
Frontotemporálna lobárna degenerácia z pohľadu nových klinicko‑patologických korelácií
Frontotemporal Lobar Degeneration from the Perspective of the New Clinical ‑ Pathological Correlations
The disease currently known as Frontotemporal Lobar Degeneration (FTLD) underwent a complicated development. From its first description by Arnold Pick and Alois Alzheimer, through the first clinical and pathological criteria introduced by David Neary and David Mann to the current perception of the disease as a complex clinical and pathological entity. At present, the Frontotemporal Lobar Degeneration is understood to be a heterogeneous clinical syndrome caused by degeneration of the frontal and temporal lobes. FTLD can manifest as any of the three clinical syndromes of frontotemporal dementia (behavioural variant of frontotemporal dementia, progressive non‑fluent aphasia and semantic dementia) as well as so called overlap syndromes encompassing corticobasal dementia and progressive supranuclear palsy. FTLD represents approximately 10% of all cases of dementia but 40% of cases of early onset dementia (between the age of 45 and 65 years). Although FTLD subtypes differ in their clinical manifestation, common denominators include behavioural disturbances and impairment of fatic, gnostic and executive functions. Mnestic and visual ‑ spatial functions are preserved until advanced stages of the disease. Compared to Alzheimer’s disease, the FTLD usually onsets at an earlier age and causes more devastating impairment of cognitive domains. Persons affected by FTLD become more quickly dependent on the help of other person or an institution. In our paper, we provide an overview of this complex entity, focusing mainly on frontotemporal dementia syndromes.
Key words:
frontotemporal lobar degeneration – frontotemporal dementia – progressive non-fluent aphasia – semantic dementia – tau protein – tautopathy – ubiquitin – TDP-43 protein – behavioural disturbances – speech disorder
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
S. Šutovský 1; M. Králová 3; B. Kollár 1; P. Šiarnik 1; J. Dragašek 2; Ľ. Izáková 3; P. Turčáni 1
Působiště autorů:
I. neurologická klinika LF UK a UN Bratislava
1; Psychiatrická klinika UPJŠ v Košiciach
2; Psychiatrická klinika LF UK a UN Bratislava
3
Vyšlo v časopise:
Cesk Slov Neurol N 2013; 76/109(6): 679-689
Kategorie:
Přehledný referát
Souhrn
Ochorenie, ktoré dnes označujeme ako frontotemporálna lobárna degenerácia (FTLD), prešlo zložitým vývojom od svojho prvého opisu Arnoldom Pickom a neskôr Aloisom Alzheimerom, cez prvé klinicko‑patologické kritériá predstavené Davidom Nearym a Davidom Mannom, až po dnešné nomenklatúrne vnímanie ako komplexnej klinicko‑patologickej entity. V súčasnosti je frontotemporálna lobárna degenerácia vnímaná ako heterogénny syndróm spôsobený progresívnou degeneráciou frontálnych a temporálnych lalokov mozgu. Klinicky sa môže prejaviť ako tri syndrómy frontotemporálnej demencie (behaviorálny variant FTD, progresívna non‑fluentná afázia a sémantická demencia), ale aj ako tzv. overlap syndrómy zahrňujúce kortikobazálnu degeneráciu a progresívnu supranukleárnu obrnu. Jej výskyt je asi 10 % spomedzi všetkých demencií a 40 % spomedzi demencií so začiatkom medzi 45. a 65. rokom života. Klinická manifestácia jednotlivých subtypov sa líši, spoločným menovateľom sú poruchy správania a postihnutie fatických, gnostických a exekutívnych funkcií. Mnestické a zrakovo ‑ priestorové funkcie zostávajú síce relatívne dlho zachované, sú však prekryté rozpadom osobnosti, fatickými alebo gnostickými poruchami. V porovnaní s Alzheimerovou chorobou má spravidla skorší vek nástupu, rýchlejší priebeh a devastujúcejšie postihnutie jednotlivých kognitívnych domén. Postihnutí sú spravidla rýchlejšie odkázaní na pomoc inej osoby alebo inštitúcie. V našom príspevku sa snažíme o súhrnný pohľad na túto výrazne heterogénnu klinicko‑patologickú entitu so zameraním sa na klinické, genetické a histopatologické špecifiká. Hlavnú pozornosť venujeme syndrómom frontotemporálnej demencie.
Kľúčové slová:
frontotemporálna lobárna degenerácia –frontotemporálna demencia – progresívna non-fluentná afázia – sémantická demencia – tau proteín – tauopatia –ubikvitin – TDP-43 proteín – poruchy správania – poruchy reči
Použité skratky
FTLD frontotemporálna lobárna degenerácia (histopatologická entita)
FTLD ‑ Tau histopatologický obraz s prítomnosťou tau proteínových inklúzií
FTLD ‑ TDP histopatologický obraz s prítomnosťou TDP ‑ 43 inklúzií
FTLD ‑ FUS histopatologický obraz s prítomnosťou FUS inklúzií
FTLD ‑ UPS histopatologický obraz s prítomnosťou UPS inklúzií
FTD frontotemporálna demencia (klinická entita)
bv ‑ FTD behaviorálny variant frontotemporálnej demencie
PNFA progresívna non‑fluentná afázia
SD sémantická demencia
PSP progresívna supranukleárna obrna
CBD kortikobazálna degenerácia
h ‑ Tau celkový tau proteín
p ‑ Tau fosforylovaný tau proteín
Ab amyloid beta
MMSE Mini Mental State Examination
C9orf72 Chromosome 9 open reading frame 72; novoobjavený gén, ktorého mutácie sú asociované s familiárnou FTLD
CDR Clinical Dementia Rating Scale
ADL Activities of Daily Living
História ochorenia a vývoj pojmu
Arnold Pick publikoval v roku 1882 v pražskom medicínskom periodiku prípad 71 - ročného muža s behaviorálnymi symptómami a progresívnou afáziou asociovanou s fokálnou atrofiou frontálnych a ľavého temporálneho laloka [1]. Nasledovali ďalšie dve práce, v ktorých Arnold Pick opisoval obdobné prípady, ktoré mali post mortem prítomnú makroskopickú atrofiu frontálnych a temporálnych lalokov, histopatologicky však neboli vyhodnotené [2,3]. Alois Alzheimer opísal histopatológiu ochorenia v roku 1911, kedy identifikoval abnormálne zväčšené achromafilné neuróny (balónové neuróny), neskôr nazvané ako Pickove bunky a argyrofilné intraneuronálne inklúzie, neskôr označené ako Pickove telieska [4]. Postupne sa od 20. rokov minulého storočia zaužíval pojem Pickova choroba. Vtedajší koncept Pickovej choroby predpokladal prítomnosť balónových neurónov a Pickových teliesok ako nevyhnutnú podmienku na stanovenie diagnózy. V neskoršom období však boli popísané prípady klinicky svedčiace pre Pickovu chorobu s prítomnosťou fokálnej frontálnej alebo temporálnej atrofie, avšak bez prítomnosti balónových neurónov alebo Pickových teliesok. Postupne narastali kontroverzie, či vedúcim príznakom pre stanovenie diagnózy má byť klinický obraz, makroskopická patológia alebo histopatológia. Takisto otvorenou otázkou ostávalo, či na stanovenie diagnózy je prítomnosť Pickových teliesok a balónových neurónov nevyhnutná. S postupom času, hlavne od 80. rokov minulého storočia, pribúdali práce opisujúce asociáciu Pickovej choroby s príznakmi parkinsonizmu a s príznakmi postihnutia motorického neurónu, ktoré podčiarkovali heterogenitu ochorenia [5 – 8]. Na označenie ochorenia bolo vystriedaných viacero synoným, postupne sa však zaužívalo označenie frontotemporálna lobárna degenerácia (FTLD). V priebehu 90. rokov minulého storočia sa postupne formoval názor, že rozhodujúcim pre stanovenie diagnózy FTLD je klinická manifestácia ochorenia, pričom histopatológia môže varírovať. V tomto zmysle boli predstavené aj tzv. Lund ‑ Manchesterské kritériá pre FTLD v roku 1994 [9], ktoré boli revidované v roku 1998 [10]. Podľa nich frontotemporálna lobárna degenerácia obsahuje tri rôzne klinické syndrómy – frontotemporálnu demenciu, progresívnu non‑fluentnú afáziu a sémantickú demenciu. V kontexte s klinickými kritériami boli neustále obnovované aj neuropatologické kritériá, ktorých zámerom bolo vniesť systém do už aj tak veľmi zložitej nomenklatúry. V súčasnosti každoročne revidovaná nomenklatúra reflektuje na aktuálny výskum neustálymi zásadnými zmenami, ktoré podčiarkujú výraznú heterogenitu histopatologických a neurochemických obrazov, ako aj výrazné napredovanie spoznávania tejto veľmi širokej klinicko‑patologickej jednotky.
Nomenklatúra a klasifikácia frontotemporálnej lobárnej degenerácie
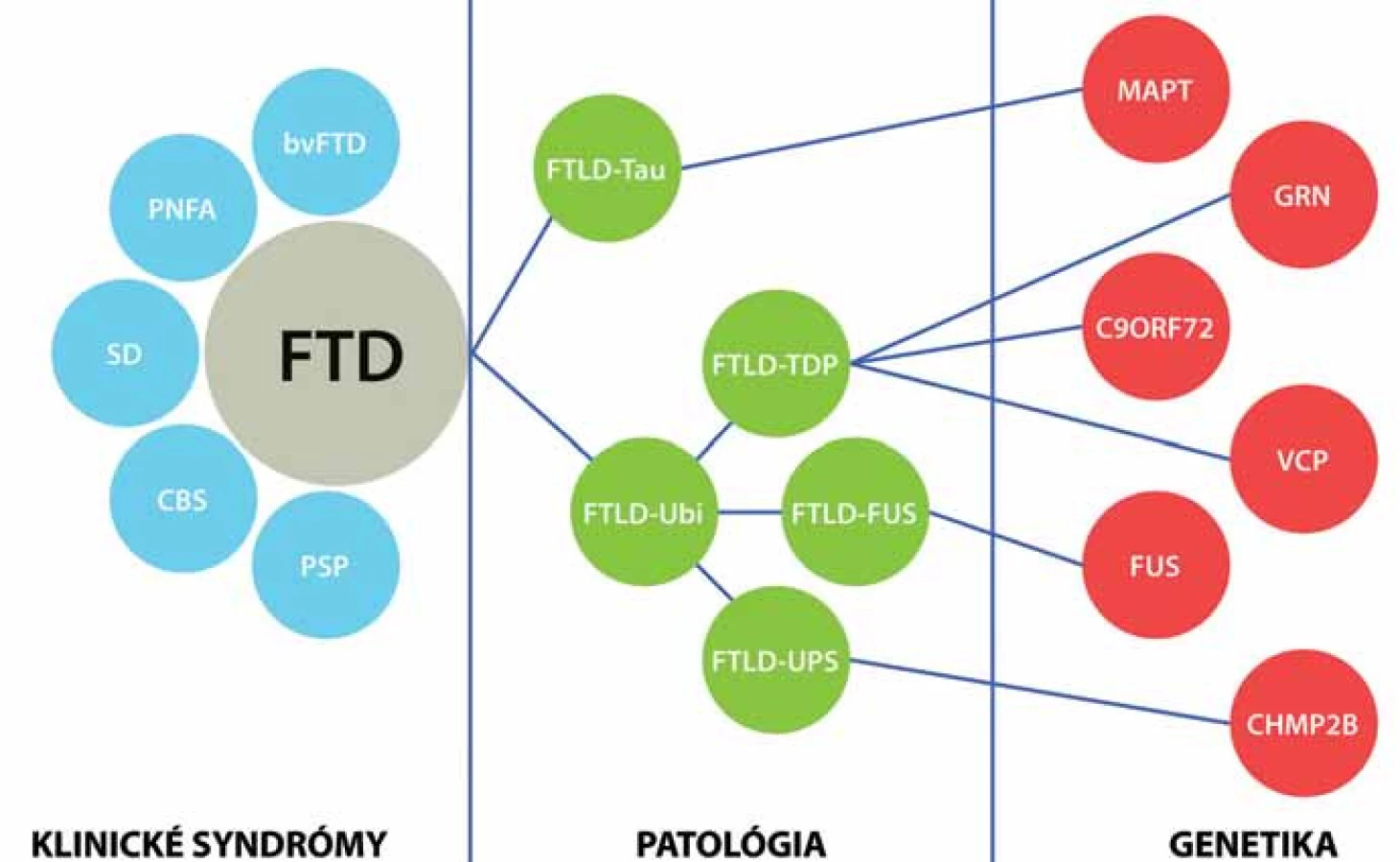
Frontotemporálna lobárna degenerácia je zastrešujúci termín zahrňujúci heterogénnu skupinu ochorení, ktorých spoločným menovateľom je degenerácia frontálnych a temporálnych lalokov. V kontexte posledných objavov je možné FTLD klasifikovať na štyri skupiny na základe proteínov obsiahnutých v inklúziách: FTLD ‑ Tau, FTLD ‑ TDP, FTLD ‑ FUS a zriedkavý variant FTLD ‑ UPS. Tieto patologické skupiny a ich špecifické patologické obrazy sú základom pre dobre definované klinické syndrómy vrátane troch variantov frontotemporálnej demencie (FTD) [behaviorálny variant frontotemporálnej demencie (bv FTD), progresívna non‑fluentná afázia (PNFA) a sémantická demencia (SD)], progresívnu supranukleárnu obrnu (PSP) a kortikobazálnu degeneráciu, resp. kortikobazálny syndróm (CBD, CBS). Progresívna supranukleárna obrna a kortikobazálna degenerácia patria do skupiny FTLD z histologického hľadiska. Z klinického hľadiska predstavujú samostatné klinické jednotky, ktoré sú často v literatúre označované ako „overlap“ syndrómy frontotemporálnej demencie, vzhľadom na to, že majú viaceré charakteristiky frontotemporálnej demencie (hlavne frontálny dysexekutívny syndróm), ale majú výrazne vyjadrené aj známky atypického parkinsonizmu. Tieto dve klinické jednotky sú preberané aj v monografiách o frontotemporálnej demencii [11], ale aj v monografiách o atypickom parkinsonizme [12,13]. Nomenklatúra a klasifikácia frontotemporálnej lobárnej degenerácie je stále v intenzívnom procese vývoja a každoročne sú publikované nové revízie a reklasifikácie. Termín frontotemporálna lobárna degenerácia označuje postihnutie frontálneho a temporálneho laloka a má hlavne histopatologický rozmer a predstavuje histopatologický termín [14]. Termín frontotemporálna demencia má hlavne klinický rozmer a označuje tri „klasické“ ochorenia (bv FTD, PNFA a SD) a dva voľne asociované „overlap“ syndrómy (PSP a CBD), ktoré stoja na pomedzí medzi frontotemporálnou demenciou a atypickým parkinsonizmom [14]. V našej publikácii sa budeme zaoberať hlavne syndrómami frontotemporálnej demencie (bv ‑ FTD, PNFA a SD).
Klinická manifestácia
Behaviorálny variant FTD (synonymá: frontálny variant; fv ‑ FTD)
Behaviorálny variant FTD (bv ‑ FTD) je charakterizovaný výraznou alteráciou osobnosti hneď od úvodných štádií, behaviorálnymi a fatickými poruchami, postihnutím exekutívnych funkcií a rôzne rýchlo progredujúcou demenciou.
Pri postihnutí dorzo ‑ mediálnych prefrontálnych štruktúr sa vyvíja tzv. apatický variant. V popredí je celková apatia, pasivita, strata záujmu o okolie, strata pracovných a interpersonálnych návykov a zanedbávanie osobnej hygieny. Na napomenutie reagujú postihnutí neprimerane a neadekvátne. S progresiou ochorenia sa prehlbuje necitlivosť a neohrabanosť celkového vystupovania a prejavu, v neskorších štádiách sa pridružujú najprv len sporadicky, neskôr čoraz častejšie afektívne poruchy. Na minimálny podnet pacienti nezriedka nepríčetne kričia, hrubo nadávajú a podobne. S progresiou ochorenia sa stupňuje apatia a abúlia, chorí spravidla nečinne presedia celé hodiny na jednom mieste, pri pokuse o premiestnenie alebo usmernenie sú reakcie hrubo neadekvátne až brachiálne agresívne.
Pri postihnutí orbitofronto ‑ a ventromediálno ‑ frontálneho kortexu sa vyvíja tzv. desinhibovaný variant. Pacienti sa stávajú desinhibovanými, detinskými, nevhodne veselými, správajú sa euforicky, strácajú sociálnu slušnosť, majú tendenciu robiť hrubé a netaktné poznámky voči ostatným. Desinhibícia môže viesť k hľadaniu konfrontácie a časom k sociopatickému správaniu, ako napr. nevhodné sexuálne zblíženia, dopravné priestupky, kradnutie v obchodoch a fyzické útoky. Pacienti sa môžu prejavovať psychomotorickou hyperaktivitou s tendenciou stáleho chodenia a blúdenia. Rečový prejav je zvýšený a naliehavý, používajú neslušné výrazy, hovoria nevhodne nahlas s tendenciou prerušovať ostatných a usmerňovať konverzáciu. Častý je zvýšený apetít a stúpajúca hmotnosť. Môžu byť prítomné bludy, ktoré sú často bizarné.
Delenie na apatický a desinhibovaný variant rozoznávajú viacerí autori [15], vo väčšine prípadov však apatia a desinhibícia ako jadrové príznaky bv ‑ FTLD koexistujú u pacientov súčasne, resp. sa v klinickom obraze striedajú alebo prelínajú [11]. Nižšie uvádzame revidované diagnostické kritériá pre behaviorálny variant FTD. Nové diagnostické kritériá dopĺňajú a v niektorých bodoch nahrádzajú pôvodné kritériá podľa Nearyho et al z roku 1998 [10].
Medzinárodný konsenzus kritérií pre bv ‑ FTD [16]
I. Neurodegeneratívne ochorenie
A. Progresívna deteriorácia správania a kognitívnych funkcií pozorovaná vyšetrením alebo objektívnou anamnézou od opatrovateľa
II. Možný bv ‑ FTD
Tri z nasledujúcich kategórií behaviorálnych a kognitívnych symptómov musia byť splnené, aby boli naplnené kritériá pre možný bv ‑ FTD. Symptómy by mali byť permanentné alebo rekurentné, nie ojedinelé alebo zriedkavé.
A. Včasná porucha správania
A1 Sociálne nevhodné správanie
A2 Strata spoločenského kódexu a dekóra
A3 Impulzívne správanie a konanie
B. Včasná apatia a inercia
B1 Apatia
B2 Inercia
C. Včasná strata empatie a pochopenia pre druhých ľudí
C1 Znížená odpoveď na potreby iných
C2 Znížený sociálny záujem
D. Včasné perseveratívne, stereotypné, kompulzívne alebo ritualistické správanie
D1 Jednoduché repetitívne pohyby
D2 Komplexné kompulzívne alebo ritualistické správanie
D3 Stereotypná reč
E. Hyperoralita a zmena dietetických návykov
E1 Zmena dietetických návykov a preferencia sladkého
E2 Prejedanie sa, zvýšená konzumácia alkoholu
E3 Hyperoralita, skúmanie predmetov ústami
F. Neuropsychologický profil – deficit exekutívnych funkcií pri relatívnom zachovaní pamäte a vizuospaciálnych funkcií
F1 Deficit exekutívnych funkcií
F2 Zachovanie pamäte
F3 Zachovanie vizuospaciálnych funkcií
III. Pravdepodobný bv ‑ FTD
Kritériá v bodoch A – C musia byť splnené:
- a) naplnenie kritérií pre možný bv ‑ FTD
- b) preukázaný signifikantný funkčný úpadok pacienta v čase (na základe informácií od opatrovateľa a dokumentovaný prostredníctvom škál funkčnosti CDR alebo ADL)
- c) nález neurozobrazovacích metód konzistentný s diagnózou bv ‑ FTD:
- c1) atrofia frontálneho laloka a/ alebo predného pólu temporálneho laloka pri CT alebo MR
- c2) hypometabolizmus frontálneho laloka a/ alebo predného pólu temporálneho laloka pri PET alebo SPECT
IV. Behaviorálny variant FTD s definitívnou FTLD patológiou
- a) spĺňa kritériá pre možnú a pravdepodobnú diagnózu bv ‑ FTLD
- b) histopatologický dôkaz FTLD patológie na základe biopsie alebo post mortem
- c) prítomnosť známych patogénnych mutácií
V. Exklúzne kritériá pre bv ‑ FTLD
A. charakter deficitu nemá charakter primárneho neurodegeneratívneho ochorenia
B. charakter poruchy správania má charakter iného psychiatrického ochorenia
C. biomarkery podporujú Alzheimerovu chorobu alebo iný neurodegeneratívny proces
Progresívna non‑fluentná afázia (PNFA)
Progresívna non‑fluentná afázia je klinicky charakterizovaná progresívnym vývojom porúch reči, ktoré sú špecifické v závislosti od typu variantu PNFA. Pri agramaticko‑nonfluentnom variante sú prvými príznakmi strata plynulosti reči, zárezy, občasné fonemické parafázie. S progresiou ochorenia sa stupňujú agramatizmy, reč je produkovaná s veľkým úsilím. Pri logopedickom variante je v úvode prítomné celkové ochudobnenie slovnej zásoby, chorý rozpráva málo a ťažko sa mu hľadajú slová. Rečový prejav je však plynulejší ako pri agramaticko‑nonfluentnom variante. V priebehu ochorenia sa môžu pridružiť známky prefrontálneho syndrómu podobné behaviorálnemu variantu (iritabilita, agitovanosť, ktorá sa môže striedať s apatiou) [17]. Ochorenie má progresívny priebeh, bez ohľadu na variant spravidla končí mutizmom a dementným syndrómom [17 – 19]. U určitého percenta pacientov sa vyvíja ochorenie motoneurónu [10,14,17]. Stále otvorenou otázkou ostáva, či ochorenie motoneurónu asociované s ktorýmkoľvek syndrómom FTD je len rozšíreným prejavom konkrétneho variantu alebo samostatnou subjednotkou. Zaujímavosťou je, že prípady FTD ‑ MND sú takmer výlučne asociované s FTLD ‑ TDP patológiou [14] (podrobnejšie viď kapitola o klinicko‑patologických koreláciách). V tab. 1 a 2 uvádzame revidované diagnostické kritériá pre PNFA.
![Diagnostické kritériá pre agramaticko-nonfluentný variant (podľa [20]).](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/a4113c136683e3e99056c59f6907f649.png)

Sémantická demencia (SD; sémantický variant PNFA, sémantická afázia a zraková agnózia)
Sémantická demencia je charakterizovaná postupnou stratou porozumenia hovorenej reči a jednotlivých slov. Rečový prejav je v úvode fluentný, prítomná je však anómia (neschopnosť pomenovania predmetu), sémantické parafázie (zámena slov z rovnakej kategórie). Stratu významu sleduje hierarchický model. Najskôr pacienti strácajú schopnosť rozlišovať medzi členmi jednej skupiny (napr. typy jabĺk), neskôr aj medzi jednotlivými skupinami (napr. jablká verzus pomaranče) a nakoniec nerozlišujú ani nadskupiny (napr. ovocie verzus zelenina). Na začiatku „pudlík“ je nazývaný „pes“, potom všetci psy sú nazývaní „zvieratá“ a nakoniec všetky zvieratá sú pomenované ako „veci“ [11]. Význam slov sa stráca napriek zachovanej schopnosti ich čítať a písať. S progresiou ochorenia je reč stále plynulá a bez námahy, ale obsahovo prázdna. Postupne pacient produkuje slovný šalát, ktorému ani sám nerozumie. Takisto je prítomná zraková asociačná agnózia (neschopnosť pomenovať videné predmety). Relatívne zachované sú elementárne vizuokonštruktívne a praktické funkcie (schopnosť obkresliť jednoduchý obrázok, spárovať podobné predmety). Popisuje sa aj paradoxná disociácia autobiografickej pamäte s ušetrením spomienok na udalosti posledných mesiacov a neschopnosť vybaviť si spomienky z detstva [21]. V pokročilých štádiách ochorenia pacient postupne prestáva rozprávať a takisto sa môžu vyskytnúť príznaky podobné dysexekutívnemu variantu, s apatickými symptómami v popredí. Prvé komplexné diagnostické kritériá pre sémantickú demenciu boli vypracované Nearym et al v roku 1998 [10]. Revidované diagnostické kritériá zahŕňajúce podporu neurozobrazovacích metód boli vypracované v roku 2011 (tab. 1) [20].
Zobrazovacie metódy
Pri neurozobrazovacích metódach u pacientov s FTLD zisťujeme atrofiu (pri vyšetrení magnetickou rezonanciou) alebo hypometabolizmus (pri vyšetrení prostredníctvom SPECT alebo PET) frontálnych a temporálnych lalokov. Pri bv ‑ FTD sú atrofiou a hypometabolizmom spravidla symetricky postihnuté frontálne laloky a predný pól temporálnych lalokov [16]. Pri progresívnej non‑fluentnej afázii je atrofia asymetrická s prevahou v dominantnej (najčastejšie ľavej) hemisfére. Pri agramaticko‑nonfluentnom variante je atrofiou a hypometabolizmom najviac postihnutá fronto ‑ inzulárna oblasť, pri logopedickom ľavá perisylviánska oblasť [20]. Pri sémantickej demencii zisťujeme atrofiu a hypometabolizmus predného pólu temporálneho laloka s prevahou v dominantnej hemisfére [20]. Viaceré štúdie sa zaoberali mierou hipokampálnej atrofie pri AD a FTLD. Van de Pol et al v roku 2006 zistili, že atrofia hipokampu pri Alzheimerovej chorobe (AD) je porovnateľná s atrofiou pri bv ‑ FTD, atrofia ľavého hipokampu je pri SD výraznejšia ako pri AD (v súlade s predominantným postihnutím ľavého temporálneho laloka pri SD) [22]. Pri PNFA nebola hipokampálna atrofia konzistentne prítomná a v prípade jej výskytu bola miernejšia ako pri AD. Je teda zrejmé, že MTA a atrofia hipokampu sa vyskytujú aj pri syndrómoch FTLD a jej nález musíme hodnotiť v kontexte s klinickou prezentáciou diagnostikovaného ochorenia.
Histopatológia a imunohistochémia
Histopatológia
Histopatológia frontotemporálnej lobárnej degenerácie je najkomplikovanejšou a najkontroverznejšou časťou celej klinicko‑patologickej jednotky. Prítomnosť Pickových teliesok a balónových neurónov bola až do 80. rokov minulého storočia nevyhnutnou podmienkou pre stanovenie diagnózy „Pickovej choroby“. V roku 1998 bol identifikovaný tau proteín ako hlavný komponent Pickových teliesok [23]. Tento objav len podčiarkol význam tau proteínu v patogenéze Pickovej choroby. Od 50. rokov minulého storočia však pribúdali prípady klinicky svedčiace pre Pickovu chorobu s prítomnosťou fokálnej frontálnej alebo temporálnej atrofie, avšak bez prítomnosti balónových neurónov alebo Pickových teliesok. Tento fakt napomáhal hľadaniu nových proteínov zúčastnených na tvorbe histologického obrazu. Prvým z týchto proteínov bol ubikvitín, čo sa odzrkadlilo v histopatologickej klasifikácii navrhnutej Nearym et al v roku 1994 [9]. Ďalším posunom bola identifikácia rozdielnych foriem tau proteínu pri jednotlivých klinicko‑patologických subjednotkách. Trojrepeatové izoformy boli nájdené pri FTLD s Pickovými telieskami, troj ‑ aj štvorrepeatové pri FTLD s parkinsonizmom s väzbou na chromozóm 17 a čisto štvorrepeatové boli identifikované pri kortikobazálnej degenerácii (CBD) a progresívnej supranukleárnej obrne (PSP), čím bola umožnená histopatologická diferenciácia medzi jednotlivými jednotkami. Postupne bolo zistené, že až 60 % všetkých FTLD prípadov obsahuje ubikvitínové telieska. Celý tento koncept sa odrazil v klasifikácii podľa konzorcia pre FTLD z roku 2007 [24]. Ubikvitinopatie však predstavovali veľmi heterogénnu histopatologickú skupinu. Postupne sa zisťovalo, že za ubikvitínovou imunoreaktivitou sa skrýva množstvo iných proteínov, ktoré boli len ubikvitínom označené na proteolýzu. Následne bol identifikovaný TDP ‑ 43 proteín a FUS ‑ proteín ako významné proteínové komponenty FTLD ubikvitinopatií [25,26]. Tento objav viedol k subklasifikácii ubikvitinopatií na FTLD ‑ TDP s obsahom TDP ‑ 43 pozitívnych inklúzií a FTLD ‑ FUS s obsahom FUS pozitívnych inklúzií [27]. Inklúzie s FUS imunoreaktivitou sa dokonca identifikovali aj v prípadoch dovtedy nezatriedených histopatologických obrazov – DLDH (Dementia Lacking Distinctive Histopathology) a v prípadoch NIFID (Neuronal Intermediate Filament Inclusion Disease), čím sa výrazne znížil počet nezatriedených histologických obrazov [27]. Určitý stupeň konfúzie do tejto klasifikácie vnáša fakt významnej vzájomnej homológie medzi TDP ‑ 43 proteínom a FUS proteínom.
Najnovšia nomenklatúra pre neuropatologické subtypy frontotemporálnej lobárnej degenerácie je uvedená v tab. 3 [27].
![Najnovšia nomenklatúra pre neuropatologické subtypy frontotemporálnej lobárnej degenerácie [27].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/7d33fdaf5bee9b69bcca27c8d73bee54.png)
Klinicko‑patologické korelácie
Doteraz boli publikované štyri veľké štúdie zaoberajúce sa klinicko‑patologickými koreláciami v rámci syndrómov FTD, CBD a PSP [28 – 31]. Štúdie zbierali dáta z viac ako 10 - ročného sledovania a korelovania klinických syndrómov a im zodpovedajúcim histopatologickým subtypom. Zhrňujúco možno povedať, že prípady behaviorálneho variantu FTD vykazovali 40% FTLD ‑ Tau, 40% FTLD ‑ TDP a 20% FTLD ‑ FUS patológiu. Prípady PNFA vykazovali FTLD ‑ Tau patológiu až v 70 %. Prípady sémantickej demencie boli asociované s FTLD ‑ TDP patológiou až v 83 %. Až 90 % prípadov PSP malo PSP patológiu, obdobne až 90 % prípadov CBD malo CBD patológiu. Najzaujímavejšiu asociáciu vykazovali prípady FTD ‑ MND, kde bola takmer výlučne FTLD ‑ TDP patológia.
Novšia štúdia Grossmana z roku 2012 [32] analyzuje klinicko‑patologické korelácie pri agramatickom a logopedickom variante PNFA a sémantickej demencii. Podľa nej agramatický variant je asociovaný s FTLD ‑ Tau patológiou (52 %), AD patológiou (25 %), FTLD ‑ TDP (19 %). Logopedický variant je asociovaný s AD patológiou (50 %), FTLD ‑ TDP (38 %) a FTLD ‑ Tau patológiou (12 %). Sémantická demencia bola asociovaná s FTLD ‑ TDP (69 %), AD (25 %) a FTLD ‑ Tau patológiou (6 %). Toto zistenie poukazuje na ďalší dôležitý aspekt pri diferenciálnej diagnostike demencií – na atypickú Alzheimerovu chorobu. Spomedzi pacientov s agramatickým variantom a sémantickou demenciou malo alzheimerovskú patológiu 25 % prípadov a u pacientov s logopedickým variantom malo alzheimerovskú patológiu až 50 % prípadov. Majú byť pacienti s logopedickým variantom a definitívnou alzheimerovskou patológiou definitívne zhodnotení ako ochorenie z okruhu FTLD alebo ochorenie z okruhu Alzheimerovej choroby? Na zodpovedanie tejto otázky je potrebné zohľadniť výsledky likvorových biomarkerov, celkový priebeh ochorenia a prítomnosť pridruženej psychiatrickej symptomatológie. Každopádne prítomnosť alzheimerovskej patológie u pacientov klinicky zhodnotených ako ochorenie z okruhu FTLD nie je zriedkavý nález a naopak pacienti klinicky zhodnotení ako Alzheimerova choroba môžu mať histopatológiu zo spektra FTLD. Má z tohto pohľadu význam hovoriť o logopedickom variante Alzheimerovej choroby a logopedickom variante PNFA? Klinický obraz oboch variantov je klinicky ťažko rozlíšiteľný, tieto dve entity možno oddiferencovať na základe likvorových biomarkerov a hlavne definitívneho histopatologického nálezu. Aj tieto zistenia podčiarkujú turbulentnosť nomenklatúry, ktorá je stále v dynamickom procese.
Genetika
Tau proteín
Skutočnosť, že frontotemporálna lobárna degenerácia môže mať familiárny výskyt, bola známa už od 20. rokov minulého storočia. Prvý veľký dokumentovaný rodokmeň s familiárnym výskytom „Pickovej choroby“ bol publikovaný v roku 1939 Sandersom et al [33]. V súčasnosti je tento rodokmeň známy ako „Dutch family 2“. Postihnutí členovia mali v popredí behaviorálne príznaky, hlavne desinhibíciu, agresivitu, obsesívne správanie a hyperoralitu. Postihnutie pamäti bolo veľmi mierne v kontraste s progresívnou rečovou poruchou spejúcou k mutizmu. Postihnutí zomierali do ôsmich rokov od objavenia sa prvých klinických príznakov. Prvý veľký posun v porozumení familiárnej podstaty FTLD bol zaznamenaný v roku 1994 a v roku 1996, kedy u dvoch veľkých rodokmeňov postihnutých FTLD bola dokázaná väzba na lokus na chromozóme 17v pozícii 17q21 – 22, ktorý zodpovedá génu pre tau proteín [34]. Následne bola táto väzba dokázaná aj u žijúcich členov „Dutch family 2“. V priebehu troch rokov bola mutácia tau proteínu dokázaná u viac ako 13 rodín s familiárnym výskytom FTLD. Postihnutí pacienti mali okrem príznakov prefrontálneho syndrómu a demencie prítomný parkinsonizmus, čo následne viedlo k etablovaniu názvu „FTD s parkinsonizmom s väzbou na chromozóm 17“ alebo v skratke FTDP ‑ 17. Doteraz bolo objavených viac ako 35 mutácií tau proteínového génu (obr. 1). Najčastejším histopatologickým obrazom u nositeľov tau proteínovej mutácie sú Pickove telieska s obsahom troj ‑ aj štvorrepeatových izoforiem tau proteínu, úbytok neurónov vo frontálnom a temporálnom kortexe, glióza a spongiformné zmeny v druhej vrstve kortexu.
![Mutácie tau proteínu (podľa [39]).](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/9b2b8f17ca84ff1aaa3d2a3ba7514574.jpg)
Progranulín
Napriek objavu mutácií tau proteínu, ako príčiny familiárnych foriem FTLD u viac ako polovici familiárnych prípadov, žiadna mutácia alebo iná genetická porucha na tau preoteínovom géne nebola identifikovaná. Hľadanie nových lokusov zapojených do patogenézy FTLD však opätovne poukazovalo na chromozóm 17, na lokus v blízkosti tau lokusu, ktorého proteínový produkt dostal názov progranulín [35]. Progranulínový gén kóduje 68,5 kDa prekurzorový glykoproteín, ktorý je elastázami štiepený na 6kDa finálne peptidy – granulíny. Progranulín je široko exprimovaný rastový faktor, ktorý hrá úlohu vo vývoji, tumorigenéze, reparácii rán, zápale a aktivácii signálnych dráh kontrolujúcich bunkový cyklus a delenie buniek. Doposiaľ bolo identifikovaných viac ako 25 mutácií progranulínového génu pri familiárnych formách FTLD.
TARDBP (TransActive Response DNA Binding Protein; TDP, TDP ‑ 43) a FUS proteín (FUsed in Sarcoma)
Mutácie TARDBP a FUS génu sú zriedkavejšou príčinou familiárnych foriem ALS a FTLD. Približne 5% familiárnych foriem ALS a rovnaké percento familiárnych foriem FTLD je nositeľom mutácie TARDBP génu. TARDBP kóduje TDP ‑ 43 proteín, ktorý patrí do rodiny RNA viažucich proteínov a vytvára jadrové ribonukleoproteínové komplexy (hnRNP komplexy) [35]. HnRNP komplexy majú úlohu v transkripcii, v alternatívnom splicingu RNA a v produkcii mikroRNA. Podobnú biologickú funkciu má aj FUS proteín. Doteraz je známych 23 mutácií FUS génu v 49 rodokmeňoch. Fenotypovým prejavom sú familiárne formy ALS a FTLD [36].
C9orf72 – nová, doteraz najfrekvenčnejšia mutácia zodpovedajúca za familiárne formy ALS a FTLD
Štúdie kandidujúcich génov, ako aj asociačné štúdie, priniesli špecifikáciu ďalšieho genetického lokusu, premietajúceho sa na chromozóm 9 do pozície 9p21. Extenzívna mutačná analýza identifikovala hexanukleotidovú expanziu G4C2 ako genetický substrát familiárnych foriem ALS a FTLD. V normálnej populácii varíruje počet repetícií od troch do 25. U postihnutých sa počet repetícií zvyšuje nad 60 [36]. Presné vyčíslenie hexanukleotidových expanzií zatiaľ na širokej vzorke pacientov nie je dostupné, vzhľadom na to, že to neumožňujú štandardné PCR techniky. Southern blot analýza u limitovaného počtu vyšetrených preukázala počet repetícií pohybujúcich sa v rozmedzí 700 – 1 600. V rozsiahlej nedávnej štúdii bolo zistených 11,4% výskyt C9orf72 repetícií na vzorke 1 381 pacientov [37]. Pri prepočte na familiárne prípady FTLD z uvedenej štúdie pripadalo až 24,8 % na nositeľov C9orf72 expanzie. Tieto údaje posúvajú C9orf72 gén na najfrekvenčnejšie postihnutý gén u pacientov s FTLD [36,37].
Familiárna FTLD môže byť takisto asociovaná s inými zriedkavými mutáciami. V jednej holandskej rodine bol fenotyp podobný FTD asociovaný s mutáciou v subjednotke endozomálneho ESCRTIII ‑ komplexu na chromozóme tri [38]. Mutácie v géne kódujúcom proteín valosin sú spojené s autozomálne dominantným FTD syndrómom asociovaným s Pagetovou chorobou a myozitídou s inklúznymi telieskami. Alela ApoE4, ako uznávaný rizikový faktor pre Alzheimerovu chorobu, sa nezdá byť rizikovým faktorom pre FTLD (tab. 4) [11,17].
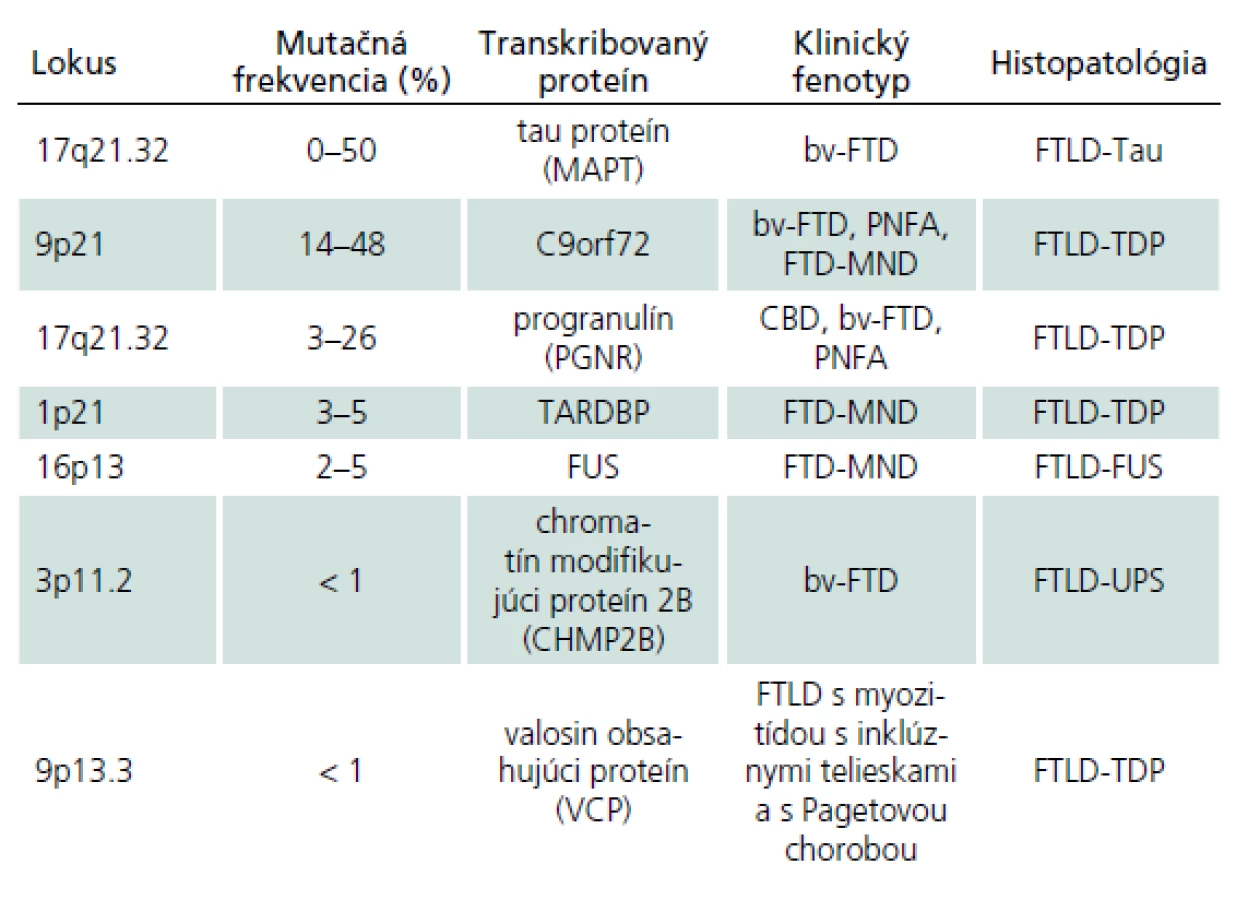
V súčasnosti prevláda názor, že približne 30 % prípadov FTLD je genetického pôvodu s autozomálne dominantnou dedičnosťou, avšak variabilnou penetranciou a fenotypovou prezentáciou. Bv ‑ FTD a FTD asociovaná s MND (FTD ‑ MND) alebo parkinsonizmom (FTDP) sa vyskytujú častejšie familiárne ako ostatné klinické syndrómy [11].
Klinicko‑patologické a klinicko‑genetické korelácie pri familiárnych FTLD
Prevažná väčšina prípadov FTLD je sporadická. Genetické pozadie je predpokladané v približne 30 % prípadov. Najdlhšie známou a najlepšie dokumentovanou príčinou familiárnych foriem FTLD sú mutácie tau proteínu. Typickým prejavom nositeľov mutácie je bv ‑ FTD so všetkými jadrovými znakmi, skorým začiatkom a často pridruženými príznakmi parkinsonizmu. Najčastejším histologickým prejavom je FTLD ‑ Tau patológia, konkrétne FTLD ‑ Tau PiD (Pickove telieska) [14,29,30,36,40]. Ďalší gén asociovaný s familiárnou FTLD je progranulín. Klinicky sa nositelia mutácie progranulínu môžu manifestovať ako CBD, PNFA alebo bv ‑ FTD v približne ekválnom rozložení [14,36]. Mutácie progranulínu sú v prevažnej väčšine prípadov asociované s FTLD ‑ TDP patológiou konkrétne so subtypom I s intranukleárnymi inklúziami [14,36,40]. Nositelia mutácie C9orf72 sa klinicky manifestujú ako bv ‑ FTD, FTD ‑ MND syndróm alebo ako PNFA. Post mortem nachádzame takmer výlučne FTLD ‑ TDP patológiu [14,36,40]. Ďalšie mutácie asociované s FTLD zahŕňajú VCP gén, CHMP2B gén, TDP ‑ 43 gén a FUS gén. Mutácie vo VCP géne, ktorý kóduje proteín valosin, sú spojené s autozomálne dominantným FTD syndrómom asociovaným s Pagetovou chorobou a myozitídou s inklúznymi telieskami. Mutácie v CHMP2B géne boli doteraz popísané len v dvoch rodokmeňoch a prejavom bol bv ‑ FTD. Histologicky bola prítomná zriedkavá FTLD ‑ UPS patológia. Mutácie v TDP ‑ 43 géne a FUS géne sú hlavne asociované s MND, asi v 30 % ich nositelia vyvinú spravidla FTD ‑ MND syndróm, ktorý je takmer výlučne asociovaný s FTLD ‑ TDP patológiou [14,36]. Takisto sú popisované prípady bv ‑ FTD bez MND s FUS patológiou (obr. 2) [14].
Molekulárne mechanizmy neurodegenerácie pri FTLD
Mutácie tau proteínu pravdepodobne vedú k ochoreniu viacerými mechanizmami.
Mutovaný proteín môže mať zvýšený sklon ku agregácii do Pickových teliesok alebo iných neurofibrilárnych inklúzií zložených z rôznych druhov insolubilného tau. Mutovaný tau proteín môže takisto viesť k zníženiu mikrotubulárnej väzby, axonálneho transportu a postihnutiu ďalších procesov závislých na tau proteíne. Na zvieracích modeloch tauopatií sa už preukázali oba tieto mechanizmy. Príkladom môžu byť mutácie v alternatívne procesovanom exóne 10, ktoré vedú k nadmernej tvorbe 4R izoforiem, vedúcej k slabej mikrotubulárnej väzbe, následnej dysfunkcii a agregácii 4R insolubilného tau. Presymptomatickí nosiči mutácií tau proteínu majú frontálny exekutívny deficit pri neuropsychologických testoch aj 10 rokov pred predpokladaným nástupom demencie [40].
Progranulín a jeho koncové metabolity s biologickou aktivitou – granulíny sú zapojené do veľkého počtu provitálnych procesov, vrátane rastu a diferenciácie, reparačných a regeneračných procesov a regulácie bunkového cyklu. Fenotypová prezentácia mutácií je väčšinou autozomálne dominantná, hypotetický patogenetický mechanizmus zahŕňa stratu funkcie, s nedostatočnou podporou rastových faktorov a následnú neurodegeneráciu. Okrem toho, progranulínové mutácie vedú k intraneuronálnym a cytoplazmatickým inklúziám zloženým z DNA viažuceho proteínu TDP ‑ 43 [14,36,40]. Presné mechanizmy vplyvu progranulínových mutácií na neurodegeneráciu nie sú zatiaľ objasnené.
TDP ‑ 43 a FUS proteín patria medzi RNA viažúce proteíny a majú úlohu v transkripcii, v alternatívnom splicingu RNA a v produkcii mikroRNA [36,41,42]. V prípade ich mutácií dochádza k poruchám na epigenetickej úrovni (procesovanie RNA, produkcia mikro‑RNA) a zásadným spôsobom sa narúša expresia génov. TDP ‑ 43 sa v rámci svojej biologickej funkcie podieľa na up ‑ regulácii 362 génov a down ‑ regulácii 239 génov [41,42].
Likvorové biomarkery pri FTLD
Likvorové biomarkery sú suverénnou súčasťou diagnostickej batérie Alzheimerovej choroby. Pri Alzheimerovej chorobe zisťujeme pokles hladiny amyloidu beta, vzostup celkového tau proteínu a vzostup fosforylovaného tau proteínu [43 – 45]. Pokles hladiny amyloidu beta sa začína rádovo roky pred objavením sa prvých príznakov kognitívneho deficitu, takže jeho stanovenie má nielen diagnostickú, ale aj prediktívnu hodnotu. Nasleduje vzostup celkového tau proteínu a fosforylovaného tau proteínu [45]. Pri FTLD je dynamika uvedených proteínov málo výpovedná pre potvrdenie diagnózy [46]. Profil amyloidu beta, celkového a fosforylovaného tau proteínu u FTLD prípadov a vekovo primeraných nedementných kontrol nedosahuje suverénne diagnosticky využiteľný rozdiel [46 – 48]. Výsledky rozsiahlejších štúdií však možno interpretovať tak, že pri všetkých troch syndrómoch FTLD hladina amyloidu beta klesá, pokles je však podstatne miernejší ako pri Alzheimerovej chorobe (tab. 5) [47,49]. Hladina celkového aj fosforylovaného tau proteínu je pri PPA mierne vyššia v porovnaní s kontrolami, pri bv ‑ FTD a SD sú hladiny tau proteínu spravidla bez signifikantného rozdielu s nedementnými kontrolami (tab. 5) [49]. Ďalšie proteínové fragmenty amyloidu beta n40, n38, n17 môžu byť nápomocné pre spresnenie diagnózy FTLD, v praxi však nemajú zásadnejší význam [48,50]. Hladina amyloidu beta (1 – 40) vykazuje výraznejší pokles pri syndrómoch FTLD ako pri Alzheimerovej chorobe [50]. V rutinnej diagnostike sa však nestanovuje. Stanovenie likvorových biomarkerov má však pomocný význam pri rozlíšení atypických foriem Alzheimerovej choroby od syndrómov FTLD. Napríklad logopedický variant môže mať histológiu AD, FTLD ‑ TDP alebo FTLD ‑ Tau. Ak sa biomarkerový profil blíži Alzheimerovej chorobe, zvyšuje pravdepodobnosť diagnózy atypickej AD. Ak sa biomarkerový profil blíži okruhu FTLD je pravdepodobný logopedický variant PNFA. Pre ilustráciu uvádzame hodnoty podľa Bibla et al 2010 [49], podobné výsledky prinášajú aj ďalšie práce [11,47].
![Hodnoty amyloidu beta, celkového tau proteínu a fosforylovaného tau proteínu pri Alzheimerovej chorobe a jednotlivých syndrómoch FTLD (podľa [49]).](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/c5899b0debbcb998cc6c851b5abac87e.png)
V súčasnosti prebieha intenzívny výskum v oblasti rozlíšenia jednotlivých histopatologických variant prostredníctvom nových likvorových biomarkerov ešte počas života pacienta. Keďže FTLD ‑ TDP, FTLD UPS aj FTLD FUS varianty môžu mať agresívnejší priebeh, výraznejšie postihnutie kognitívnych domén a kratší čas prežívania pacientov, ich odlíšenie od FTLD ‑ Tau varianty má diagnostický aj prognostický význam [51]. Ako najsľubnejšie biomarkery sa javia interleukín 17 (IL‑17), eotaxín ‑ 3, adrenokortikotropný hormón (ACTH), agouti related peptid a fas, ktorých výskyt pri FTLD ‑ TDP, FUS aj UPS variante je signifikantne vyšší ako pri FTLD ‑ Tau variante [51]. Rozlíšenie na základe uvedených biomarkerov má 85,7% senzitivitu a 77,8% špecificitu. Akurátnosť rozlíšenia sa teda pohybuje okolo 82 %.
Terapia
Terapeutické ovplyvnenie frontotemporálnej lobárnej degenerácie je zatiaľ veľmi obmedzené. Bol dokázaný deficit v serotonínovom a dopamínovom neurotransmiterovom systéme, zatiaľ čo acetylcholínergný systém zostáva relatívne intaktný. Inhibítory spätného vychytávania serotonínu (SSRI) môžu zmierniť depresiu, apatiu, desinhibíciu, túžbu po sacharidoch, prípadne i ďalšie behaviorálne prejavy chorých s FTLD, nebolo však popísané zlepšenie kognitívnych funkcií [52]. Dvojito zaslepená štúdia s trazodonom preukázala efekt na behaviorálne symptómy, táto práca však zahrňovala iba 26 pacientov. Okrem toho bola liečba trazodonom limitovaná nežiaducimi účinkami (hlavne únavou a ospalosťou) [52]. Atypické antipsychotiká, hlavne olanzapín a kvetiapín možno podávať na zmiernenie agitovanosti, agresivity a neprimeraného správania sa, hlavne u pacientov s frontálnym variantom FTLD [53]. Niektoré práce prinášajú pozitívny vplyv rivastigmínu na zmiernenie progresie rečovej poruchy u pacientov s progresívnou nonfluentnou afáziou a mierne zlepšenie kognitívneho deficitu u pacientov s bv ‑ FTD [54]. Najnovšia multicentrická placebom kontrolovaná štúdia analyzujúca účinok memantínu na kognitívne a exekutívne funkcie nepreukázala žiadny efekt [55]. Aktuálne prebieha multicentrická, randomizovaná placebom kontrolovaná štúdia zameraná na liečbu bv ‑ FTD prostredníctvom inhibítora tau proteínovej agregácie s označením TRx0237 [56]. Je to prvá klinická štúdia, ktorej endpointom by malo byť spomalenie alebo zastavenie patologického procesu.
Záver
Frontotemporálna lobárna degenerácia je heterogénna klinicko‑patologická entita, ktorá je v skupine demencií so skorým začiatkom (pred 65. rokom života) približne rovnako častá ako Alzheimerova choroba. Má spravidla rýchlejšiu progresiu a devastujúcejšie poškodenie postihnutých kognitívnych domén v porovnaní s Alzheimerovou chorobou. Stanovenie diagnózy sa spravidla opiera o vedúcu klinickú symptomatiku, výsledky zobrazovacích vyšetrení a vývoj ochorenia. Pri bv ‑ FTD je v popredí porucha sociálneho správania, pri PNFA expresívna non‑fluentná afázia a pri SD fluentná senzorická afázia. V pokročilejších štádiách ochorenia sa uvedené symptómy kombinujú v rámci jednotlivých syndrómov FTD.
Histopatologicky podľa morfológie a typu proteínov obsiahnutých v inklúziách rozoznávame štyri hlavné subtypy FTLD – FTLD ‑ Tau, FTLD ‑ TDP, FTLD ‑ UPS a FTLD ‑ FUS. Jednotlivé histopatologické varianty sa líšia rýchlosťou progresie ochorenia a dobou prežívania pacientov. Tauopatie majú variabilnú rýchlosť progresie. Popisované sú prípady pomalej progresie (viac ako 10 rokov), hlavne PNFA, aj prípady rýchlej progresie (tri až päť rokov), hlavne fv ‑ FTD, CBD, PSP, ale aj PNFA [11,13,14]. Ostatné histologické formy (FTLD ‑ TDP, FTLD ‑ UPS a FTLD ‑ FUS) sú agresívnejšie s dobou prežívania dvoch až piatich rokov [11,13,14,51].
V súčasnosti je snaha rozlíšiť uvedené subtypy FTLD počas života, hlavne na základe likvorových biomarkerov. Likvorový amyloid beta a tau proteín nemajú dostatočnú rozlišovaciu hodnotu, a preto sa hľadajú nové likvorové biomarkery. Ako najnádejnešie sa javia interleukín 17,eotaxín ‑ 3, ACTH, agouti related peptid a fas, ktorých výskyt je pri FTLD ‑ TDP aFTLD ‑ FUS variante signifikantne vyšší ako pri FTLD ‑ Tau variante.
Magnetická rezonancia je síce štandardnou súčasťou diagnostickej batérie, v úvodných v štádiách však očakávaný vzorec atrofie mozgu nemusí byť prítomný. V štádiu plne rozvinutých symptómov spravidla už nachádzame typickú lokalizáciu atrofie mozgu – pri bv ‑ FTD atrofiu frontálnych lalokov, pri SD predilekčnú atrofiu predného pólu temporálneho laloka a pri PNFA atrofiu perisylviánskej oblasti a asymetrickú atrofiu mozgu s prevahou v ľavej hemisfére. Atrofia hipokampu býva prítomná pri všetkých syndrómoch FTLD, neslúži však ako rozlišovací znak.
Najproblematickejšou oblasťou je terapia syndrómov FTLD, ktorá je obmedzená len na liečbu sprievodných, hlavne behaviorálnych symptómov. Antidepresíva zo skupiny SSRI sú súčasťou všeobecných doporučení, takisto bolo publikovaných viacero prác opisujúcich pozitívny efekt AchE inhibítorov, hlavne na kognitívne funkcie a na rečové schopnosti pri PNFA. Memantín nepreukázal účinnosť na žiadny syndróm FTD.
V našom príspevku sme sa snažili čitateľovi priblížiť frontotemporálnu lobárnu degeneráciu ako komplexnú a v klinickej praxi často poddiagnostikovanú a nedocenenú entitu. Jej príznaky môžu byť hlavne v úvodných štádiách nesprávne hodnotené ako atypická depresia alebo poruchy správania v séniu. Vzhľadom na to a v kontexte zlepšovania diagnostiky a stratifikácie demencií si FTLD zasluhuje osobitú pozornosť, aby sa správne rozpoznala aj v úvodných štádiách a v prípadoch menej typickej klinickej prezentácie.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Stanislav Šutovský, PhD.
I. neurologická klinika
LF UK a UN Bratislava
Mickiewiczova 13
813 67 Bratislava
e-mail: nilusuto@gmail.com
Publikácia vznikla v rámci projektu „Kognitívno-komunikačné poruchy u pacientov s miernou kognitívnou poruchou a demenciou“, spolufinancovaného z grantu APVV-0048-11.
Prijaté k recenzii: 27. 9. 2012
Prijaté do tlače: 19. 2. 2013
Zdroje
1. Pick A. Über die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Med Wochenschr 1882; 17 :165 – 167.
2. Pick A. Senile Hirnatrophie als Grundlage von Herderscheinungen. Wien Klin Wochenschr 1901; 14 : 403 – 404.
3. Pick A. Zur Symptomatologie der linksseitigen Schlafenlappenatrophie. Monatschr Psychiatr Neurol 1904; 16 : 378 – 388.
4. Alzheimer A. Über eigenartige Krankheitsfälle des späteren Alters. Z Gesamte Neurol Psychiatr 1911; 4 : 356 – 385.
5. Hudson AJ. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurologic disorders: a review. Brain 1981; 104(2): 217 – 247.
6. Mitsuyama Y. Presenile dementia with motor neuron disease in Japan: clinical pathological review of 26 cases. J Neurol Neurosurg Psychiatr 1984; 47 : 953 – 959.
7. Maurita K, Kaiya H, Ikeda T, Namba M. Presenile dementia combined with amyotrophy: a review of 34 Japanese cases. Arch Gerontol Geriatr 1987; 6(3): 263 – 277.
8. Neary D, Snowden JS, Mann DM, Northen B, Goulding PJ, Macdermott N. Frontal lobe dementia and motor neuron disease. J Neurol Neurosurg Psychiatr 1990; 53(1): 23 – 32.
9. Neary D, Snowden JS, Mann D. Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry 1994; 57(4): 416 – 418.
10. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51(6): 1546 – 1554.
11. Kipps CM, Knibb JA, Hodges JR. Clinical presentation of frontotemporal dementia In: Hodges JR (ed). Frontotemporal dementia syndromes. Cambridge: Cambridge University Press 2007 : 38 – 79
12. Litvan I. Cortocobasal degeneration. In: Atypical Parkinsonian Disorders – Clinical and Research Aspects. Humana Press 2005 : 309 – 335.
13. Litvan I. Progressive supranuclear palsy. In: Litvan I (ed). Atypical Parkinsonian Disorders – Clinical and Research Aspects. New York: Humana Press 2005 : 287 – 309.
14. Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011; 122(2): 137 – 153.
15. Snowden JS, Bathgate D, Varma A, Blackshaw A,Gibbons ZC, Neary D. Distinct behavioural profiles in frontotemporaldementia and semantic dementia. J Neurol Neurosurg Psychiatry 2001; 70(3): 323 – 332.
16. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134(Pt 9): 2456 – 2477.
17. Šutovský S, Malík M, Traubner P, Turčáni P. Primárna progresívna afázia – zriedkavá alebo poddiagnistikovaná. Neurol Prax 2007; 8(3): 170 – 173.
18. Rektorová I. Frontotemporální lobární degenerace – diagnóza z neuro‑psychiatrického pomezí. Neurol Prax 2006; 7(4): 208 – 211.
19. Rektorová I. Neurodegenerativní demence. Cesk Slov Neurol N 2009; 72/ 105(2): 97 – 109.
20. Gorno ‑ Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF et al. Classification of primary progressive aphasia and its variants. Neurology 2011; 76(11): 1006 – 1014.
21. Vyhnálek M, Škoda D, Varjassyová A, Hort J. Sémantická demence – důkaz mnohotvárnosti paměťových procesů. Neurol Prax 2005; 6(6): 316 – 318.
22. van de Pol LA, Hensel A, van der Flier WM, Visser PJ, Pijnenburg YA, Barkhof F et al. Hippocampal atrophy on MRI in frontotemporal lobar degeneration and Alzheimer‘s disease. J Neurol Neurosurg Psychiatry 2006; 77(4): 439 – 442.
23. Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol 1998; 8(2): 387 – 402.
24. Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration consortium for rontotemporal lobar degeneration. Acta Neuropathol 2007; 114(1): 5 – 22.
25. Mackenzie IR, Foti D, Woulfe J, Hurwitz TA. Atypical frontotemporal lobar degeneration with ubiquitin‑positive, TDP ‑ 43 – negative neuronal inclusions. Brain 2008; 131(Pt 5): 1282 – 1293.
26. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 2009; 117(1): 15 – 18.
27. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 2010; 119(1): 1 – 4.
28. Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M,Bak TH et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol 2004; 56(3): 399 – 406.
29. Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations andPSP. Neurology 2006; 66(1): 41 – 48.
30. Kertesz A, McMonagle P, Blair M, Davidson W,Munoz DG. The evolution and pathology of frontotemporal dementia. Brain 2005; 128(Pt 9): 1996 – 2005.
31. Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol 2007; 114(1): 31 – 38.
32. Grossman M. The non‑fluent/ agrammatic variant of primary progressive aphasia. Lancet Neurol 2012; 11(6): 545 – 555.
33. Sanders J, Schenk VW, van Veen PF et al. A family with Pick disease. Amsterdam: Veerhandelingen de Koninklijike Nederlandese Akademie van Wetenschappen 1939.
34. Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A et al. High prevalence of mutations in the microtubule‑associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 1999; 64(2): 414 – 421.
35. Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M,Adamson J et al. Mutations in progranulin are a major cause of ubiquitin‑positive frontotemporal lobar degeneration. Hum Mol Genet 2006; 15(20): 2988 – 3001.
36. Sieben A, Van Langenhove T, Engelborghs S, Martin JJ, Boon P, Cras P et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 2012; 124(3): 353 – 372.
37. Majounie E, Renton AE, Mok K, Dopper EG, Waite A,Rollinson S et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross ‑ sectional study. Lancet Neurol 2012; 11(4): 323 – 330.
38. Mackenzie IR, Neumann M, Cairns NJ, Munoz DG, Isaacs AM. Novel types of frontotemporal lobar degeneration: beyond tau and TDP ‑ 43. J Mol Neurosci 2011; 45(3): 402 – 408.
39. Goedert M, Spillantini MG. Pathogenesis of the tauopathies. J Mol Neurosci 2011; 45(3): 425 – 431.
40. Snowden JS, Thompson JC, Stopford CL, Richardson AM, Gerhard A, Neary D et al. The clinical diagnosis of early ‑ onset dementias: diagnostic accuracy and clinicopathological relationships. Brain 2011; 134(Pt 9): 2478 – 2492.
41. Polymenidou M, Lagier ‑ Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY et al. Long pre‑mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP ‑ 43. Nat Neurosci 2011; 14(4): 459 – 468.
42. Lagier ‑ Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC et al. Divergent roles of ALS‑linked proteins FUS/ TLS and TDP ‑ 43 intersect in processing long pre‑mRNAs. Nat Neurosci 2012; 15(11): 1488 – 1497.
43. Ressner P, Hort J, Rektorová I, Bartoš A, Rusina R, Línek V et al. Doporučené postupy pro diagnostiku Alzheimerovy nemoci a dalších onemocnění spojených s demencí. Cesk Slov Neurol N 2008; 71/ 104(4): 494 – 501.
44. Hort J, Glosová L, Vyhnálek M, Bojar M, Škoda D,Hladíková M. Tau protein a beta amyloid v likvoru u Alzheimerovy choroby. Cesk Slov Neurol N 2007; 70/ 103(1): 30 – 36.
45. Dubois B, Feldman HH, Jacova C, Cummings JL, Dekosky ST, Barberger ‑ Gateau P et al. Revising the definition of Alzheimer‘s disease: a new lexicon. Lancet Neurol 2010; 9(11): 1118 – 1127.
46. Pijnenburg YA, Schoonenboom NS, Rosso SM, Mulder C, Van Kamp GJ, Van Swieten JC et al. CSF tau and Abeta42 are not useful in the diagnosis of frontotemporal lobar degeneration. Neurology 2004; 62(9): 1649.
47. Schoonenboom NS, Reesink FE, Verwey NA, Kester MI, Teunissen CE, van de Ven PM et al. Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology 2012; 78(1): 47 – 54.
48. Sorbi S, Hort J, Erkinjuntti T, Fladby T, Gainotti G,Gurvit H et al. EFNS Scientist Panel on Dementia and Cognitive Neurology. EFNS ‑ ENS Guidelines on the diagnosis and management of disorders associated with dementia. Eur J Neurol 2012; 19(9): 1159 – 1179.
49. Bibl M, Mollenhauer B, Lewczuk P, Esselmann H, Wolf S, Otto M et al. Cerebrospinal fluid tau, p ‑ tau 181 and amyloid ‑ b38/ 40/ 42 in frontotemporal dementias and primary progressive aphasias. Dement Geriatr Cogn Disord 2011; 31(1): 37 – 44.
50. Verwey NA, Kester MI, van der Flier WM, Veerhuis R,Berkhof H, Twaalfhoven H et al. Additional value of CSF amyloid‑beta 40 levels in the differentiation between FTLD and control subjects. J Alzheimers Dis 2010; 20(2): 445 – 452.
51. Hu WT, Chen ‑ Plotkin A, Grossman M, Arnold SE, Clark CM, Shaw LM et al. Novel CSF biomarkers for frontotemporal lobar degenerations. Neurology 2010; 75(23): 2079 – 2086.
52. Freedman M. Frontotemporal dementia: recommendations for therapeutic studies, designs, and approaches. Can J Neurol Sci 2007; 34 (Suppl 1): S118 – S124.
53. Seltman RE, Matthews BR. Frontotemporal lobar degeneration: epidemiology, pathology, diagnosis and management. CNS Drugs 2012; 26(10): 841 – 870.
54. Lampl Y, Sadeh M, Lorberboym M. Efficacy of acetylcholinesterase inhibitors in frontotemporal dementia. Ann Pharmacother 2004; 38(11): 1967 – 1968.
55. Boxer AL, Knopman DS, Kaufer DI, Grossman M, Onyike C, Graf ‑ Radford N et al. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double‑blind, placebo ‑ controlled trial. Lancet Neurol 2013; 12(2): 149 – 156.
56. New Drugs Online [on‑line]. Available from URL: http:/ / www.ukmi.nhs.uk/ applications/ ndo/ record_view_open.asp?newDrugID=5763.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2013 Číslo 6
Nejčtenější v tomto čísle
- Frontotemporálna lobárna degenerácia z pohľadu nových klinicko‑patologických korelácií
- Tuberózní skleróza u dětí sledovaných od novorozeneckého věku pro prenatální nález rhabdomyomů srdce – dvě kazuistiky
- Expanze pineální krajiny
- Zlomeniny kondylu okciputu