
První český pacient s deficitem aminoacylázy 1
Authors:
D. Procházková 1,2; R. Borská 3; P. Chrastina 4; L. Fajkusová 1,2; P. Konečná 1; K. Slabá 1; J. Šenkyřík 5; K. Pešková 4; P. Jabandžiev 1; T. Honzík 4
Authors‘ workplace:
Pediatrická klinika LF MU a FN Brno
1; Ústav lékařské genetiky a genomiky, LF MU a FN Brno
2; Interní hemato-onkologická klinika, Centrum molekulární biologie a genetiky, LF MU a FN Brno
3; Klinika pediatrie a dědičných poruch metabolizmu 1. LF UK a VFN v Praze
4; Klinika dětské radiologie LF MU a FN Brno
5
Published in:
Cesk Slov Neurol N 2023; 86(1): 83-85
Category:
Letter to Editor
doi:
https://doi.org/10.48095/cccsnn202383
Vážená redakce,
deficit aminoacylázy 1 (ACY1D) (MIM 609924) patří mezi vzácné dědičné poruchy metabolizmu a je charakterizován zvýšeným vylučováním N-acetylovaných aminokyselin do moči (serin, kyselina glutamová, alanin, methionin, glycin, leucin a valin). Porucha se dědí autozomálně recesivně, gen ACY1 (104620.0001) je lokalizován v oblasti 3p21.2. Nemoc byla poprvé popsána v roce 2005 Van Costerem et al [1,2]. Dosud bylo na světě detekováno asi 20 pacientů. Enzym aminoacyláza 1 (EC 3.5.1.14) se exprimuje v lidském těle nejvíce v ledvinách a v mozku. Jedná se o homodimerický zinek vázající metaloenzym, který je lokalizován v cytosolu.
Katalyzuje hydrolýzu (deacetylaci) N-acetylovaných aminokyselin na L-aminokyseliny a acetylovanou skupinu. Poslední studie ukazují, že gen ACY1 hraje patrně významnou roli ve vzniku, vývoji a růstu různých typů zhoubného nádorového bujení u člověka [3].
Fenotyp probandů je variabilní. Onemocnění se nejčastěji manifestuje neurologickým a psychiatrickým postižením, psychomotorickou retardací, poruchou svalového tonu, častěji svalovou hypotonií a slabostí, křečemi, retardací expresivní složky řeči, růstovou retardací, sensorineurální hluchotou, tiky, hyperaktivitou a poruchami autistického spektra. Byly popsány opožděná myelinizace a demyelinizační změny v bílé hmotě, atrofie mozku a mozečku. Probandi mohou být stigmatizováni v obličeji, nejčastěji se jedná o hypertelorizmus a široký kořen nosu [4–8].
V naší práci popisujeme prvního českého dětského pacienta s deficitem aminoacylázy 1. Rodiče jsou nepříbuzní Romové. V rodu probanda se nevyskytují závažná onemocnění. Jedná se o chlapce z 1. gravidity, porod v termínu, sekcí, porodní hmotnost 2 800 g, porodní délka 47 cm. Poporodní adaptace v normě. Od kojeneckého věku sledován neurologem pro hypotonii, retardaci vertikalizace a retardaci expresivní složky řeči. Pro tiky a dětský autismus v péči psychiatra, na medikaci risperidonem. Pacient trpí bizarními neúčelnými pohyby, nenavazuje sociální a verbální kontakt, je hluboce autistický, jen ojediněle vydává zvuk. Jeho stav je hodnocen jako těžká mentální retardace (IQ 30).
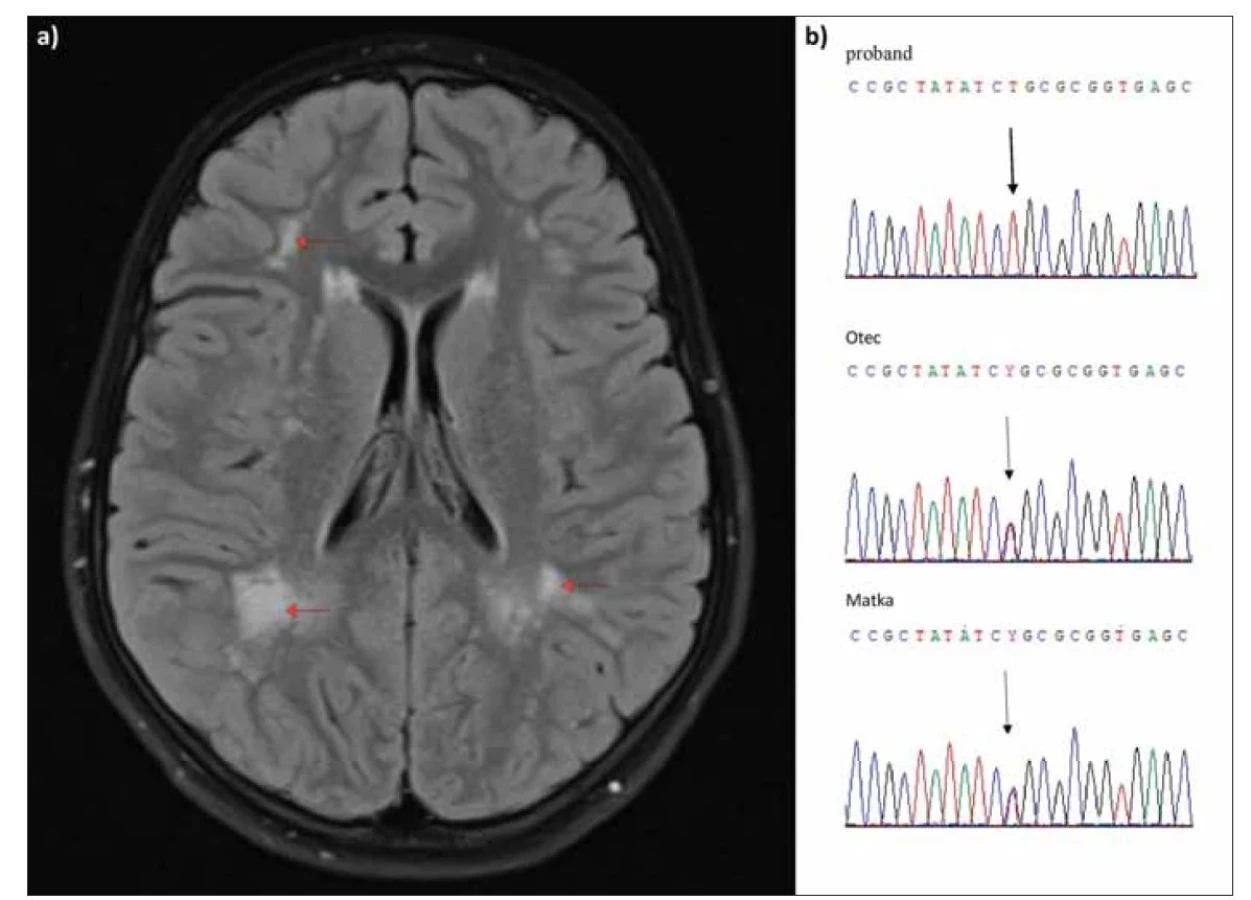
V 9 letech věku byl přijat do FN Brno pro akutní gastroenteritidu s výraznou iontovou dysbalancí. Jeho antropometrické parametry byly v normě (výška 132 cm, hmotnost 27 kg, body mass index [BMI] 15,5, tj. 25.–50. percentil, výška k věku 25.–50. percentil), stejně jako kardiologický nález. Dítě bylo stigmatizované (hypertelorizmus, široký kořen nosu – obr. 1), hypotonické, nemluvilo, nedodržovalo tělesnou čistotu, mělo pleny a nenavazovalo oční kontakt. Bylo vysloveno podezření na poruchu sluchu. Vyšetření sluchu (brainstem evoked response audiometry; BERA) potvrdilo sensorineurální (percepční) hluchotu středně těžkého charakteru. Byla provedena MR mozku, nativní vyšetření, která odhalila v bílé hmotě mozku nahodilá mnohočetná ložiska demyelinizačního charakteru, která byla lokalizována převážně v paraventrikulární a juxtakortikální zóně, zejména vpravo parietookcipitálně splývala ve větší okrsky. V těle levé postranní komory byla detekována cysta cca 3 mm (obr. 2a). Vyšetření očního pozadí bylo v normě. V rámci diferenciální diagnostiky byla zvažována možnost dědičné poruchy metabolizmu a proveden metabolický screening. Z výsledků krevních vyšetření: alaninaminotransferáza (ALT) 0,28 µkat/l (norma 0,25–0,26), aspartátaminotransferáza (AST) 0,41 µkat/l (0,2–0,63), kreatinkináza (CK) 1,42 µkat/l (0,2–2,27), laktátdehydrogenáza (LD) 2,6 g/l (2–5), laktát z nezatažené končetiny 2,9 mmol/l (0,6–2,1), mírná hyperlaktacidémie, laktát v moči v normě.
Fig. 1. Hypertelorism, wide root of the
nose.

FLAIR – fluid attenuated inversion recovery
Fig. 2. (a) MRI scan, FLAIR sequence in the axial plane, multiple demyelinating foci of white matter of the brain with predominance in
paraventricular ditribution (arrows). (b) Sanger sequencing confirmed the pathogenic sequential variant of missense type c.1057C>T p.
(Arg353Cys) in the ACY1 gene (NM_000666.2) in the homozygous state in the proband and in the heterozygous state in the parents.
FLAIR – fluid attenuated inversion recovery
Ultrazvuk svalových skupin lýtek v normě, EMG a EEG nebyly provedeny pro nespolupráci pacienta.
Byl stanoven profil organických kyselin v moči extrakcí do ethylacetátu s ethoxim-trimethylsilyl derivatizací a následnou plynovou chromatografií s hmotnostně spektrometrickou detekcí [9]. V profilu organických kyselin v moči bylo zjištěno zvýšené vylučování N-acetyl-alaninu (27 mg/gKr), N-acetyl-glycinu (23 mg/gKr), N-acetyl-methioninu (78 mg/gKr), N-acetyl-glutamátu (45 mg/gKr) a N-acetyl-valinu (12 mg/gKr), které se za fyziologických podmínek v moči nedetekují. Tento překvapující nález vedl k vyslovení podezření na možný deficit enzymu ACY1D.
V Centru molekulární biologie a genetiky Interní hemato-onkologické kliniky FN Brno byla provedena analýza DNA genu ACY1 pomocí panelového sekvenování nové generace (target sequencing) s využitím SeqCap EZ Choice Library (Roche Nimble-Gene, Madison, WI, USA) na platformě Illumina. Nalezená sekvenční varianta NM_000666.2, c.1057C>T p. (Arg353Cys) byla potvrzena pomocí Sangerova sekvenování u probanda v homozygotním stavu a u rodičů v heterozygotním stavu (obr. 2b). Uvedená mutace typu missense je uvedena v databázi HGMD jako kauzální, spojená s ACY1D. Poprvé byla popsána Van Costerem et al [1] v roce 2005 a je dosud nejčastěji popsanou patogenní sekvenční variantou pro ACY1D.
ACY1D patří mezi velmi vzácné dědičné poruchy metabolizmu. První dítě bylo popsáno v roce 2005 Van Costerem et al [1]. Jednalo se o novorozence s encefalopatií, která nastoupila 3. den po porodu. Dítě bylo hypotonické, zvracelo, mělo dechové potíže, apnoe a percepční hluchotu.
Námi popisované romské dítě s ACY1D trpí stigmatizací v obličeji, hypotonií, mentálním postižením, poruchou autistického spektra, tiky, percepční hluchotou středně těžkého stupně a demyelinizačními změnami bílé hmoty mozku. Romské dítě s ACY1D bylo popsáno v roce 2007 Sassem et al [2]. Trpělo multifokální farmakorezistentní epilepsií od 1 roku věku, bylo hyperaktivní a mělo lehkou mentální retardaci. Bylo stigmatizováno v obličeji stejně jako náš proband (hypertelorizmus a široký kořen nosu).
Fenotyp dalších probandů byl značně heterogenní. Jsou popisovány atrofie mozečku, syringomyelie, febrilní křeče, tranzitorní hemiplegie, hypotonie, hypertonie, opistotonus, dystonie [7] a retardace expresivní složky řeči [1,2,6]. V roce 2010 popsali Tylki-Szymanska et al probanda s poruchou autistického spektra [4], stejně jako Alessandri et al v roce 2018 [8].
Byla popsána probandka s mírným neurologickým postižením ve smyslu horší pohybové koordinace (nemotornost) a impulzivním chováním a hyperaktivitou [5].
Objasnění diagnózy našeho probanda ukazuje, že v nejasných případech je třeba takové pacienty vést v evidenci, pomýšlet na možnost výskytu velmi vzácné dědičné poruchy metabolizmu a využívat všechny diagnostické možnosti k objasnění kauzální příčiny postižení. Dosud nejvíce probandů s ACY1D bylo popsáno v souvislosti s dětmi, které podstoupily selektivní metabolický screening pro opoždění psychomotorického vývoje a výskyt křečí v anamnéze. Diagnóza byla stanovena pomocí gas chromatography-mass spectrometry (GC-MS) s vyšetřením organických kyselin v moči a potvrzena molekulárně genetickým vyšetřením genu ACY1 a detekcí snížené enzymové aktivity enzymu ACY1 v Epstein-Barr virus-transformovaných lymfoblastech nebo fibroblastech.
Kauzální léčba nemoci není známa. Možnost provést molekulárně genetické potvrzení diagnózy ACY1D má zásadní význam pro rodinu postiženého probanda. Vyšetření genu ACY1 postižení potvrdí, umožní vyšetřit další členy rodiny a provést možnou prenatální, resp. preimplantační diagnostiku.
Vzhledem k nízkému počtu diagnostikovaných případů dosud nelze předvídat klinický průběh nemoci a prognóza pacientů zůstává nejasná.
Etické aspekty
Fotografie probanda uveřejněna se souhlasem rodičů.
Grantová podpora
Práce vznikla za podpory projektu MZ ČR RVO FNB 65269705 a MZ ČR RVO VFN 64165.
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem práce nemají žádný konflikt zájmů.
doc. MUDr. Dagmar Procházková, Ph.D.
Pediatrická klinika
LF MU a FN Brno
Černopolní 9
613 00 Brno
e-mail: prochazkova.dagmar@fnbrno.cz
Přijato k recenzi: 24. 6. 2022
Přijato do tisku: 1. 12. 2022
Sources
1. Van Coster RN, Gerlo EA, Giardina TG et al. Aminoacylase I deficiency: a novel inborn error of metabolism. Biochem Biophys Res Commun 2005; 338 (3): 1322–1326. doi: 10.1016/j.bbrc.2005.10.126.
2. Sass JO, Mohr V, Olbrich H et al. Mutations in ACY1, the gene encoding aminoacylase 1, cause a novel inborn error of metabolism. Am J Hum Genet 2006; 78 (3): 401–409. doi: 10.1086/500563.
3. Chen H, Wang W, Xiao C et al. ACY1 regulating PTEN/PI3K/AKT signaling in the promotion of non-small cell lung cancer progression. Ann Transl Med 2021; 9 (17): 1378. doi: 10.21037/atm-21-3127.
4. Tylki-Szymanska A, Gradowska W, Sommer A et al. Aminoacylase 1 deficiency associated with autistic behavior. J Inherit Metab Dis 2010; 33 (Suppl 3): S211–214. doi: 10.1007/s10545-010-9089-3.
5. Alessandri MG, Casarano M, Pezzini I et al. Isolated mild intellectual disability expands the aminoacylase 1 phenotype spectrum. JIMD Rep 2014; 16: 81–87. doi: 10.1007/8904_2014_323.
6. Ferri L, Funghini S, Fioravanti A et al. Aminoacylase I deficiency due to ACY1 mRNA exon skipping. Clin Genet 2014; 86 (4): 367–372. doi: 10.1111/cge.12297.
7. Sass JO, Vaithilingam J, Gemperle-Britschgi C et al. Expanding the phenotype in aminoacylase 1 (ACY1) deficiency: characterization of the molecular defict in a 63-year-old woman with generalized dystonia. Metab Brain Dis 2016; 31 (3): 587–592. doi: 10.1007/s11011-015-9778-6.
8. Alessandri MG, Milone R, Casalini C et al. Four years follow up study of ACY1 defitient patient and pedigree study. Brain Dev 2018; 40 (7): 570–575. doi: 10.1016/j.braindex.2018.03.009.
9. Chalmers RA, Lawson AM. Organic acid in man, analytical chemistry, biochemistry and diagnosis of the organic acidurias. Chapman and Hall Ltd.: London 1982.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2023 Issue 1
Most read in this issue
- Progresivní roztroušená skleróza ve světle nejnovějších poznatků
- Dietní přístupy specifické pro pacienty s roztroušenou sklerózou
- Doporučení pro strukturální zobrazení MR mozku v diagnostice epilepsie
- Specifické nástroje pro hodnocení kvality života související se zdravím u mladých pacientů po ischemické cévní mozkové příhodě