
Okulofaryngeální muskulární dystrofie v populaci České republiky
Oculopharyngeal Muscular Dystrophy in the Population of the Czech Republic
Oculopharyngeal muscular dystrophy is an autosomal dominant muscular dystrophy due to amplification of the GCG triplet in the polyadenylate binding protein nuclear 1 gene (PABPN1) located on chromosome 14q11.2–q13. The first symptoms, usually facial muscle weakness, frequently occur in the fourth or sixth decade; other symptoms include dysphagia, progressive ptosis of the upper eyelids as well as weakness of limb-girdle muscles and sometimes anal or urethral sphincter weakness. The prevalence of the mutation varies depending on geographic region (the highest prevalence was found in Bukchara Jews in Izrael 1 : 600 and in Quebec 1 : 1,000 and very low in France 1 : 200,000). This mutation is very stable and shows no anticipation phenomenon. We correlated the phenotype and genotype of 36 patients from five independent families and we found no correlation between the onset of symptoms, progression, severity of phenotype and number of triplet GCG repetitions. The prevalence of OPMD in the Czech Republic can only be estimated (35/10 million, i.e. 1 : 285,700) and carriers of mutation and permutation, respectively, are substantially more common, because the majority of young relatives of probands with 50% genetic risk refused presymptomatic testing.
Key words:
oculopharyngeal muscle dystrophy – PABPN1 protein – trinucleotid repeat expansion – ptosis – dysphagia – limb-girdle myopathy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
R. Mazanec 1; P. Seeman 2,3; E. Seemanová 4
Authors‘ workplace:
Neurologická klinika 2. LF UK a FN v Motole, Praha
1; DNA laboratoř Kliniky dětské neurologie 2. LF UK a FN v Motole, Praha
2; Centrum lékařské genetiky Gennet, Praha
3; Oddělení klinické genetiky Ústavu biologie a lékařské genetiky 2. LF UK a FN v Motole, Praha
4
Published in:
Cesk Slov Neurol N 2013; 76/109(6): 717-722
Category:
Short Communication
Overview
Okulofaryngeální muskulární dystrofie (OPMD) je autozomálně dominantně dědičná svalová dystrofie v důsledku amplifikace tripletu GCG v polyadenylate binding protein nuclear 1 (PABPN1) genu na chromozomu 14q11.2– q13. Choroba se obvykle manifestuje ve 4. až 6. dekádě, nejčastěji slabostí obličejových svalů (hypomimie), ptózou, dysfagií a svalovou slabostí, zejména pelvifemorálních svalů, někdy mohou být postiženy i sfinktery. OPMD se vyznačuje variabilním výskytem v závislosti na geografických faktorech (nejvyšší prevalence je mezi bucharskými Židy v Izraeli 1 : 600 nebo v kanadské provincii Quebec 1 : 1 000, nízká je ve Francii 1 : 200 000). Mutace – zvýšený počet tripletů GCG – vykazuje vysokou stabilitu v PABPN1 genu a nebyl pozorován fenomén anticipace. V České republice dosud nebyl podán systematický přehled o výskytu této svalové dystrofie. Z nám dostupné databáze českých pacientů s OPMD jsme porovnali fenotyp s genotypem u 36 pacientů z pěti nezávislých rodin a nepotvrdili jsme závislost mezi věkem nástupu příznaků, rychlostí progrese, závažností projevů a počtem repetic tripletů GCG v exonu 1 PAPBN1 genu. Prevalenci OPMD v České republice lze jen odhadnout (35/ 10 mil., tj. 1 : 285 700), neboť nositelů mutace je mnohem více, avšak v rodinách našich probandů presymptomatické genetické testování odmítla většina mladších příbuzných s 50% rizikem manifestace OPMD. Prevalenci mutace v naší populaci tudíž nelze upřesnit.
Klíčová slova:
okulofaryngeální svalová dystrofie –PABPN1 protein – trinukleotidová expanze –ptóza – dysfagie – pletencová myopatie
Úvod
Okulofaryngeální muskulární dystrofie (OPMD) je abiotrofické progresivní svalové onemocnění s autozomálně dominantním typem dědičnosti a pozdním nástupem klinických symptomů. Termín abiotrofie označuje odloženou manifestaci klinických příznaků, tj. nemoc probíhá řadu let asymptomaticky a po klinické manifestaci dále progreduje. OPMD patří do skupiny více než 25 chorob, které vznikají důsledkem specifického typu mutace – zvýšeného počtu tripletů, někdy také označované jako amplifikace trinukleotidů. Příčinou nemoci je zmnožení tripletu GCG nad normální počet, tj. šest opakování, v exonu 1 PolyAdenylate Binding Protein Nuclear 1 genu (PABPN1) v lokusu 14q11.2– q13 [1]. Gen kóduje jaderný poly(A)binding protein 1, který se vyskytuje ve všech tkáních, nejvíce v kosterních svalech. Vyšší počet tripletů GCG než šest má za následek tvorbu polyalaninových makromolekul v buněčném jádře, jež jsou odolné vůči proteozomální degradaci. Během trvání choroby dochází k jejich kumulaci a k formování jaderných intranukleárních inkluzí. V určitém okamžiku je buněčné jádro již natolik naplněno těmito inkluzemi, že dochází k poruše jaderných funkcí, které vedou k předčasnému zániku buňky (vliv na další jaderné proteiny a funkci mRNA). OPMD je první choroba, u níž byl potvrzen toxický vliv polyalaninu důsledkem zmnožení trinukleotidovů (GCG)n. Opakování tripletu (GCG)7 je považováno za premutaci a v heterozygotním stavu zřejmě nevede ke klinickým projevům, nicméně u složených heterozygotů s premutací (GCG)7 a plnou mutací (GCG)9– 11 je popisován časný nástup příznaků již ve 3. a 4. dekádě života a rychlejší progrese [2].
Cílem práce je upozornit na zřejmé opomíjení této afekce v naší populaci a korelovat fenotyp s genotypem v rodinách našich probandů.
Epidemiologická data
Populační frekvence této choroby není v České republice známa. Ve světě existují významné geografické rozdíly, např. v populaci bucharských Židů v Izraeli byla zjištěna mutace u 1 : 600 osob [3], stejně vysoká je v kanadské provincii Quebec 1 : 1 000 [4], zatímco ve Francii, odkud emigrovali do Quebecu předci všech známých kanadských pacientů, byla zjištěna jen u 1 : 200 000. Mutace je velmi stabilní, jak potvrzují údaje z Quebecu a od bucharských Židů, což vysvětluje odložená manifestace závažné poruchy do vyššího věku, překvapivá je však stabilita počtu zmnožení po 3– 4 generace v rodině [5]. Epidemiologická data jsou do určité míry ovlivněna i tzv. dekádově specifickou penetrancí příznaků choroby u nosičů např. dominantně dědičné mutace (GCG)9 – 1 % (do 40 let), 6 % (40– 49 let), 31 % (50– 59 let), 63 % (60– 69 let) a 99 % (nad 69 let), čili choroba se manifestuje vždy u osob starších 70 let [6].
Klinický obraz choroby
OPMD je choroba s pozdním nástupem prvních příznaků až v 5.– 6. dekádě. Nejčastějším prvním příznakem je ptóza víček a slabost obličejových svalů, které dávají obličeji maskovitý výraz již před začátkem subjektivních obtíží. Kromě ptózy se může objevit i diplopie jako důsledek poruchy extraokulárních svalů, častěji ve směru vertikálním než horizontálním. Progrese choroby vede záhy ke vzniku dysfagie, nejprve tuhých jídel a později i tekutin. Příčinou dysfagie je oslabení faryngeálních svalů, které nejsou schopny překonat bariéru v oblasti faryngoezofageálního přechodu. Kromě této dysfunkce se na dysfagii podílí i oslabení a atrofie jazyka, jež jsou patrny u 82 % nemocných, a ochablost svalstva měkkého patra vede k rinofonii u 67 % [7]. Pokročilá dysfagie vede k opakovaným aspiracím s rizikem aspirační pneumonie, je příčinou hubnutí až kachektizace nemocných a představuje rozhodující faktor morbidity a mortality. Kromě těchto dvou hlavních příznaků nastupují v 6. a 7. dekádě života myopatické obtíže ze svalové slabosti pletencových svalů, častěji pelvifemorálních (71 %), méně i skapulohumerálních (38 %) [7]. Na horních končetinách jsou postiženy hlavně deltové svaly a bicepsy, na dolních končetinách flexory a extenzory kyčle, dále adduktory a abduktory stehen. Důsledkem slabosti těchto svalových skupin jsou poruchy chůze a stability, hrozí riziko pádu a narůstá závislost na zdravotních pomůckách (hole, vozík) i na druhé osobě. Kromě postižení kosterních svalů byly u 10 ze 13 homozygotů s 13x (GCG) expanzí, v úzké komunitě uzbeckých Židů a francouzských Kanaďanů, popsány kognitivní a psychické poruchy (deprese, paranoidní psychóza, agrese). U těchto pacientů se první symptomy OPMD objevují již ve 35 letech a délka života je zřetelně zkrácená (umírají v 6. dekádě) [9]. Tato studie ukazuje, že OPMD u homozygotů (expanze GCG tripletů na obou alelách) není nemoc vázaná pouze na kosterní svaly, ale postihuje i centrální nervový systém. Naopak periferní neuropatie do obrazu OPMD nejspíše nepatří [10,11]. Z pomocných vyšetření zjišťujeme zvýšené hodnoty kreatinkinázy dle tíže postižení kosterních svalů (u našich pacientů v rozmezí 6,5– 43,2 µkat/ l) a volného sérového myoglobinu (117– 390 µg/ l). Jehlová elektromyografie prokazuje normální spontánní aktivitu (bez fibrilací, pozitivních ostrých vln) a při volním úsilí rychlý nábor nízkých a úzkých potenciálů motorických jednotek (myogenní lézi) (tab. 1).
![Základní klinická kritéria pro dominantní OPMD [7,8].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/76024f68c5f6254ddbf3cb833d8b11e0.png)
Současná diagnostika OPMD je dnes snadno dostupná molekulárněgenetickými testy (viz dále). Tyto testy nahradily svalovou biopsii, dříve z diagnostických důvodů indikovanou. Pro OPMD byly ve svalové biopsii typické intranukleární inkluze a tzv. rimmed vakuoly (obr. 1) [12].

Diferenciální diagnostika
V diferenciální diagnostice OPMD připadají v úvahu další klinické jednotky, které jsou spojené s ptózou, okohybnými poruchami, hypomimií a dysfagií. Na prvním místě sem patří vzhledem k věku při manifestaci okulární forma myasthenie gravis. Pro myasthenii svědčí spíše náhlý začátek potíže než chronický průběh, fluktuace příznaků se zhoršením během dne a k večeru a negativní rodinná anamnéza. V objektivních testech je důležitý Coganův test, kdy usilovný pohled očima dolů a následný rychlý návrat bulbů do původní polohy vyprovokuje u myasteniků výraznou ptózu. Z dalších jednotek je nutné zvažovat mitochondriální myopatie, hlavně chronickou progresivní extraokulární oftalmoplegii (CPEO) a Kearnsův‑ Sayreův syndrom, které se však manifestují mnohem dříve a brzy vedou k dalším komplikacím. Třetí jednotka spojená s ptózou a dysfagií je myotonická dystrofie 1 typu (MD1). Myotonické příznaky, převaha distální svalové slabosti a katarakta pomohou odlišit MD1 od OPMD (tab. 2).

Soubor pacientů
V genetické ambulanci jsme v průběhu 10 let konzultovali pět rodin s podezřením na okulofaryngeální muskulární dystrofii. V každé z pěti rodin byl nejméně se dvěma členy sestaven čtyř‑ až pětigenerační rodokmen. Nejstarší generace dokumentuje první známé postižené v 19. století. Novorozená dívka je nejmladším jedincem v riziku. Onemocnění bylo klinicky ověřeno lékařskými zprávami např. z neurologie, ortopedie, oftalmologie, kardiologie. U již zemřelých příbuzných bylo onemocnění hodnoceno nejen anamnesticky, ale i za pomoci rodinné fotodokumentace (ptóza víček, maskovitý obličej). V genealogii jsme zjistili onemocnění u 35 osob. Molekulárně genetickou analýzu jsme indikovali u osmi klinicky manifestních nemocných a 56 jejich příbuzných s 50% pravděpodobností přenosu mutace od rodiče. Možnosti presymptomatické diagnostiky nevyužilo 39 osob, přičemž 16 jich žije v zahraničí.
Molekulárněgenetická diagnostika
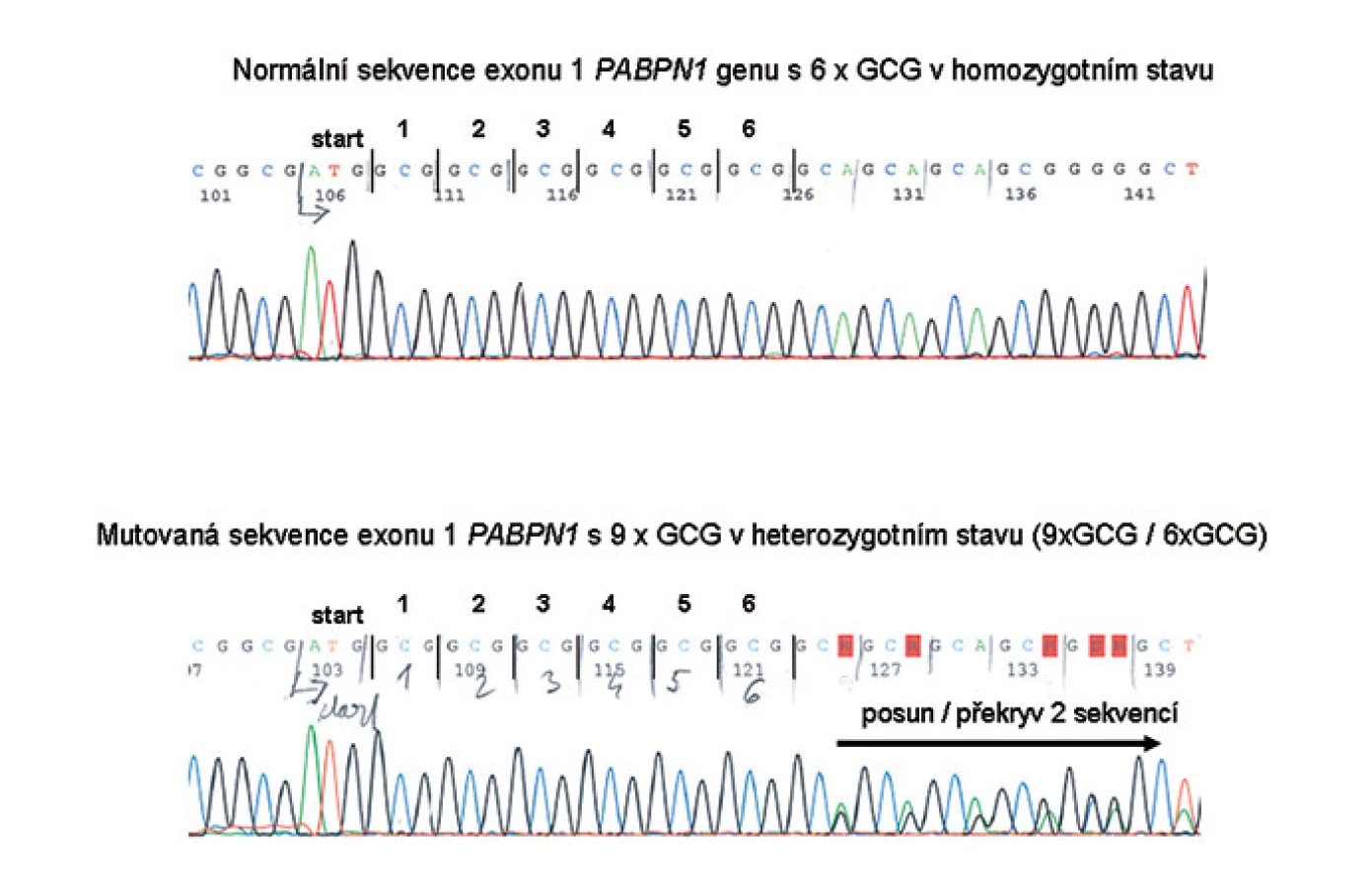
DNA diagnostika OPMD: zpočátku byly používány zjednodušené metody fragmentační analýzy délky PCR produktů obsahující oblast s repeticí a ze zvýšené délky úseku bylo usuzováno na počet tripletů. Později bylo ale zjištěno, že u některých osob je v této oblasti s triplety GCG vložena navíc jiná sekvence či jiné base, které repetici přerušují. Proto je v současnosti již používáno sekvenování kritické oblasti exonu 1 PABPN1 genu. Sekvenování části exonu 1 PABPN1 genu jsme tudíž použili i pro vyšetření rodin B a F a využívá se v DNA diagnostice OPMD v centru Gennet v Praze pro indikované případy k potvrzení nebo vyloučení zvýšeného počtu tripletů GCG (obr. 2).
Autoři prohlašují, že studie na lidských subjektech popsaná v manuskriptu byla provedena v souladu s etickými standardy příslušné komise (institucionální a národní) odpovědné za provádění klinických studií a Helsinskou deklarací z roku 1975, revidovanou v r. 2000.
Výsledky
Molekulárněgenetické vyšetření mohlo být provedeno u 25 jedinců (osm nemocných a 17 dosud zdravých jedinců s 50% rizikem přenosu autozomálně dominantní OPMD). Diagnóza OPMD byla potvrzena DNA vyšetřením u pěti probandů, u dalších tří klinicky nemocných příbuzných a u 12 dosud asymptomatických příbuzných. U pěti osob v riziku DNA vyšetření vlohu pro OPMD vyloučilo.
Genealogická analýza umožnila následná zjištění:
- dokumentovat přenos choroby v přímé linii ve všech generacích. V žádné rodině nebyla zjištěna mutace de novo vzniklá,
- potvrdit autozomálně dominantní typ přenosu choroby – onemocnění děděné i z otce na syna a poměr zdravých a nemocných potomků pacientů byl 37 ze 47 (tj. po odečtení probandů 2 : 3), což by mohlo svědčit pro genetickou výhodu gamet s amplifikací GCG tripletů při fertilizaci,
- odhadnout frekvenci OPDM v naší populaci. První příznaky v souboru našich nemocných se projevily hypomimií až maskovitým obličejem, ptózou očních víček po 40. roce věku (tab. 3), a proto jsme pro odhad frekvence expanze GCG tripletu zvolili rok 1970. Tito jedinci jsou již v 5. deceniu a rodina si všímá prvních příznaků, které zná od již starších nemocných. Z našeho souboru v roce 1970 žilo v pěti rodinách 35 pacientů. Nejnižší odhad frekvence OPMD v České republice tedy odpovídá 1 : 285 700,
- odhadnout, že OPMD pacienta sice dlouhodobě invalidizuje, avšak nezkracuje délku života a kardiální postižení jsou vzácná,
- amplifikace tripletu GCG v lokusu 14q11– q13 je pro rodinu charakteristická a u jednotlivých členů rodiny vždy stejná a nezměněná. V rodině K je expandovaná alela 9x (GCG), v rodině F 10x (GCG), rodině B 11x (GCG), v rodině Z 8x (GCG) a v rodině D 12x (GCG). Nálezy potvrzují známou stabilitu mutace,
- usuzovat, že námi vyšetřované rodiny nejsou ani v dávné minulosti spojeny pokrevním příbuzenstvím, neboť každá rodina má odlišnou mutaci s odlišně zvýšeným počtem tripletů GCG expanzi, která je pro každou z rodin charakteristická,
- usuzovat, že u OPDM genotyp nekoreluje s fenotypem. Klinicky nejzávažnější z hlediska začátku onemocnění a rychlosti progrese do pohybových obtíží se jeví rodina F, jejíž amplifikovaná alela představuje devět opakování GCG. Mezi příslušníky dalších rodin není podstatnější rozdíl v začátku a postupu progrese příznaků, i když zmnožení tripletů je v každé rodině jiné. V tab. 3 uvádíme začátek a rychlost progrese jednotlivých příznaků a počet kopií GCG v PABPN1 genu v lokusu 14q11.2– q13.
Diskuze
Autozomálně dominantně dědičná okulofaryngeální muskulární dystrofie je abiotrofické dlouhodobě invalidizující progresivní svalové onemocnění. V evropské populaci se zřejmě jedná o vzácně se vyskytující afekci. V České republice je náš nejnižší odhad 1 : 285 700 srovnatelný s údajem o frekvenci onemocnění ve Francii (1 : 200 000), i když v izolovaných populacích bucharských Židů v Izraeli a Francouzů v Quebecu je známa frekvence nositelů mutace až 1 : 600– 1 000 [2,4,5]. Předávání závažné choroby z generace na generaci má příčinu v abiotrofickém charakteru afekce, klinická manifestace je odložena do středního věku, kdy nositel mutace má děti, mnohdy i vnoučata. Zajímavé je vysoké riziko přenosu mutace do další generace v našich rodinách (poměr 2 : 3 oproti očekávanému 1 : 1) v našem souboru. Pravděpodobný je vliv malých čísel našeho souboru, nicméně nelze vyloučit ani selekční výhodu mutace, jak je dokumentován u jiné afekce z amplifikace trinukleotidů – syndromu fragilního X chromozomu – v tzv. Shermanové paradoxu – , jehož podstata není objasněna. O stabilitě genu kromě abiotrofie svědčí i konstantní počet opakování tripletů GCG u jednotlivých členů ve stejných rodinách [4]. V našich pěti rodinách je počet opakování charakteristický pro jednotlivé rodiny, a lze tedy soudit, že nejsou pokrevně příbuzné (founder efekt). Zkušenosti s OPMD z našich pěti rodin ukazují, že kardiální porucha je ve srovnání s jinými myopatiemi vzácná.
Léčba
OPMD je vzácně se vyskytující choroba, jejíž kauzální léčba není dostupná a k dispozici je pouze léčba symptomatická. Pacient se musí naučit snižovat rizika aspirace. Nejprve nastupují dietní opatření (např. krájení jídla na malé kousky, kašovitá strava, vysoký obsah proteinů ve stravě), později zavedení sondy do žaludku perkutánní endoskopickou gastrostomií nebo cricoezofageální myotomií. Nově se zkouší dilatace jícnu nebo aplikace botulotoxinu do horního ezofageálního sfinkteru. Ptózu víček koriguje nemocný nejprve záklonem a polohováním hlavy, poté plastickou operací víček. Nevýhodou jsou však recidivy ptózy i po operaci během progrese choroby. U pacientů s rozvinutým myopatickým syndromem jsou na místě zdravotní pomůcky. Progresivní a dlouholetý průběh nemoci vede k postižení různých svalových skupin a vyžaduje pro pacienty zajištění mezioborové péče – neurologem, gastroenterologem, nutricionistou, oftalmologem, plastickým chirurgem, foniatrem, fyzioterapeutem, příp. dalšími specialisty. První příznaky mohou být pozorovány spíše okolím, aniž by pacienta obtěžovaly (hypomimie, maskovitý obličej, rinofonie), ale již tehdy by zvláště v rodinách s výskytem OPMD měly přivést pacienta k neurologovi.
V současnosti jsou v literatuře udávány experimentální zkušenosti s inhibitory proteotoxických účinků polyalaninu ve svalových vláknech (sirtuin), pokud je léčba zahájena co nejdříve [13]. V experimentálních modelech transgenních myší byl potvrzen příznivý efekt doxycyklinu, který je schopen redukovat počet intranukleárních agregátů, a tím brzdit jejich toxicitu u OPMD. Tento objev se může v budoucnu uplatnit nejen u OPMD, ale i u jiných s agregáty asociovaných chorob [14]. Uvažuje se rovněž o využití vektorů kombinujících „gene silencing“ techniky mutovaného genu a jeho náhradu „zdravým“ genem, podobně jako u jiných svalových dystrofií.
Národní registry vzácných chorob neuromuskulárních
Kvalita zdravotní péče a potenciální možnosti moderní terapie této závažné svalové dystrofie jsou závislé na záchytu jednotlivých případů v České republice a jejich evidenci v národním registru. V roce 2011 byl ve spolupráci s MU (Brno) a díky aktivitě neuromuskulární sekce ČNS JEP spuštěn národní registr závažných svalových dystrofií (Duchenneova a Beckerova progresivní dystrofinopatie, myotonické dystrofie a facioskapulohumerální svalové dystrofie). Rovněž OPMD by měla být, vzhledem k závažnosti průběhu, do tohoto registru zařazena. Pouze dokonalá evidence nemocných se vzácnými chorobami v národních registrech totiž umožní rozvíjet národní programy např. v rámci České asociace pro vzácná onemocnění (ČAVO) a využít i potenciální možnosti finanční i léčebné, které nabízí pod záštitou Evropské unie komise Evropského výboru expertů pro vzácná onemocnění (EUCERD).

Autoři děkují pacientům a jejich rodinám za spolupráci při sběru dat a rodině B za poskytnutí rodinné fotografie.
Podpořeno OPPK CZ.2.16/3.1.0024022 a MZ ČR‑RVO, FN v Motole 00064203.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
as. MUDr. Mazanec Radim, Ph.D.
DNA laboratoř Kliniky dětské neurologie
2. LF UK a FN v Motole
V Úvalu 84, 150 06 Praha 5
e-mail: radimmaz@hotmail.com
Přijato k recenzi: 8. 3. 2013
Přijato do tisku: 24. 4. 2013
Sources
1. Brais B, Xie ZG, Sanson M, Morgan K, Weissenbach J, Korcyn AD et al. The oculopharyngeal muscular dystrophy locus maps to the region of the cardiac alpha and beta myosin heavy chain genes on chromosome 14q11.2– q13. Hum Mol Genet 1995; 4(3): 429– 434.
2. McKusick VA. Mendelian inheritance in man [on-line]. Mendelian Inheritance in Man. Available from URL: http:/ / OMIM.org.
3. Blumen SC, Nisipeanu P, Sadeh M, Asherov A, Blumen N, Wirguin Y et al. Epidemiology and inheritance of oculopharyngeal muscular dystrophy in Israel. Neuromuscular Disord 1997; 7 (Suppl 1): S38– S40.
4. Brais B, Bouchard JP, Jomphe M. When genetics and history converge: the fine mapping and North American introduction and diffusion of the French Canadian oculopharyngeal muscular dystrophy mutation. Am J Hum Genet 1998; 63: 229.
5. Blumen SC, Korczyn AD, Lavoie H, Medynski S, Chapman J, Asherov A et al. Oculopharyngeal muscular dystrophy among Bukhara Jews is due to a founder (GCG)9 mutation in the PABP2 gene. Neurology 2000; 55(9): 1267– 1270.
6. Bouchard JP, Brais B, Brunet D, Gould PV, Rouleau GA. Recent studies on oculopharyngeal muscular dystrophy in Quebec. Neuromuscular Disord 1997; 7 (Suppl 1): S22– S29.
7. Dionne A, Bouchard JP. Oculopharyngeal Muscular Dystrophy. In: Tawil RN, Venance S (eds). Neuromucular Disorders. 1st ed. Chichester: Willey‑ Blackwell 2011: 87– 90.
8. Brais B, Rouleau GA. Oculopharyngeal muscular dystrophy. In: Katirji B, Kaminski HJ, Ruff RL et al (eds). Neuromuscular Disorders in Clinical Practice. 1st ed. Newton, MA: Buttenworth‑ Heinemann 2002: 1101– 1105.
9. Blumen SC, Bouchard JP, Brais B, Carasso JL, Paleacu D, Drory VE et al. Cognitive impairment and reduced life span of oculopharyngeal muscular dystrophy homozygotes. Neurology 2009; 73(8): 596– 601.
10. Jones LK jr, Harper CM. Clinical and electrophysiologic features of oculopharyngeal muscular dystrophy: lack of evidence for an associated peripheral neuropathy. Clin Neurophysiol 2010; 121(6): 870– 873.
11. Piccolo G, Cortese A, Tavazzi E, Piccolo L, Sassone J, Ciammola A et al. Late onset oculopharyngeal muscular dystrophy with prominent neurogenic features and short GCG trinucleotide expansion. Muscle Nerve 2011; 43(1): 141– 142.
12. Tomé FM, Chateau D, Hlebling‑ Leclerc A, Fardeau M.Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul Disord 1997; 7 (Suppl 1): S63– S69.
13. Catoire H, Pasco MY, Abu‑ Baker A, Holbert S, Tourette C, Brais B et al. Sirtuin inhibition protects from the polyalanine muscular dystrophy protein PABPN1. Hum Mol Genet 2008; 17(14): 2108– 2117.
14. Davies JE, Wang L, Garzia‑ Oroz L, Cook LJ, Vacher C, O’Donovan DG et al. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat Med 2005; 11(6): 672– 677.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 6
Most read in this issue
- Frontotemporálna lobárna degenerácia z pohľadu nových klinicko‑patologických korelácií
- Tuberózní skleróza u dětí sledovaných od novorozeneckého věku pro prenatální nález rhabdomyomů srdce – dvě kazuistiky
- Expanze pineální krajiny
- Zlomeniny kondylu okciputu