease. J Neuropathol Exp Neurol 1975; 34(2): 119– 132.
9. Paulus W, Peiffer J, Becker I, Roggendorf W, Schumm F. Adult‑ onset rod disease with abundant intranuclear rods. J Neurol 1988; 235(6): 343– 347.
10. Yamaguchi M, Robson RM, Stromer MH, Dahl DS,Oda T. Actin filaments form the backbone of nemaline myopathy rods. Nature 1978; 271(5642): 265– 267.
11. Shimomura C, Nonaka I. Nemaline myopathy: comparative muscle histochemistry in the severe neonatal, moderate congenital, and adult‑ onset forms. Pediatr Neurol 1989; 5(1): 25– 31.
12. Engel WK, Resnick JS. Late rod myopathy: a newly recognized acquired and progressive disease. Neurology 1966; 16: 308– 309.
13. Engel AG. Late‑ onset rod myopathy (a new syndrome?): light and electron microscopic observations in two cases. Mayo Clin Proc 1966; 41(11): 713– 741.
14. Milone M, Katz A, Amato AA, Soderland CA, Segarceanu M, Young NP et al. Sporadic late onset nemaline myopathy responsive to IVIg and immunotherapy. Muscle Nerve 2010; 41(2): 272– 276. doi: 10.1002/ mus.21504.
15. Wengert O, Meisel A, Kress W, Dekomien G, Angstwurm K, Heppner FL et al. Progressive external ophthalmoplegia as initial manifestation of sporadic late‑ onset nemaline myopathy. J Neurol 2011; 258(5): 915– 917. doi: 10.1007/ s00415– 010– 5819– 6.
16. Chahin N, Selcen D, Engel AG. Sporadic late onset nemaline myopathy. Neurology 2005; 65(8): 1158– 1164.
17. Agrawal PB, Strickland CD, Midgett C, Morales A, Newburger DE, Poulos MA et al. Heterogeneity of nemaline myopathy cases with skeletal muscle alpha‑ actin gene mutations. Ann Neurol 2004; 56(1): 86– 96.
18. Lomen‑ Hoerth C, Simmons ML, Dearmond SJ, Layzer RB. Adult‑ onset nemaline myopathy: another cause of dropped head. Muscle Nerve 1999; 22(8): 1146– 1150.
19. Voermans NC, Minnema M, Lammens M, Schelhaas HJ, Kooi AV, Lokhorst HM et al. Sporadic late‑ onset nemaline myopathy effectively treated by melphalan and stem cell transplant. Neurology 2008; 71(7): 532– 534. doi: 10.1212/ 01.wnl.0000310814.54623.6f.
20. Danon MJ, Giometti CS, Manaligod JR, Perurena OH,Skosey JL. Adult‑ onset nemaline rods in a patient treated for suspected dermatomyositis. Study with two‑dimensional electrophoresis. Arch Neurol 1981; 38(12): 761– 766.
21. Cape CA, Johnson WW, Pitner SE. Nemaline structures in polymyositis. A nonspecific pathological reaction of skeletal muscles. Neurology 1970; 20(5): 494– 502.
22. Martinez AJ, Hooshmand H, Faris AA. Acute alcoholic myopathy. Enzyme histochemistry and electron microscopic findings. J Neurol Sci 1973; 20(3): 245– 252.
23. Reyes MG, Tal A, Abrahamson D, Schwartz M. Nemaline myopathy in an adult with primary hypothyroidism. Can J Neurol Sci 1986; 13(2): 117– 119.
24. Konno H, Iwasaki Y, Yamamoto T, Inosaka T. Nemaline bodies in spinal progressive muscular atrophy. An autopsy case. Acta Neuropathol 1987; 74(1): 84– 88.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2014 Číslo 2
Nejčtenější v tomto čísle
- Autonomní dysreflexie – závažná komplikace u pacientů po poranění míchy
- Syndrom Dravetové: těžká myoklonická epilepsie v časném dětství – kazuistiky
- Normativní hodnoty parametrů vedení pro nervus ulnaris a nervus medianus měřené standardizovaným způsobem
- Neuromodulace