
Pletencové svalové dystrofie
Limb girdle muscular dystrophies
Termín pletencové svalové dystrofie (limb girdle muscular dystrophy; LGMD) byl poprvé použit v roce 1954 J. N. Waltonem a F. Nattrassem. Autoři se jím snažili vymezit další klinickou jednotku vedle častější X-vázané Duchennovy muskulární dystrofie a autozomálně dominantně dědičných myotonické a facioskapulohumerální svalové dystrofie (FSHD). V dalších letech přibývalo poznatků a publikací popisujících jednotlivé LGMD nejen s autozomálně recesivním, ale také dominantním typem dědičnosti. Bylo zjevné, že LGMD nebude jedním onemocněním, nýbrž zastřešujícím termínem pro celou skupinu velmi variabilních klinických jednotek s různým genetickým i patofyziologickým podkladem. Prudký rozvoj molekulární genetiky (zejména technik sekvenování nové generace) vedl k objevení velkého množství nových asociovaných genů. Nová klasifikace z roku 2018 definuje více než 30 subtypů LGMD a je koncipována s předpokladem, že i v budoucnosti budou přibývat další. Tato publikace přináší stručný přehled dostupných informací o LGMD a jejich epidemiologii, patogenezi, fenotypických znacích vč. popisu nejčastějších klinických jednotek, diagnostice, diferenciální diagnostice a dostupných a vyvíjených možnostech terapie.
Keywords:
limb girdle muscular dystrophies – myopathies
Autoři:
L. Mensová; D. Baumgartner; V. Potočková; R. Mazanec
Působiště autorů:
Neurologická klinika 2. LF UK a FN Motol, Praha
Vyšlo v časopise:
Cesk Slov Neurol N 2022; 85(6): 435-448
Kategorie:
Minimonografie
doi:
https://doi.org/10.48095/cccsnn2022435
Souhrn
Termín pletencové svalové dystrofie (limb girdle muscular dystrophy; LGMD) byl poprvé použit v roce 1954 J. N. Waltonem a F. Nattrassem. Autoři se jím snažili vymezit další klinickou jednotku vedle častější X-vázané Duchennovy muskulární dystrofie a autozomálně dominantně dědičných myotonické a facioskapulohumerální svalové dystrofie (FSHD). V dalších letech přibývalo poznatků a publikací popisujících jednotlivé LGMD nejen s autozomálně recesivním, ale také dominantním typem dědičnosti. Bylo zjevné, že LGMD nebude jedním onemocněním, nýbrž zastřešujícím termínem pro celou skupinu velmi variabilních klinických jednotek s různým genetickým i patofyziologickým podkladem. Prudký rozvoj molekulární genetiky (zejména technik sekvenování nové generace) vedl k objevení velkého množství nových asociovaných genů. Nová klasifikace z roku 2018 definuje více než 30 subtypů LGMD a je koncipována s předpokladem, že i v budoucnosti budou přibývat další. Tato publikace přináší stručný přehled dostupných informací o LGMD a jejich epidemiologii, patogenezi, fenotypických znacích vč. popisu nejčastějších klinických jednotek, diagnostice, diferenciální diagnostice a dostupných a vyvíjených možnostech terapie.
Klíčová slova:
myopatie – pletencové svalové dystrofie
Úvod
Pletencové svalové dystrofie (limb girdle muscular dystrophy; LGMD) jsou širokou skupinou vzácných klinicky i geneticky heterogenních svalových onemocnění. Termín LGMD byl poprvé použit v roce 1954 J. N. Waltonem a F. Nattrassem [1] a autoři se jím snažili vymezit další klinickou jednotku vedle častější X-vázané Duchennovy muskulární dystrofie a autozomálně dominantně dědičných myotonické a facioskapulohumerální svalové dystrofie (FSHD). Jako základní charakteristiky LGMD definovali:
a) začátek příznaků v první, druhé nebo třetí dekádě, někdy až ve středním věku;
b) počátek svalové slabosti v oblasti pažního nebo pánevního pletence;
c) přenos obvykle autozomálně recesivně;
d) relativně pomalý průběh onemocnění často vedoucí k těžké invaliditě či předčasnému úmrtí [1].
V dalších letech přibývalo poznatků a publikací popisujících jednotlivé LGMD nejen s autozomálně recesivním, ale také dominantním typem dědičnosti. Bylo zjevné, že LGMD nebude jedním onemocněním, nýbrž zastřešujícím termínem pro celou skupinu velmi variabilních klinických jednotek s různým genetickým i patofyziologickým podkladem. V roce 1995 byla konsorciem ENMC (European Neuromuscular Centre Consortium) navržena nová ucelená klasifikace LGMD vymezující 2 podskupiny dle typu dědičnosti – autozomálně dominantní LGMD1 a autozomálně recesivní LGMD2. Jednotlivým klinickým jednotkám pak bylo přiřazeno písmeno abecedy dle pořadí objevení asociovaného genu např. LGMD2A [2]. Prudký rozvoj molekulární genetiky, zejména technik sekvenování nové generace (next generation sequencing; NGS), vedl k objevení velkého množství nových asociovaných genů. Již nějakou dobu před popsáním LGMD 2Z asociované s genem POGLUT1 [3] bylo zjevné, že bude třeba vytvořit novou klasifikaci zohledňující geny přibývající rychlým tempem a s nimi asociované fenotypy. Zároveň s klasifikací byla navržena i nová definice LGMD (tab. 1). Byla publikována v květnu 2018. Podle ní je „pletencová svalová dystrofie geneticky podmíněné onemocnění, primárně postihující kosterní svaly, vedoucí k progresivní, především proximální svalové slabosti způsobené ztrátou svalových vláken”.

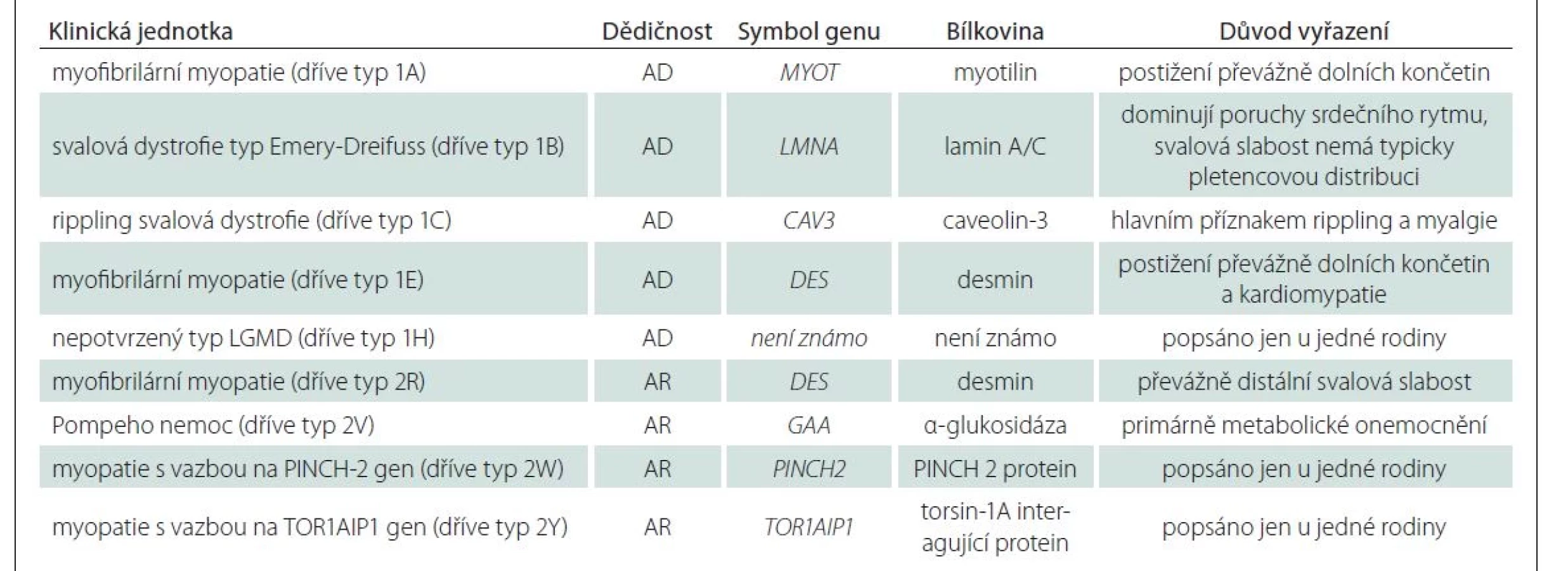
Nová klasifikace znovu rozděluje dvě základní podskupiny s dominantním a recesivním typem dědičnosti, nově označené jako LGMD D a LGMD R. Součástí názvu jsou pak číselné označení dle pořadí objevení asociovaného genu a název proteinu, jehož dysfunkce je patofyziologickým podkladem onemocnění, např. LGMD R1 calpain3-related [4] (tab. 2). Deset jednotek dříve řazených mezi LGMD nesplnilo kritéria vymezená novou definicí a bylo z klasifikace vyřazeno (tab. 3).
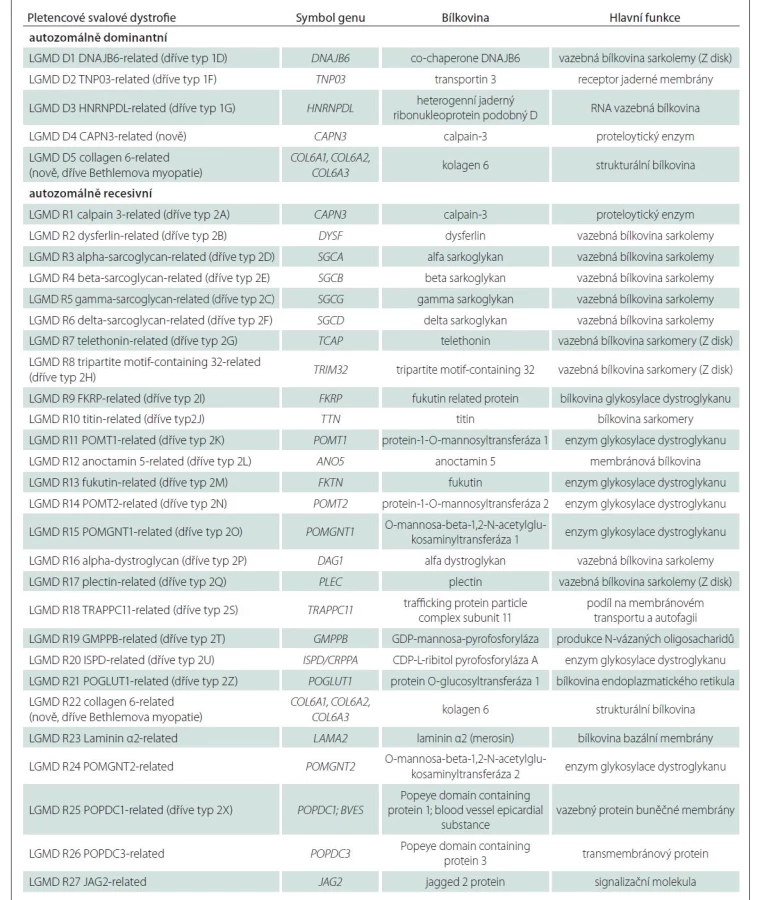
Epidemiologie
Pletencové svalové dystrofie jsou čtvrtou nejčastější geneticky podmíněnou příčinou myopatie (po dystrofinopatii, myotonických dystrofiích a FSHD1). Prevalence je odhadována na 1: 14 500 až 1: 45 000 [4–6], podle jiných zdrojů až 1: 123 000 [7] a liší se v závislosti na etnicitě a geografickém původu. Relativně vyšší prevalence některých jinak velmi vzácných typů LGMD v určitých populacích [8,9] poukazuje na možný efekt mutace zakladatele (zvláštní případ genetického driftu, ke kterému dochází, když se malá skupina odštěpí od původní populace a vytvoří populaci novou). Nová populace pak může mít menší genetickou variabilitu než původní a v případě, že mezi zakládajícími členy populace bylo vyšší zastoupení nositelů určité mutace (typicky několik postižených rodin), bude i v nové populaci prevalence nositelů mutace vyšší než v původní populaci [8]. Častější je autozomálně recesivní typ dědičnosti – u 90 % LGMD, autozomálně dominantní je pak popisován jen u 5–10 % [5,7,10,11]. S ohledem na autozomální dědičnost by měly LGMD postihovat obě pohlaví shodně, ale při zobrazení MR byly popsány rozdíly mezi pohlavími v míře poškození svalů [12]. U LGMD R12 anoctamin5-related bývají muži postiženi častěji [13]. Nejčastějším typem LGMD je podle četných publikací LGMD R1 calpain3-related (26,5 – 30 % všech LGMD) a LGMD R9 FKRP-related (19 %). ČR v tomto není výjimkou. Nejčastějšími typy LGMD jsou R1 calpain3-related (32,6 %), LGMD R9 FKRP-related (9 %) a dále LGMD R3 a-sarcoglycan-related (2,8 %) a LGMD R12 anoctamin5-related (1,4 %) [6,14,15].
Patogeneze
Neexistuje patofyziologický mechanizmus typický pro skupinu LGMD, který by je nějak odlišoval od ostatních svalových dystrofií. Kauzální geny kódují proteiny s nejrůznějšími funkcemi v sarkolemě, cytosolu i v jádře svalového vlákna. Jejich variabilita se do jisté míry odráží ve variabilitě fenotypických projevů [16]. Mechanizmy, které se v rozvoji poškození myocytu uplatňují, zahrnují nestabilitu membrány svalového vlákna (např. u sarkoglykanopatií [17]), funkční i strukturální poruchy kontraktilního aparátu (např. u plektinopatie [18]), poruchy mechanizmů remodelace a regenerace svalového vlákna (např. u kalpainopatie či dysferlinopatie [19]) nebo chyby v utváření funkčního dystroglykanového komplexu [20–22]. Následkem těchto změn se u většiny LGMD projevují nestabilita svalového vlákna, vzestup intracelulární koncentrace kalcia a postupná degenerace myocytu [23,24]. V různé míře se uplatňují vliv zánětlivé úklidové reakce a nahrazení odumřelých svalových vláken satelitními buňkami. Po vyčerpání jejich rezervy je finálním histopatologickým nálezem náhrada svalové tkáně tkání vazivovou či tukovou (end stage muscle) [25].
Důležité je rovněž mít na paměti, že mutace v genech asociovaných s LGMD způsobují i celou řadu dalších svalových (distální, pseudometabolické, kongenitální myopatie) či jiných onemocnění (kardiomyopatie, artrogrypózy, …)
Klinický obraz a diagnostika
Z definice LGMD vyplývá, že společným jmenovatelem jednotlivých subjednotek je progresivní, převážně proximální svalová slabost. Nicméně nacházíme obrovskou variabilitu v nástupu a tíži příznaků. První příznaky se mohou objevit kdykoli od raného dětství po důchodový věk. Fenotypické projevy pak zahrnují celé spektrum od asymptomatické hyperCKémie či sotva znatelné pomalu progredující svalové slabosti po těžký rychle invalidizující Duchenne-like obraz. Variabilitu projevů nacházíme nejen mezi jednotlivými subtypy LGMD, ale i v rámci jedné diagnózy. Typickým příkladem je LGMD R1 calpain3-related [15]. U jednotek s počátkem v dětství bývá výrazněji vyjádřena pelvifemorální slabost, u těch s pozdním nástupem jsou často srovnatelně postižené pánevní i pažní pletenec [26]. Svalová slabost má převážně symetrický charakter, i když u některých subjednotek, např. LGMD R2 dysferlin-related a LGMD R12 anoctamin5-related, nacházíme častěji asymetrii a různý stupeň distálního postižení [16,26–28]. Slabost obličejových a bulbárních svalů není pro LGMD charakteristická, extraokulární svaly bývají typicky ušetřeny. Pro získání komplexního obrazu je nutno cíleně pátrat po dalších symptomech, jako jsou scapula alata, hypertrofie lýtek, přítomnost kontraktur a jejich lokalizace, deformity páteře, makroglosie. Pozdními komplikacemi, ale i prvním projevem onemocnění mohou být rozvoj respirační insuficience nebo projevy kardiální dekompenzace na podkladu kardiomyopatie či převodních poruch. Intelekt a kognitivní funkce zpravidla nebývají postiženy, ale jejich poruchy byly popsány např. v souvislosti s LGMD R11 POMT1-related a LGMD R14 POMT2-related [26]. Myalgie nejsou pro LGMD typické, ale zejména v pokročilejších fázích onemocnění mohou pacienti popisovat myoskeletální bolesti v souvislosti s přetěžováním některých svalových skupin, svalovými dysbalancemi a s rozvojem kontraktur.
S ohledem na vzácný výskyt LGMD a jejich variabilitu jsou zásadní a stále aktuální studie přirozeného průběhu onemocnění, které pomáhají mapovat fenotypické projevy v korelaci s genotypem a získávat zásadní klinimetrická data. Tyto výstupy jsou pak nezbytným zdrojem poznatků pro vytváření standardů péče a definování vhodných biomarkerů účinnosti terapie v rámci klinického hodnocení léčiv. Hlavním zdrojem dat o přirozeném průběhu onemocnění jsou národní a globální registry pacientů.
Vedle klinického vyšetření specialistou na neuromuskulární onemocnění je nezbytné doplnit další testy. Zejména biochemické vyšetření – stanovení hladin CK a myoglobinu, EMG a MR svalů. U většiny LGMD nacházíme jen mírné zvýšení CK, výjimkami jsou sarkoglykanopatie, dysferlinopatie a LGMD R9 FKRP-related s hodnotami typicky v řádu 10násobku normy a vyšších [14]. Obecně také platí, že u autozomálně recesivních LGMD bývají hodnoty CK výrazně vyšší než u forem dominantních [29]. U pokročilých forem LGMD s převahou fibrotické a tukové přestavby svalové tkáně jsou pak hodnoty CK zpravidla již v normě či jen mírně elevované. V rámci diferenciální diagnostiky LGMD mohou být zvažovány i metabolické myopatie, proto by součástí diagnostického procesu měl být i screeningový test suché krevní kapky na Pompeho nemoc [30,31], v případě rekurentních myoglobinurií či nápadné či vícefázové intolerance fyzické zátěže pak konzultace s Ústavem dědičných poruch metabolizmu Všeobecné fakultní nemocnice Praha 2.
Jehlová EMG hraje roli v odlišení neurogenní a myogenní léze a při detekci myotonických výbojů. Volní aktivita je spojená v pozdějších stádiích s časným náborem motorických jednotek (motor unit potential; MUP) a náborová křivka vykazuje nízkou amplitudu. Při kvantitativní analýze motorických jednotek obvykle registrujeme MUP nízké amplitudy a krátkého trvání [32].
Důležitou součástí diagnostického procesu i následné péče o pacienty s LGMD se v posledních letech staly zobrazovací metody, zejména MR svalů. Umožňuje neinvazivní zobrazení všech svalových skupin ve vysokém rozlišení, posouzení svalové atrofie a při využití vhodných sekvencí také detekci tkáňových změn charakteru edému či tukové přestavby. Hlavní přínos MR spočívá nejen ve stanovení rozsahu a stupně postižení jednotlivých svalů, ale také v možnosti identifikovat vzorec postižení svalů (pattern of involvement), který je charakteristický pro určitou klinickou jednotku. Získaných informací lze dále využít při indikaci a navigaci svalové biopsie (spíše při podezření na získanou myopatii) či cíleného genetického vyšetření. Korelace nálezu MR a fenotypu pacienta hraje roli rovněž v objasňování nalezených mutací nejasného významu.
Role svalové biopsie v diagnostice myopatií se v uplynulých letech změnila a toto nákladné a invazivní vyšetření přestalo být metodou první volby. U hereditárních myopatií je indikována v případě, kdy molekulárně genetické vyšetření neodhalí kauzální mutaci, případně odhalí tzv. varianty nejasného významu. Svalová biopsie může být dalším krokem k upřesnění kauzální mutace. Histologický nález u LGMD není specifický, může zahrnovat změny charakteru atrofie, hypertrofie či regenerace svalových vláken. V počátečních fázích onemocnění dominují zejména edém s různou mírou zánětlivé infiltrace či nekróza, v pozdějších fázích onemocnění fibróza a tuková přestavba [33]. Výtěžnost zásadně zvyšuje využití imunohistochemické analýzy a analýzy Western blot k detekci přítomnosti/deficitu/absenci konkrétních proteinů. Pro dosažení maximální výtěžnosti svalové biopsie by standardem měl být odběr ze svalu se středním stupněm svalového postižení, tedy takového svalu, kde již došlo k rozvoji patologických změn ve tkáni, ale fibrózní změny či tuková přestavba nejsou rozsáhlé a získaný vzorek bude obsahovat dostatek svalových vláken k analýze. U klinických jednotek s multifokálním a nehomogenním postižením svalů, kterými LGMD jsou, je bezpochyby přínosné užití zobrazovací metody před svalovou biopsií [33]. Metodou volby je MR svalů, která zároveň přináší informaci o přítomnosti vzorce postižení; případně lze užít sonografii [34–36]. Zásadní je hodnocení svalové biopsie zkušeným specialistou v laboratořích s odpovídajícím vybavením (FN Motol, FN Brno).
Definitivní diagnózu LGMD lze stanovit jen molekulárně genetickým vyšetřením. U genů spojených s LGMD (v současnosti jich je 32) byly popsány různé typy patogenních sekvenčních variant – mutace typu missence, nonsence, změny sestřihu mRNA, delece/duplikace/inzerce na úrovni několika nukleotidů či celých exonů i delece/duplikace na úrovni celých genů. Obecně převažují sekvenční varianty malého rozsahu [37]. Patogenní varianty v genech spojených s LGMD (stejně jako varianty pravděpodobně patogenní, benigní, pravděpodobně benigní a varianty nejasného významu) jsou evidovány v databázi HGMD (Human Gene Mutation Database). Pro představu o komplexnosti problematiky u genu CAPN3 se jedná o 471 patogenních variant, u genu DYSF o 521 a u genu FKRP o 52 variant. Jen některé z nich jsou vedeny v databázi ve spojení s fenotypem LGMD. Vzhledem k množství asociovaných genů je molekulární diagnostika LGMD komplikovaný proces, v současné době založený na technikách NGS. U pacienta s podezřením na LGMD se zpravidla testují nejen geny spojené přímo s LGMD, ale i ostatní geny v rámci panelu neuromuskulárních onemocnění, protože klinické projevy těchto onemocnění se mohou do značné míry překrývat. Zásadní jsou potom správná interpretace nálezu a spolupráce mezi klinickým genetikem a neuromuskulárním specialistou, a to zejména v nejednoznačných případech, kdy NGS odhalí pouze varianty pravděpodobně patogenní či nejasného významu. Nálezy je nutné znovu konfrontovat s fenotypem pacienta či výsledky MR svalů, případně doplnit svalovou biopsii nebo rozšířit genetické vyšetření o vyšetření rodinných příslušníků. Molekulárně genetická diagnostika následně může pokračovat NGS celého genomu, případně RNA na úrovni vybraného souboru genů nebo celého transkriptomu. Vzhledem k velkým finančním nárokům, a hlavně pro velmi náročnou interpretaci identifikovaných variant, nejsou tyto metody v klinické praxi rozšířené.
Nejčastější pletencové svalové dystrofie
LGMD R1 calpain 3-related, LGMD D4 calpain 3-related
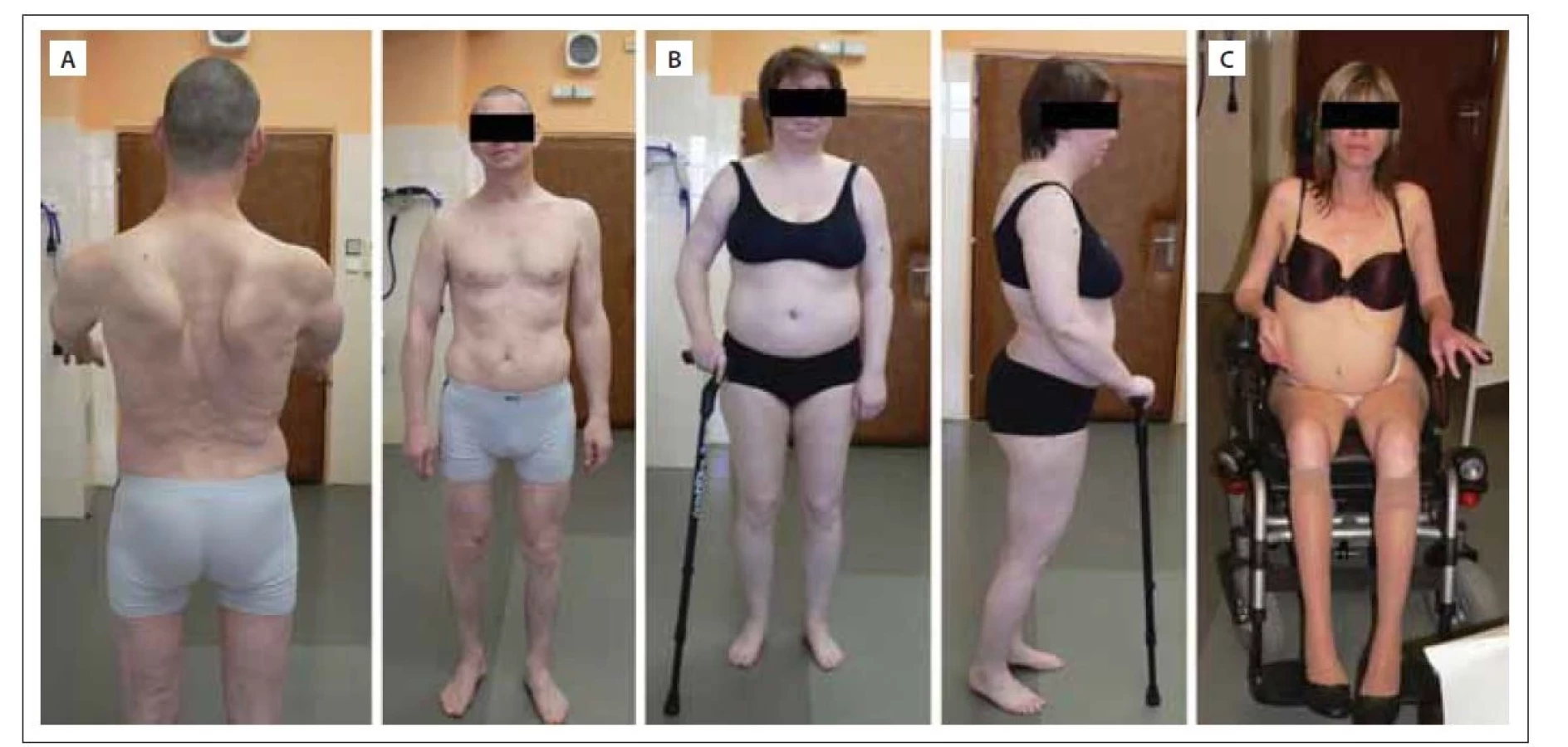
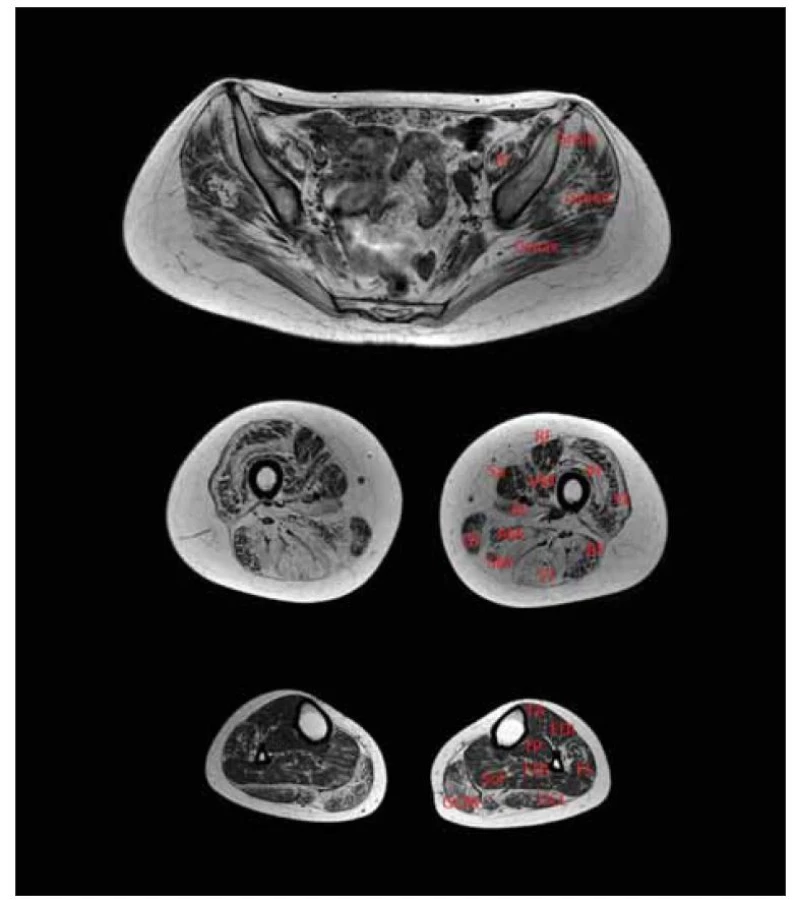
Calpain 3 je svalově specifická vápníkem aktivovaná neutrální proteáza zapojená do remodelace sarkomery, jež váže titin. Gen CAPN3 je prvním genem, který byl v příčinné souvislosti s LGMD popsán a LGMD R1 calpain 3-related je první popsanou jednotkou, u které k rozvoji svalové dystrofie dochází v souvislosti s defektem enzymu a nikoli strukturálního proteinu [24]. Později byly popsány případy s autozomálně dominantní dědičností, dle nové klasifikace LGMD D4 calpain 3-related. LGMD R1 calpain 3-related je celosvětově pravděpodobně nejčastější subtyp LGMD [14,15]. Jedná se o genotypicky velmi variabilní klinickou jednotku s nástupem příznaků nejčastěji mezi 2.–40. rokem věku a ztrátou chůze nejčastěji mezi 5.–39. rokem (obr. 1). Dominuje zpravidla pelvifemorální slabost, ale popisovány jsou i případy izolované slabosti pažního pletence a axiální svalové slabosti a prominentní slabosti bránice [38]. Časně a hojně dochází k oboustrannému rozvoji kontraktur Achillovy šlachy. V pozdních stádiích může dojít k rozvoji respirační insuficience a k deformitám páteře. Kardiální komplikace nejsou s touto klinickou jednotkou asociovány [39]. Hodnoty CK zpravidla dosahují 3–20násobek normy. Na MR svalů nacházíme dystrofické změny v oblasti pažního pletence a zadního kompartmentu stehna s ušetřením m. sartorius a m. vastus medialis [34] (obr. 2).
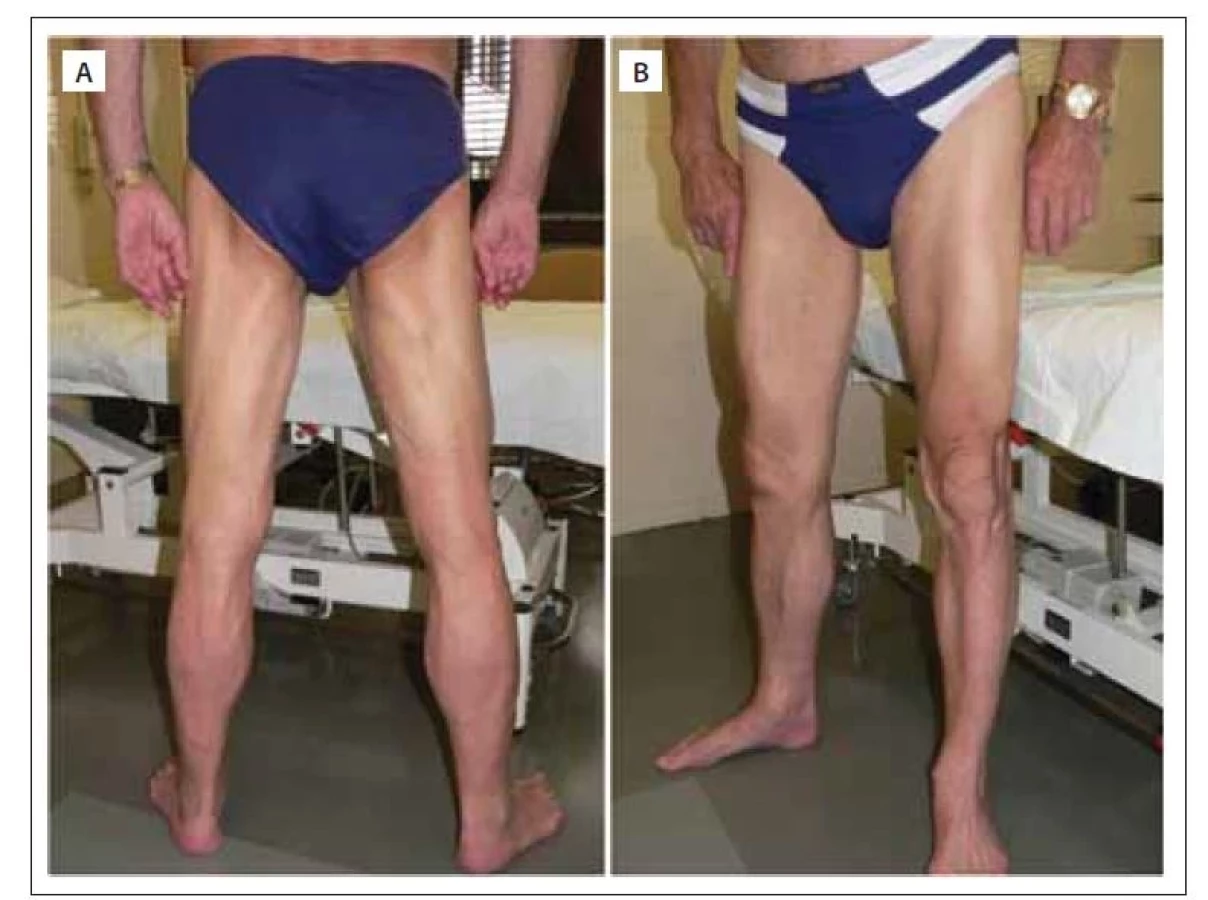
(A) Muž (50 let) nástup příznaků ve 22 letech – pelvifemorální slabost, v čase vzniku fotky myopatický syndrom vyjádřený proximálně na horních
i dolních končetinách, kontraktury Achillovy šlachy, schopen chůze bez opory, dřep s lehkou dopomocí horních končetin.
(B) Žena (40 let) nástup příznaků v 15 letech – slabost pelvifemorální, v čase vzniku fotky myopatický syndrom vyjádřený proximálně na horních
i dolních končetinách, kontraktury Achillovy šlachy, chůze jen s oporou, dřep nesvede od 30 let, nutná asistence druhé osoby v běžných
denních aktivitách.
(C) Žena (48 let) nástup příznaků ve 4 letech – zakopávání a pády, v čase vzniku fotky těžký generalizovaný myopatický syndrom s maximem
proximálně na dolních končetinách, kontraktury Achillovy šlachy, flexorů kolene, flexorů lokte oboustranně, imobilní, ztráta chůze v 26 letech,
dřep nelze od 18 let, plně závislá na péči druhé osoby
Fig. 1. Patients with LGMD R1 calpain 3-related. Photo from the author´s archive.
(A) Male (50-year-old) with onset of symptoms at the age of 22 years – pelvifemoral weakness; at the time of the photo, myopathic syndrome
was expressed proximally on the upper and lower limbs, he had Achilles tendon contractures, was able to walk without support, and
do squat with light upper limbs support.
(B) Female (40-year-old) with onset of symptoms at the age of 15 years – pelvifemoral weakness; at the time of the photo, myopathic syndrome
was expressed proximally on the upper and lower limbs, she had Achilles tendon contractures, was ambulant only with support, was
unable to squat since her 30s, and second person assistance was needed in normal activities of daily living.
(C) Female (48-year-old) with onset of symptoms at the age of 4 years – stumbling and falls; at the time of the photo, she had severe generalized
myopathic syndrome with maximum proximal on the lower limbs, had contractures of Achilles tendon, knee flexors on both sides, elbow
flexors, was non-ambulant since 26 years of age, she cannot squat from the age of 18, and was fully dependent on the care of another
person.
AL – m. adductor longus; AM – m. adductor magnus; BF – m. biceps femoris (caput longum);
EDL – m. extensor digitorum longus; FDL – flexor digitorum longus; GCL – m. gastrocnemius
lateralis; GCM – m. gastrocnemius medialis; Gmin – m. gluteus minimus; Gmed – m. gluteus
medius; Gmax – m. gluteus maximus; Gr – m. gracilis; IP – m. iliopsoas; LGMD – pletencové
svalové dystrofie; PL – m. peroneus longus; RF – m. rectus femoris; Sa – m. sartorius;
SM – m. semimembranosus; ST – m. semitendinosus; VI – m. vastus intermedius;
TP – m. tibialis posterior; VL – m. vastus lateralis; VM – m. vastus medialis
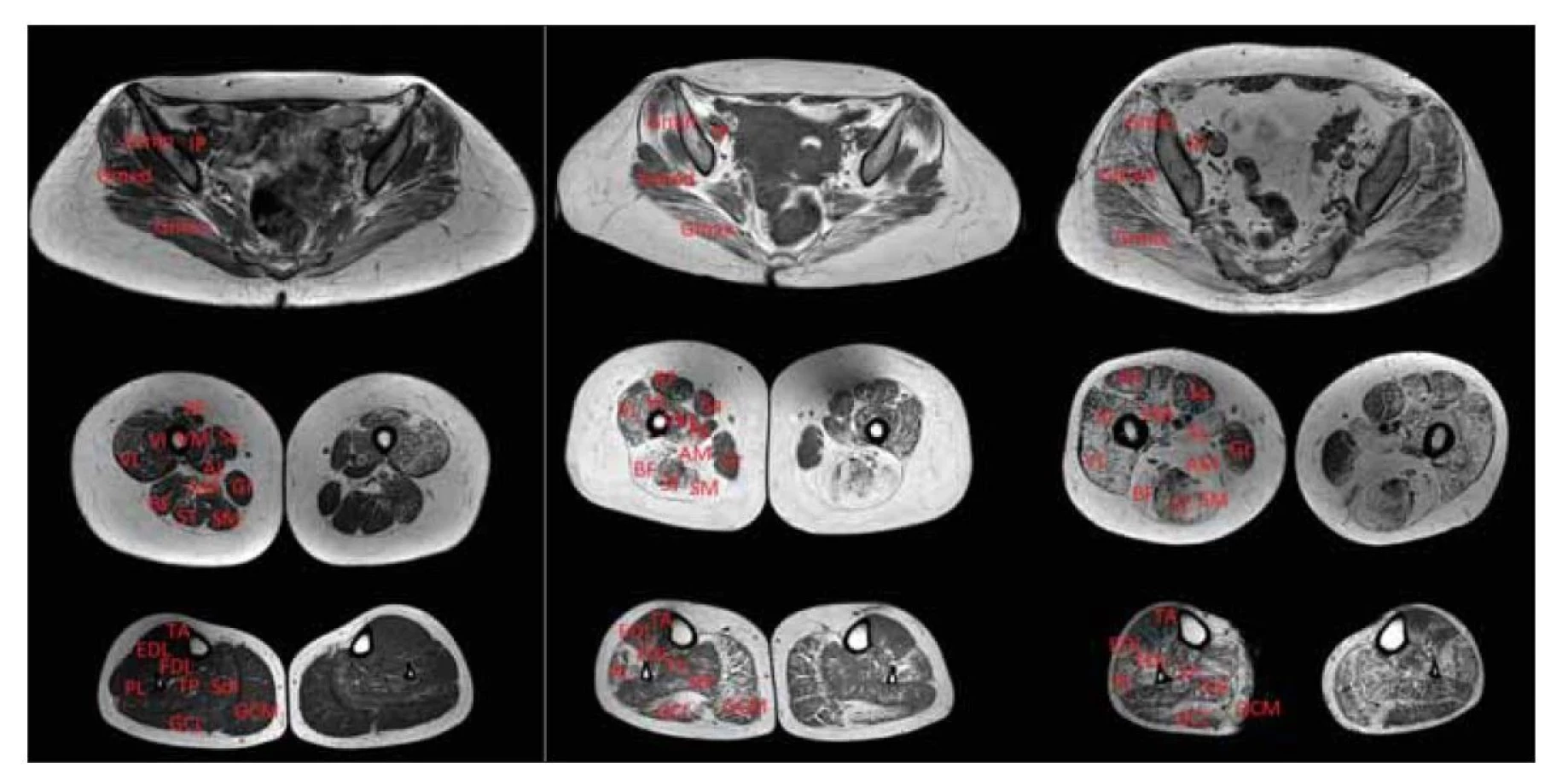
Fig. 2. T1-weighted MRI of lower limb muscles. Patient with LGMD R1 calpain 3-related.
Symmetrical fibrosis and fat replacement in the pelvic area especially in the gluteal
muscles; in the thighs, the maximal changes were in the posterior compartment (semitendinosus
muscle, semimembranosus muscle, m. biceps femoris), which relatively spared
the rectus femoris, sartorius; in the calves, mainly m. gastrocnemii were affected.
AL – adductor longus muscle; AM – adductor magnus muscle; BF – biceps femoris muscle
(caput longum); EDL – extensor digitorum longus muscle; FDL – flexor digitorum longus
muscle; GCL – gastrocnemius lateralis muscle; GCM – gastrocnemius medialis muscle;
Gmin – gluteus minimus muscle; Gmed – gluteus medius muscle; Gmax – gluteus maximus
muscle; Gr – gracilis muscle; IP – iliopsoas muscle; LGMD – limb girdle muscular dystrophy;
PL – peroneus longus muscle; RF – rectus femoris muscle; Sa – sartorius muscle; SM – semimembranosus
muscle; ST – semitendinosus muscle; VI – vastus intermedius muscle;
TP – tibialis posterior muscle; VL – vastus lateralis muscle; VM – vastus medialis muscle
LGMD R9 FKRP-related a skupina dystroglykanopatií
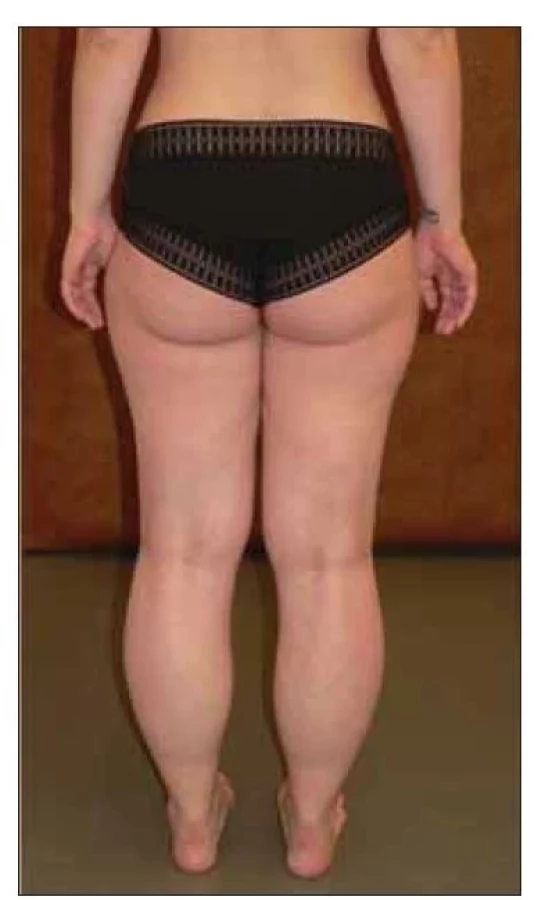
Pojem dystroglykanopatie zahrnuje několik klinických jednotek v rámci skupiny LGMD, jedná se o autozomálně recesivně dědičné LGMD asociované s mutacemi v genech FKRP, POMT1, POMT2, POMGnT1, DAG1, GMPPB a CRPPA. Společným patofyziologickým mechanizmem rozvoje těchto typů LGMD je porucha funkce dystroglykanového komplexu, nejčastěji na podkladě abnormální glykosylace. Dystroglykanový komplex zajišťuje vazbu mezi extracelulární matrix a cytoskeletem a má zásadní vliv na správnou funkci myocytu a stabilitu membrán [20,40]. Fenotypické projevy jsou v rámci skupiny velmi variabilní, s nástupem příznaků nejčastěji mezi 1. a 4. dekádou. Jednoznačně nejčastějším subtypem je LGMD R9 FKRP-related. Klinicky jsou v různé míře vyjádřeny proximální svalová slabost a atrofie, typické jsou hypertrofie lýtek (obr. 3), lumbální hyperlordóza, scapula alata a makroglosie. Hladina CK bývá typicky násobně zvýšená, a to někdy i do extrémních hodnot (50násobek normy) [41]. Časté jsou asociace s kardiomyopatií a rozvoj respirační insuficience, které se mohou vyskytnout již v časných fázích onemocnění a jejich závažnost nemusí korelovat s rozsahem svalové slabosti [42]. Kvalita života pacientů je ovlivněna myalgiemi, které se v průběhu onemocnění vyskytnou u většiny z nich. Třetina pacientů má v průběhu onemocnění myoglobinurii. Svalová biopsie ukáže při imunohistochemické analýze redukci barvení na alfa-dystroglykan, často bývá popisován bohatý zánětlivý úklidový infiltrát. Při MR svalů bývají nejvíce postiženými svaly m. gastrocnemius, soleus a zadní skupiny svalů stehna (m. semitendinosus, m. semimebranosus, m. biceps femoris), naopak typicky ušetřeny zůstávají m. vastus lateralis, m. gracilis a m. sartorius [12] (obr. 4).
LGMD – pletencové svalové dystrofie
Fig. 3. Female patient with LGMD R9
FKRP-related – typical pseudohypertrophy
of the calves.
LGMD – limb girdle muscular dystrophy;
AL – m. adductor longus; AM – m. adductor magnus; BF – m. biceps femoris (caput longum); EDL – m. extensor digitorum longus;
FDL – flexor digitorum longus; GCL – m. gastrocnemius lateralis; GCM – m. gastrocnemius medialis; Gmin – m. gluteus minimus; Gmed –
m. gluteus medius; Gmax – m. gluteus maximus; Gr – m. gracilis; IP – m. iliopsoas; LGMD – pletencové svalové dystrofie; PL – m. peroneus
longus; RF – m. rectus femoris; Sa – m. sartorius; SM – m. semimembranosus; ST – m. semitendinosus; VI – m. vastus intermedius; TP –
m. tibialis posterior; VL – m. vastus lateralis; VM – m. vastus medialis
Fig. 4. T1-weighted MRI of the lower limb muscles. From left to right, female (26 years), female (30 years) and male (38 years) with
LGMD R9 FKRP-related; all show typical pseudohypertrophy of the calves with different proportions of fibrosis and fat conversion;
in the thighs, relatively less affected areas are the sartorius, gracilis and semimembranosus.
AL – adductor longus muscle; AM – adductor magnus muscle; BF – biceps femoris muscle (caput longum); EDL – extensor digitorum longus
muscle; FDL – flexor digitorum longus muscle; GCL – gastrocnemius lateralis muscle; GCM – gastrocnemius medialis muscle; Gmin – gluteus
minimus muscle; Gmed – gluteus medius muscle; Gmax – gluteus maximus muscle; Gr – gracilis muscle; IP – iliopsoas muscle; LGMD – limb
girdle muscular dystrophy; PL – peroneus longus muscle; RF – rectus femoris muscle; Sa – sartorius muscle; SM – semimembranosus muscle;
ST – semitendinosus muscle; VI – vastus intermedius muscle; TP – tibialis posterior muscle; VL – vastus lateralis muscle; VM – vastus medialis
muscle
LGMD R3 α-sarcoglycan-related a skupina sarkoglykanopatií
Sarkoglykany (a, b, g, d, e a ζ) jsou skupinou transmembránových proteinů, které spoluvytvářejí dystrofinový glykoproteinový komplex (a-sarkoglykan byl původně popsán jako dystrofin asociovaný glykoprotein, DAG [43]). Rolí tohoto komplexu je ukotvení a vzájemné propojení cytoskeletu a extracelulární matrix, a tím stabilizace sarkolemy a její ochrana při svalovém přepětí [44]. Nestabilita membrány je pak příčinou násobné elevace CK charakteristické pro tuto skupinu LGMD. Mutace v genech pro sarkoglykanové řetězce jsou příčinou LGMD v 5–10 % [42,45,46]. Většina pacientů se sarkoglykanopatií má závažné klinické příznaky podobné fenotypu Duchennovy muskulární dystrofie (pletencová svalová slabost na dolních i horních končetinách, rozvoj kontraktur, ztráta chůze v dětském věku), ale byly popsány i mírnější projevy se ztrátou chůze až v dospělém věku [47] či fenotyp charakteru intolerance zátěže a rekurentní rhabdomyolýzy [47–50]. Relativně častým příznakem je i makroglosie [47]. Velká variabilita fenotypu i v rámci postižených rodin nasvědčuje roli nejen genetických, ale i epigenetických faktorů a vlivu prostředí na rozvoj klinických příznaků [51]. S progresí onemocnění se zpravidla objevují respirační a kardiální komplikace (dilatační kardiomyopatie, převodní poruchy). Ty mají závažný, až život ohrožující charakter zejména u typů LGMD R4 b-sarcoglycan-related a R6 d-sarcoglycan-related, patofyziologickým podkladem je zastoupení b a d-sarkoglykanu nejen v kosterním, ale také v srdečním svalu. Naopak u nejčastější ze sarkoglykanopatií LGMD R3 a-sarcoglycan-related [14,52,53] jsou kardiální komplikace raritní, neboť a-sarkoglykan je v myokardu nahrazen sarkoglykanem e [54]. MR svalů prokazuje časné postižení skupiny adduktorů, hýžďových svalů a zadní skupiny svalů stehna, charakteristické je minimum změn v oblasti lýtka (na rozdíl od dystrofinopatie typu Duchenne [DMD] nebo typu Becker [BMD], kde nacházíme obraz pseudohypertrofie) a distálního m. quadriceps [55,56]. Pokud je provedena svalová biopsie, zpravidla vykazuje dystrofické změny a sníženou imunoreaktivitu při barvení na sarkoglykan [47].
LGMD R12 anoctamin5-related
Anoctamin 5 je integrální membránový protein endoplazmatického retikula exprimovaný zejména v kosterním a srdečním svalu, kde ovlivňuje transport kalcia. Jedná se o autozomálně recesivní formu pletencové svalové dystrofie relativně nejvíce rozšířenou zejména v severní Evropě a Kanadě [57]. Nástup příznaků bývá nejčastěji v dospělém věku (3. dekáda a později). Onemocnění postihuje častěji muže, ale důvod tohoto jevu nebyl dosud objasněn [13,26]. V klinickém obraze bývá popisována dominující slabost dolních končetin, proximálně zejména postižení m. quadriceps a zadní skupiny svalů stehna (obr. 5, 6), distálně pak oslabení a atrofie zejména m. gastrocnemius medialis, na horních končetinách pak oslabení m. biceps. Dle nejnovějších publikací nálezů MR u pacientů s mutacemi v genu ANO5 se zdá, že LGMD a distální myopatie jsou jen různými póly klinického spektra jednoho onemocnění [58,59]. Onemocnění má zpravidla velmi pomalou progresi, kdy si pacienti uchovávají schopnost chůze. Kardiální a respirační komplikace nebyly v souvislosti s touto diagnózou popsány.
(A) Atrofie hamstringů, zejména dlouhé hlavy m. biceps femoris při relativně ušetřené krátké
hlavě. (B) Typická selektivní atrofie m. vastus medialis a lateralis, na lýtkách nápadná atrofie
m. gastrocnemius medialis oboustranně symetricky.
LGMD – pletencové svalové dystrofie
Fig. 5. Patient with LGMD R12 anoctamin 5-related.
(A) Hamstring atrophy, especially of the long head of the biceps femoris with a relative
sparing of the short head. (B) Typical selective atrophy of the medial and lateral vastus
muscles, and on the calves, conspicuous atrophy of the medial gastrocnemius symmetrically
on both sides.
LGMD – limb girdle muscular dystrophy
AL – m. adductor longus; AM – m. adductor magnus; BF – m. biceps femoris (caput longum);
EDL – m. extensor digitorum longus; FDL – flexor digitorum longus; GCL – m. gastrocnemius
lateralis; GCM – m. gastrocnemius medialis; Gmin – m. gluteus minimus; Gmed – m. gluteus
medius; Gmax – m. gluteus maximus; Gr – m. gracilis; IP – m. iliopsoas; LGMD – pletencové
svalové dystrofie; PL – m. peroneus longus; RF – m. rectus femoris; Sa – m. sartorius; SM –
m. semimembranosus; ST – m. semitendinosus; VI – m. vastus intermedius; TP – m. tibialis
posterior; VL – m. vastus lateralis; VM – m. vastus medialis
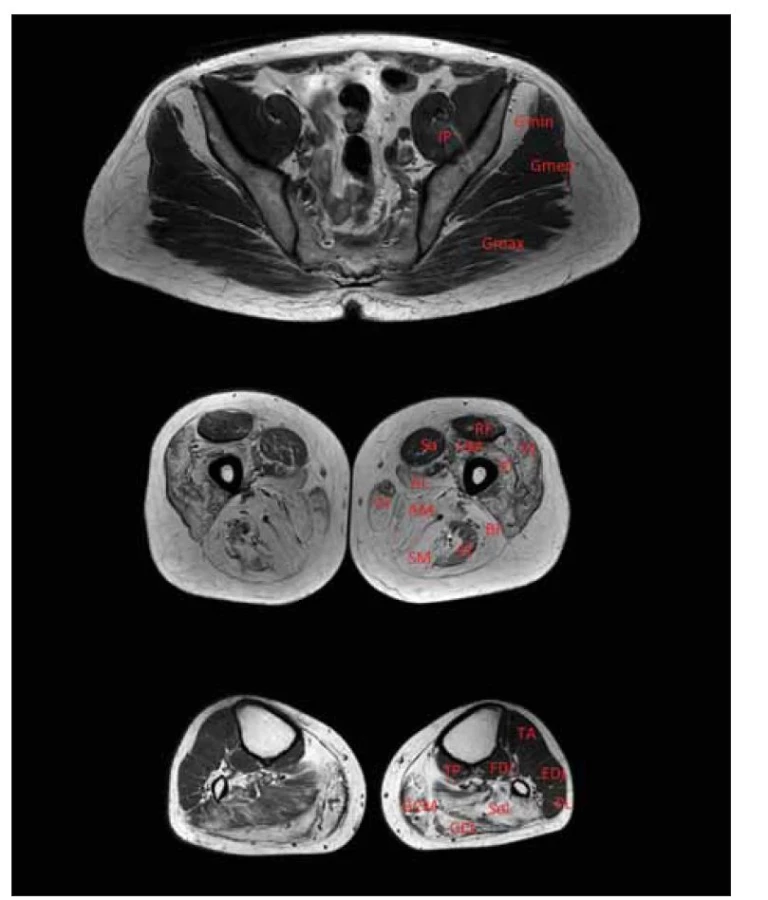
Fig. 6. T1-weighted MRI of the lower limb muscles. Patient with LGMD R12 anoctamine
5-related. Relatively minor involvement of the gluteal muscles; maximal dystrophic
changes in the thigh area in the posterior compartment, relatively spared rectus femoris
and sartorius, and maximal changes in the calf area in m. triceps.
AL – adductor longus muscle; AM – adductor magnus muscle; BF – biceps femoris muscle
(caput longum); EDL – extensor digitorum longus muscle; FDL – flexor digitorum longus
muscle; GCL – gastrocnemius lateralis muscle; GCM – gastrocnemius medialis muscle;
Gmin – gluteus minimus muscle; Gmed – gluteus medius muscle; Gmax – gluteus maximus
muscle; Gr – gracilis muscle; IP – iliopsoas muscle; LGMD – limb girdle muscular dystrophy;
PL – peroneus longus muscle; RF – rectus femoris muscle; Sa – sartorius muscle; SM – semimembranosus
muscle; ST – semitendinosus muscle; VI – vastus intermedius muscle; TP –
tibialis posterior muscle; VL – vastus lateralis muscle; VM – vastus medialis muscle
Diferenciální diagnostika
Diferenciální diagnostika LGMD, respektive primárně proximální svalové slabosti, má velmi široké možnosti a zahrnuje celou řadu hereditárních i získaných svalových chorob, ale i primárně neurogenní onemocnění (tab. 4). Diferenciálně diagnostický proces by měl postupovat systematicky od relativně častějších klinických jednotek k těm raritním.

Hereditární svalové choroby
Dystrofinopatie (DMD, BMD, symptomatické přenašečky) jsou častější než LGMD a jejich projevy mohou být shodné (proximální svalová slabost, hypertrofie lýtek, násobná elevace CK). Zásadní pro jejich odlišení je molekulárně genetické vyšetření genu DMD. V případě DMD fenotypu a negativního molekulárně genetického vyšetření dystrofinového genu je vhodné pomyslet zejména na LGMD R9 FKRP-related a na skupinu sarkoglykanopatií.
Facioskapulohumerální svalová dystrofie je na rozdíl od LGMD charakterizovaná postižením obličejových svalů a často asymetrickou svalovou slabostí. Pouze ve vzácných případech absence postižení obličejových svalů může FSHD imitovat LGMD. Na rozdíl od FSHD2 není častější forma FSHD1 diagnostikována v rámci vyšetření panelu genů asociovaných s neuromuskulárními onemocněními a je nutno ji indikovat zvlášť.
Svalová dystrofie Emery-Dreifuss způsobená mutacemi v genech EMD, FHL1 a LMNA je kromě převážně proximální svalové slabosti charakterizována přítomností kardiálních komplikací, hlavně u X vázané formy (kardiomyopatie, převodní poruchy) a časným rozvojem kontraktur (flexory loktů, Achillovy šlachy). Může se dědit gonozomálně recesivně, autozomálně dominantně i recesivně. Kauzální geny jsou testovány v rámci panelu genů asociovaných s neuromuskulárními onemocněními.
Pacienti s patogenními variantami v genu LMNA a pletencovou či humeroperoneální slabostí byli v minulosti klasifikováni jako skupina trpící autozomálně dominantní pletencovou svalovou dystrofií LGMD 1B, v rámci nové klasifikace byla však i těmto pacientům přisouzena diagnóza svalové dystrofie Emery-Dreifuss.
Pompeho choroba, dříve řazená mezi LGMD jako LGMD 2V, byla v rámci nové klasifikace vyloučena a je řazena mezi metabolické myopatie. Její příčinou je deficit enzymu alfa-glukosidázy. Její juvenilní a adultní formy charakterizované zejména pletencovou svalovou slabostí, slabostí dýchacích svalů, méně pak přítomností kardiomyopatie, mohou jako LGMD imponovat. Důležitým příznakem Pompeho nemoci je slabost bránice, která u LGMD není nápadná. Zásadní je provedení screeningového testu suché krevní kapky a následně doplnění enzymologického vyšetření. Alfa-glukosidázu kódující GAA gen je testován v rámci panelu genů asociovaných s neuromuskulárními onemocněními. Časná diagnostika Pompeho nemoci je zásadní zejména z důvodu dostupnosti enzymové substituční terapie.
Získaná svalová onemocnění – endokrinní, toxické, autoimunitní myopatie
Již při odběru anamnézy je dobré si všímat údajů o autoimunitních a revmatologických onemocněních, endokrinních komorbiditách (tyreopatie), užívání potenciálně myotoxických léků (statiny, fibráty, kortikoidy, chlorochin) a mimosvalových symptomů – kožních změn, artritidy, elevace laboratorních zánětlivých parametrů (FW). Zásadním vyšetřením při podezření na získanou myopatii je svalová biopsie. Svalové biopsii by měla předcházet MR svalů. Důležitá je správná interpretace nálezu v korelaci s anamnestickými a klinickými údaji, protože histologické změny ve svalové biopsii charakteru zánětlivého infiltrátu nacházíme nejen u zánětlivých myopatií. Velký podíl zánětlivého infiltrátu je přítomen i u celé řady LGMD (kalpainopatie, sarkoglykanopatie, R9 FKRP-related, R11 anoctamin5-related). U pacientů s myozitidami refrakterními na léčbu by naopak mělo být zváženo, zda se nemůže jednat o svalovou dystrofii [60–62].
Onemocnění primárně neurogenní
Pacienti se spinální svalovou atrofií (SMA) typu 3 a 4 se rovněž prezentují proximální svalovou slabostí, přítomna může být i mírná elevace CK (do 5násobku normy). Zásadní pro odlišení neurogenní léze je elektromyografické vyšetření. Kondukční studie zpravidla prokazují sníženou amplitudu sumačního svalového akčního potenciálu při normální rychlosti vedení, jehlová EMG pak vykazuje typicky redukovaný interferenční vzorec, akční potenciály s vyšší amplitudou a prodlouženou dobou trvání [63]. Definitivní diagnózu stanoví molekulárně genetické vyšetření genu SMN1. Potvrzení kauzální mutace umožní zahájit léčbu SMA.
Terapie
V současné době je péče o pacienty se vzácnými svalovými onemocněními, mezi která LGMD patří, organizovaná v síti neuromuskulárních center na celém území ČR. Seznam Neuromuskulárních center v ČR je k dispozici na webových stránkách Neuromuskulární sekce ČNS [64]. Základním cílem je udržení maximální mobility, soběstačnosti a kvality života. Schválená kauzální či chorobu modifikující terapie není dosud k dispozici u žádné z forem LGMD. Dostupná terapie je založená na režimových a podpůrných opatřeních a na prevenci a řešení komplikací. Centrová péče umožňuje komplexní přístup k řešení zdravotních, sociálních i psychologických problémů pacientů v rámci multioborového týmu. Jeho součástí by kromě neurologa, fyzioterapeuta a sociálního pracovníka měl být v závislosti na konkrétním typu LGMD i kardiolog, pneumolog, logoped, ortoped, případně ergoterapeut a psycholog.
V rámci základních režimových opatření jsou zásadní vyvážená strava s dostatkem bílkovin a pravidelná aerobní fyzická aktivita [65]. Dle menší dánské studie se jeví přínosný i odborně vedený silový trénink se submaximální zátěží [66]. Jakákoli fyzická aktivita by měla být prováděna s vědomím, že pacienti se svalovou dystrofií jsou náchylnější k zátěžovému svalovému postižení, a proto by měli cvičit po důkladném rozehřátí a jen do mírné únavy, dostatečně se hydratovat a po aktivitě se protáhnout. Nedoporučuje se supramaximální zátěž ani postizometrická kontrakce (výdrže, mrtvý tah). Doba regenerace po výkonu by neměla přesáhnout 24 h, v opačném případě je nutné zátěž snížit, a to i v případě fyzioterapie [7].
V rámci fyzioterapie jsou hlavními strategiemi prevence špatných pohybových stereotypů i přetěžování určitých svalových skupin a s tím související rozvoj myoskeletálních bolestí a dále prevence kontraktur. Zatímco klasický strečink (opakované několikavteřinové protažení svalů, šlach) nemá v prevenci kontraktur význam [66], určitý efekt může mít strečink poziční [67], tedy pasivní polohování končetin do protažení např. vleže na břiše při zkrácení flexorů kyčle, s elevovanými nataženými dolními končetinami při zkrácení extenzorů kolene. Specifickou formou pozičního strečinku je pak dlahování.
V určitých situacích mohou pacienti profitovat z péče ortopeda – v případě těžkých deformit páteře (hyperkyfózy, skoliózy) a kontraktur [7]. Výkonu by měla předcházet multioborová konzultace, následovat by měla péče fyzioterapeuta se zkušeností s neuromuskulárními pacienty.
Všichni pacienti s podezřením na LGMD by měli v rámci diagnostického procesu podstoupit kardiologické vyšetření zahrnující EKG a zobrazovací metodu (echokardiografie nebo MR srdce). U pacientů s abnormálním nálezem či anamnestickým či klinickým podezřením na arytmii je doporučeno doplnit dlouhodobou monitoraci. Pacienti s typy LGMD s častým výskytem kardiálních komplikací (LGMD D1 DNAJB6-related, LGMD R9 FKRP-related, sarkoglykanopatie), kteří jsou asymptomatičtí s normálním kardiologickým nálezem, by měli být dispenzarizování kardiologem s frekvencí kontrol 1× za 2 roky, pacienti s abnormálním kardiologickým nálezem pak minimálně 1× ročně [68]. Terapie srdečního selhání se řídí obecnými principy, doporučeno je časné nasazení inhibitorů angiotenzin konvertujícího enzymu (ACEI) [69]. Rolí kardiologa je rovněž indikace vhodné protidestičkové, případně antikoagulační terapie a indikace implantací kardiostimulátorů (KS), implantabilních kardioverterů-defibrilátorů (implantable cardioverter-defibrillator; ICD) a srdeční resynchronizační léčby (SRL). Přínos a rizika výkonu by vždy měly být zvažovány v multioborovém týmu ošetřujících specialistů. U pacientů s LGMD R9 FKRP-related, kteří dospěli k městnavému srdečnímu selhání a nemají závažné respirační komplikace, se zvažuje transplantace srdce [69–71]. Pravidelná dispenzární péče kardiologa prokazatelně zlepšuje prognózu pacientů a vede ke snížení počtu hospitalizací [72].
Dalším ze základních vyšetření prováděných v rámci diagnostiky a následné dispenzární péče je vyšetření plicních funkcí, které by u pacientů s abnormálním nálezem a pacientů s typem LGMD asociovaným s relativně častějším postižením dýchacích svalů (např. LGMD R9 FKRP-related) mělo být prováděno minimálně 1× ročně. Základními screeningovými metodami jsou spirometrie a noční monitorace pulzním oxymetrem [73]. V případě abnormálního nálezu je indikováno vyšetření pneumologem. Na jeho posouzení je pak indikace vhodné formy non-invazivní či invazivní plicní ventilace, případně pomocných metod (např. kašlací asistent). Součástí neurologických kontrol by měly být cílené dotazy na přítomnost příznaků noční hypoventilace – nekvalitní spánek, nadměrná únava a denní spavost, ranní bolest hlavy. Pomoci může rovněž Epsworthská škála spavosti. V případě klinického podezření na noční hypoventilaci je indikována polysomnografie.
Komplikací pokročilých stádií některých typů LGMD může být také dysfagie a s ní spojené komplikace z podvýživy či aspirace. Pacienti neschopní dostatečného perorálního příjmu potravy, referující polykací obtíže či signifikantní váhový úbytek, by měli podstoupit logopedické vyšetření a vyšetření polykacího aktu. Individuálně a dle stavu by pak měl být zvažován nácvik polykání, v závažných případech jiné formy enterální výživy, nejčastěji perkutánní endoskopická gastrostomie (PEG).
Výzkum
Navzdory již několik desetiletí probíhajícímu výzkumu není kauzální či chorobu modifikující terapie LGMD v tuto chvíli k dispozici. Pro její vznik jsou zásadní objasnění patogenetických mechanizmů vzniku onemocnění a detailní znalost fenotypu a přirozeného průběhu onemocnění. S pomocí studií na tkáňových kulturách a zvířecích modelech byly vytyčeny čtyři základní strategie možného terapeutického zásahu:
1. genová terapie;
2. potlačení zánětlivé reakce;
3. redukce fibrózy a expanze tukové tkáně;
4. podpora růstu svalů.
Příčinou rozvoje LGMD je zpravidla chybějící či narušená funkce některé ze strukturálních, signálních či enzymatických bílkovin. Genová terapie je zaměřená na obnovení exprese funkčního proteinu 1) vnesením nové kopie mutovaného genu do cílových buněk, 2) opravením mutace, 3) přeskočením mutace. K vnášení nemutovaných genů se využívají virové vektory se svalově specifickým promotorem zajišťujícím selektivní expresi v cílové tkáni. Vývoj těchto částic je převážně ve fázi preklinických studií (na myších modelech a tkáňových kulturách), publikována byla data u genů CAPN3 [74], SGCA [75], SGCG [76], DYSF [77], FKRP [78,79].
Univerzální reakcí lidského organizmu na poškození je zánět. Ten nacházíme v různé míře také v dystrofické svalové tkáni. Velký podíl zánětlivého infiltrátu ve svalové biopsii bývá přítomen zejména u LGMD R2 dysferlin-related a LGMD R9 FKRP-related. Potlačením masivní zánětlivé reakce lze zpomalit ztrátu svalových vláken a oddálit nevratné změny charakteru fibrózy. Základní skupinou léků s tímto mechanizmem účinku jsou glukokortikoidy. Zatímco u Duchennovy svalové dystrofie byl prokázán pozitivní vliv léčby glukokortikoidy a tato je součástí terapeutických standardů [80], u LGMD není k dispozici dostatek validních dat. Vzhledem k vzácnosti jednotlivých podtypů LGMD byl publikován efekt léčiva jen na malých skupinách nebo kazuistikách např. u dvou sourozenců s LGMD R4 b-sarcoglycan-related [81]. V současnosti probíhají dvě klinické studie: dvojitě zaslepená placebem kontrolovaná studie testující terapii deflazacortem u pacientů s LGMD R9 FKRP-related (NCT03783923) a studie testující bezpečnost a efekt podávání prednisonu 1× týdně pacientům s LGMD (NCT-04054375).
Z dalších možných zásahů do patofyziologie se zkouší např. blokáda myostatinu [82,83] s cílem podpořit růst svalové tkáně, blokáda fibrogeneze s využitím monoklonálních protilátek [84] a mnohé další.
U LGMD je stejně jako u ostatních vzácných onemocnění pro získání dostatečného množství validních dat nezbytná mezinárodní spolupráce, jakou zaštiťuje např. Evropská referenční síť pro vzácná onemocnění (European Reference Network; ERN) či iniciativa TREAT-NMD, která propojuje základní a aplikovaný výzkum, vědce, lékaře, zástupce pacientských organizací i farmaceutických firem. Informace z globálních registrů mají zásadní roli ve výzkumu přirozeného průběhu onemocnění a vývoji spolehlivých specifických biomarkerů pro hodnocení efektů terapeutických zásahů. Od června roku 2020 v ČR funguje v rámci platformy REaDY (Registr svalových dystrofií) také registr REaDY LGMD, který je k dispozici na adrese [85]. Data do něj zadávají lékaři neuromuskulárních center a v současné době obsahuje záznamy 93 pacientů.
Závěr
Skupina LGMD je velmi variabilní a dynamickou skupinou diagnóz. Jen od publikování nové klasifikace v roce 2018 byly popsány další tři nové klinické jednotky splňující kritéria LGMD a lze očekávat, že se skupina bude dále rozrůstat. Velmi nízká prevalence a vysoká rozmanitost klinických projevů LGMD ztěžují jejich diagnostiku. Pacienti s důvodným podezřením na svalovou dystrofii (klinické příznaky, elevace CK, EMG) by měli být konzultováni v neuromuskulárních centrech. Rolí neuromuskulárních specialistů je indikace dalších vyšetření (zejména MR svalů), ve spolupráci s klinickým genetikem pak indikace vhodného molekulárně genetického vyšetření a jeho interpretace. Přesná znalost kauzální mutace je nezbytná nejen pro genetické poradenství, ale i pro možnost případné genové terapie, jejíž klinické testování lze u některých subtypů LGMD v příštích letech očekávat. I přes prudký rozvoj molekulárně genetických metod však zůstává část pacientů s fenotypem odpovídajícím LGMD bez definitivní genetické diagnózy. U těchto pacientů je – po vyloučení ostatních příčin v rámci diferenciální diagnostiky a konzultaci s genetikem – vhodné molekulárně genetické vyšetření s odstupem opakovat. Pro rozšíření našich znalostí o těchto velmi vzácných onemocněních, nastavení standardů péče a vyhledávání vhodných biomarkerů pro hodnocení účinku léčiv v klinických studiích má zásadní význam sběr dat prostřednictvím registrů.

Poděkování
Snímky MR v této publikaci byly použity s laskavým svolením Kliniky JL – MR, s. r. o.
Konflikt zájmů
Autoři nemají v souvislosti s touto minimonografií žádný konflikt zájmů.

MUDr. Lívie Mensová
Promovala v roce 2017 na 2. LF UK a nastoupila na Neurologickou kliniku 2. LF UK a FN Motol, kde působí dodnes. Je studentkou postgraduálního studia v oboru Neurovědy se zaměřením na problematiku pletencových svalových dystrofií. Pracuje v Neuromuskulárním centru v Motole, které je součástí Evropské referenční sítě pro vzácná neuromuskulární onemocnění ERN-NMD. Je členkou pracovní skupiny ERN-NMD v sekci Neuromuscular Imaging. Aktivně se zúčastnila několika zahraničních kurzů (MYO-MRI, Summer School of Myology, European LGMD Masterclass). Je garantkou Národního registru pletencových svalových dystrofií LGMD REaDY a hlavní koordinátorkou projektu Celonárodní screening Pompeho nemoci. Pravidelně přednáší na domácích i mezinárodních kongresech (ICNMD, Český a slovenský neuromuskulární kongres,…) se zaměřením na svalové choroby. Je autorkou nebo spoluautorkou několika zahraničních i domácích publikací s tématikou dědičných myopatií. V rámci postgraduálního studia vyučuje na 2. LF UK a podílí se na přípravě kurzů pro lékaře v předatestační přípravě.
MUDr. Lívie Mensová
Neurologická klinika
2. LF UK a FN Motol
V Úvalu 84
150 06 Praha
e-mail: mensova.livie@gmail.com
Přijato k recenzi: 1. 6. 2022
Přijato do tisku: 3. 11. 2022
Zdroje
1. Walton NJ, Nattrass FJ. On the classification, natural history and treatment of the myopathies. Brain 1954; 77 (2): 169–231. doi: 10.1093/brain/77.2.169.
2. Bushby KMD. Diagnostic criteria for the limb-girdle muscular dystrophies: report of the ENMC consortium on limbgirdle dystrophies. Neuromuscular Disord 1995; 5 (1): 71–74. doi: 10.1016/0960-8966 (93) e0006- g.
3. Servián‑Morilla E, Takeuchi H, Lee TV et al. A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol Med 2016; 8 (11): 1289–1309. doi: 10.15252/emmm.201505815.
4. Straub V, Murphy A, Udd B et al. 229th ENMC international workshop: limb girdle muscular dystrophies – Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscular Disord 2018; 28 (8): 702–710. doi: 10.1016/j.nmd.2018.05.007.
5. van der Kooi AJ, Barth PG, Busch HF et al. The clinical spectrum of limb girdle muscular dystrophy. A survey in the Neatherlands. Brain 1996; 119 (Pt 5): 1471–1480. doi: 10.1093/brain/119.5.1471.
6. Norwood FLM, Harling C, Chinnery PF et al. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009; 132 (Pt 11): 3175–3186. doi: 10.1093/brain/awp236.
7. Narayanaswami P, Weiss M, Selcen D et al. Evidence-based guideline summary. Neurology 2014; 83 (16): 1453–1463. doi: 10.1212/wnl.0000000000000892.
8. Mojbafan M, Bahmani R, Bagheri SD et al. Mutational spectrum of autosomal recessive limb-girdle muscular dystrophies in a cohort of 112 Iranian patients and reporting of a possible founder effect. Orphanet J Rare Dis 2020; 15 (1): 14. doi: 10.1186/s13023-020-1296-x.
9. Polavarapu K, Mathur A, Joshi A et al. A founder mutation in the GMPPB gene [c.1000G > A (p.Asp334Asn) ] causes a mild form of limb-girdle muscular dystrophy/congenital myasthenic syndrome (LGMD/CMS) in South Indian patients. Neurogenetics 2021; 22 (4): 271–285. doi: 10.1007/s10048-021-00658-1.
10. Bushby K. Report on the 12th ENMC sponsored international workshop – the “limb-girdle” muscular dystrophies. Neuromuscular Disord 1992; 2 (1): 3–5. doi: 10.1016/0960-8966 (92) 90019-3.
11. Angelini C, Giaretta L, Marozzo R. An update on diag- nostic options and considerations in limb-girdle dystrophies. Expert Rev Neurother 2018; 18 (9): 693–703. doi: 10.1080/14737175.2018.1508997.
12. Willis TA, Hollingsworth KG, Coombs A et al. Quantitative magnetic resonance imaging in limb-girdle muscular dystrophy 2I: a multinational cross-sectional study. PLoS One 2014; 9 (2): e90377. doi: 10.1371/journal.pone.0090377.
13. Sarkozy A, Hicks D, Hudson J et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Hum Mutat 2013; 34 (8): 1111–1118. doi: 10.1002/humu.22342.
14. Stehlíková K, Skálová D, Zídková J et al. Autosomal recessive limb-girdle muscular dystrophies in the Czech Republic. BMC Neurol 2014; 14: 154. doi: 10.1186/s12883-014-0154-7.
15. Richard I, Roudaut C, Saenz A et al. Calpainopathy – a survey of mutations and polymorphisms. Am J Hum Genetics 1999; 64 (6): 1524–1540. doi: 10.1086/302426.
16. Murphy AP, Straub V. The classification, natural history and treatment of the limb girdle muscular dystrophies. J Neuromuscul Dis 2015; 2 (2): S7–S19. doi: 10.3233/jnd-150105.
17. Hack AA, Groh ME, McNally EM. Sarcoglycans in muscular dystrophy. Microsc Res Tech 2000; 48 (3–4): 167–180. doi: 10.1002/ (SICI) 1097-0029 (20000201/15) 48: 3/4<167:: AID-JEMT5>3.0.CO; 2-T.
18. Zrelski MM, Kustermann M, Winter L. Muscle-related plectinopathies. Cells 2021; 10 (9): 2480. doi: 10.3390/cells10092480.
19. Liu J, Aoki M, Illa I et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 1998; 20 (1): 31–36. doi: 10.1038/1682.
20. Liewluck T, Milone M. Untangling the complexity of limb‑girdle muscular dystrophies. Muscle Nerve 2018; 58 (2): 167–177. doi: 10.1002/mus.26077.
21. Kawahara G, Guyon JR, Nakamura Y et al. Zebrafish models for human FKRP muscular dystrophies. Hum Mol Genet 2010; 19 (4): 623–633. doi: 10.1093/hmg/ddp528.
22. Herrmann R, Straub V, Blank M et al. Dissociation of the dystroglycan complex in caveolin-3-deficient limb girdle muscular dystrophy. Hum Mol Genet 2000; 9 (15): 2335–2340. doi: 10.1093/oxfordjournals.hmg.a018926.
23. Alderton JM, Steinhardt RA. How calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. Trends Cardiovasc Med 2000; 10 (6): 268–272. doi: 10.1016/s1050-1738 (00) 00075-x.
24. Fanin M, Nascimbeni AC, Fulizio L et al. Loss of calpain-3 autocatalytic activity in LGMD2A patients with normal protein expression. Am J Pathol 2003; 163 (5): 1929–1936. doi: 10.1016/s0002-9440 (10) 63551-1.
25. Wallace GQ, McNally EM. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu Rev Physiol 2009; 71: 37–57. doi: 10.1146/annurev.physiol.010908.163216.
26. Darras BT, Nordli DR, Shefner JM. Limb-girdle muscular dystrophy. [online]. Available from URL: https: //www.uptodate.com/contents/limb-girdle-muscular-dystrophy.
27. Paradas C, Llauger J, Diaz-Manera J et al. Redefining dysferlinopathy phenotypes based on clinical findings and muscle imaging studies. Neurology 2010; 75 (4): 316–323. doi: 10.1212/wnl.0b013e3181ea1564.
28. Magri F, Bo RD, D’Angelo MG et al. Frequency and characterisation of anoctamin 5 mutations in a cohort of Italian limb-girdle muscular dystrophy patients. Neuromuscul Disord 2012; 22 (11): 934–943. doi: 10.1016/j.nmd.2012.05.001.
29. Bushby K. Limb-girdle muscular dystrophies. [online]. Dostupné z: https: //rarediseases.org/rare-dis- eases/limb-girdle-muscular-dystrophies/.
30. Parmová O, Mensová L, Voháňka S et al. Celonárodní screening Pompeho nemoci u pacientů s nespecifikovanou svalovou slabostí, hyperCKémií a respirační insuficiencí. Neurol Praxi 2020; 21 (Suppl B): 3–9 doi: 10.36290/neu.2020.064.
31. Kishnani PS, Steiner RD, Bali D et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8 (5): 267–288. doi: 10.1097/01.gim.0000218152.87434.f3.
32. Mazanec R, Mensová L, Baumgartner D et al. Diagnostický algoritmus u svalových dystrofií. Neurol Praxi 2019; 20 (3): 190–194 doi: 10.36290/neu.2019.017.
33. Joyce NC, Oskarsson B, Jin L-W. Muscle biopsy evaluation in neuromuscular disorders. Phys Med Rehabil Clin N Am 2012; 23 (3): 609–631. doi: 10.1016/j.pmr.2012.06.006.
34. Mercuri E, Bushby K, Ricci E et al. Muscle MRI findings in patients with limb girdle muscular dystrophy with calpain 3 deficiency (LGMD2A) and early contractures. Neuromuscular Disord 2005; 15 (2): 164–171. doi: 10.1016/j.nmd.2004.10.008.
35. Mercuri E, Pichiecchio A, Allsop J et al. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magn Reson Imaging 2007; 25 (2): 433–440. doi: 10.1002/jmri.20804.
36. Zaidman CM, Holland MR, Anderson CC et al. Calibrated quantitative ultrasound imaging of skeletal muscle using backscatter analysis. Muscle Nerve 2008; 38 (1): 893–898. doi: 10.1002/mus.21052.
37. Fajkusová L, Zídková J. Pletencové svalové dystrofie. Neurol Praxi 2021; 22 (2): 100–103. doi: 10.36290/neu. 2020.107.
38. Angelini C. LGMD. Identification, description and classification. Acta Myol 2020; 39 (4): 207–217. doi: 10.36185/2532-1900-024.
39. Quick S, Schaefer J, Waessnig N et al. Evaluation of heart involvement in calpainopathy (LGMD2A) using cardiovascular magnetic resonance. Muscle Nerve 2015; 52 (4): 661–663. doi: 10.1002/mus.24717.
40. Campbell DEMP, Campbell KP. Dystrophin-glycoprotein comples: post-translational processing and dystroglycan function. J Biol Chem 2003; 278 (18): 15457–15460. doi: 10.1074/jbc.r200031200.
41. Mercuri E, Brockington M, Straub V et al. Phenotypic spectrum associated with mutations in the fukutin‑related protein gene. Ann Neurol 2003; 53 (4): 537–542. doi: 10.1002/ana.10559.
42. Wicklund MP, Kissel JT. The limb-girdle muscular dystrophies. Neurol Clin 2014; 32 (3): 729–749. doi: 10.1016/ j.ncl.2014.04.005.
43. Matsumura K, Tomé FMS, Collin H et al. Deficiency of the 50K dystrophin-associated glycoprotein in severe childhood autosomal recessive muscular dystrophy. Nature 1992; 359 (6393): 320–322. doi: 10.1038/359320a0.
44. Tarakci H, Berger J. The sarcoglycan complex in skeletal muscle. Front Biosci 2016; 21 (4): 744–756. doi: 10.2741/4418.
45. Nallamilli BRR, Chakravorty S, Kesari A et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol 2018; 5 (12): 1574–1587. doi: 10.1002/acn3.649.
46. Wicklund MP. The limb-girdle muscular dystrophies. Continuum 2019; 25 (6): 1599–1618. doi: 10.1212/ con.0000000000000809.
47. Kirschner J, Lochmüller H. Sarcoglycanopathies. Handb Clin Neurol 2011; 101: 41–46. doi: 10.1016/b978-0-08-045031-5.00003-7.
48. Cagliani R, Comi GP, Tancredi L et al. Primary beta-sarcoglycanopathy manifesting as recurrent exercise-induced myoglobinuria. Neuromuscul Disord 2001; 11 (4): 389–394. doi: 10.1016/s0960-8966 (00) 00207-8.
49. Mongini T, Doriguzzi C, Bosone I et al. Alpha-sarcoglycan deficiency featuring exercise intolerance and myoglobinuria. Neuropediatrics 2002; 33 (2): 109–111. doi: 10.1055/s-2002-32374.
50. Pena L, Kim K, Charrow J. Episodic myoglobinuria in a primary gamma-sarcoglycanopathy. Neuromuscul Disord 2010; 20 (5): 337–339. doi: 10.1016/j.nmd.2010.02.015.
51. Angelini C, Fanin M, Menegazzo E et al. Homozygous a‑sarcoglycan mutation in two siblings: one asymptomatic and one steroid‑responsive mild limb-girdle muscular dystrophy patient. Muscle Nerve 1998; 21 (6): 769–775. doi: 10.1002/ (sici) 1097-4598 (199806) 21: 6<769:: aid-mus9>3.0.co; 2-5.
52. Fanin M, Nascimbeni AC, Aurino S et al. Frequency of LGMD gene mutations in Italian patients with distinct clinical phenotypes. Neurology 2009; 72 (16): 1432–1435. doi: 10.1212/wnl.0b013e3181a1885e.
53. Fanin M, Duggan DJ, Mostacciuolo ML et al. Genetic epidemiology of muscular dystrophies resulting from sarcoglycan gene mutations. J Med Genet 1997; 34 (12): 973–977. doi: 10.1136/jmg.34.12.973.
54. Lancioni A, Rotundo IL, Kobayashi YM et al. Combined deficiency of alpha and epsilon sarcoglycan disrupts the cardiac dystrophin complex. Hum Mol Genet 2011; 20 (23): 4644–4654. doi: 10.1093/hmg/ddr398.
55. Tasca G, Monforte M, Díaz-Manera J et al. MRI in sarcoglycanopathies: a large international cohort study. J Neurology Neurosurg Psychiatry 2018; 89 (1): 72–77. doi: 10.1136/jnnp-2017-316736.
56. Lodi R, Muntoni F, Taylor J et al. Correlative MR imaging and 31P-MR spectroscopy study in sarcoglycan deficient limb girdle muscular dystrophy. Neuromuscul Disord 1997; 7 (8): 505–511. doi: 10.1016/s0960-8966 (97) 00108-9.
57. Penttilä S, Palmio J, Suominen T et al. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology 2012; 78 (12): 897–903. doi: 10.1212/wnl.0b013e31824c4682.
58. Vázquez J, Lefeuvre C, Escobar RE et al. Phenotypic spectrum of myopathies with recessive anoctamin-5 mutations. J Neuromuscul Dis 2020; 7 (4): 443–451. doi: 10.3233/jnd-200515.
59. Christiansen J, Güttsches A-K, Schara-Schmidt U et al. ANO5-related muscle diseases: from clinics and genetics to pathology and research strategies. Genes Dis 2022; 9 (6): 1506–1520. doi: 10.1016/j.gendis.2022.01.001.
60. Vinit J, Samson M, Gaultier J-B et al. Dysferlin deficiency treated like refractory polymyositis. Clin Rheumatol 2009; 29 (1): 103–106. doi: 10.1007/s10067-009-1273-1.
61. Barresi R. From proteins to genes: immunoanalysis in the diagnosis of muscular dystrophies. Skelet Muscle 2011; 1 (1): 24. doi: 10.1186/2044-5040-1-24.
62. Rowin J, Meriggioli MN, Cochran EJ et al. Prominent inflammatory changes on muscle biopsy in patients with Miyoshi myopathy. Neuromuscular Disord 1999; 9 (6–7): 417–420. doi: 10.1016/s0960-8966 (99) 00041-3.
63. Rosenfeld A. Spinal muscular atrophy. [online]. Available from URL: https: //emedicine.medscape.com/article/1181436-overview.
64. Neuromuskulární sekce České neurologické společnosti. [online]. Dostupné z URL: https: //www.neuromuskularni-sekce.cz/
65. Sveen M-L, Jeppesen TD, Hauerslev S et al. Endurance training. Neurology 2007; 68 (1): 59–61. doi: 10.1212/01.wnl.0000250358.32199.24.
66. Sveen M, Andersen SP, Ingelsrud LH et al. Resistance training in patients with limb‑girdle and becker muscular dystrophies. Muscle Nerve 2013; 47 (2): 163–169. doi: 10.1002/mus.23491.
67. Harvey LA, Katalinic OM, Herbert RD et al. Stretch for the treatment and prevention of contractures. Cochrane Database Syst Rev 2017; 1 (1): CD007455. doi: 10.1002/14651858.cd007455.pub3.
68. Feingold B, Mahle WT, Auerbach S et al. Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation 2017; 136 (13): e200–231. doi: 10.1161/cir.0000000000000526.
69. Norwood F, Visser M de, Eymard B et al. EFNS guideline on diagnosis and management of limb girdle muscular dystrophies. Eur J Neurol 2007; 14 (12): 1305–1312. doi: 10.1111/j.1468-1331.2007.01979.x.
70. Margeta M, Connolly AM, Winder TL et al. Cardiac pathology exceeds skeletal muscle pathology in two cases of limb‑girdle muscular dystrophy type 2I. Muscle Nerve 2009; 40 (5): 883–889. doi: 10.1002/mus. 21432.
71. D’Amico A, Petrini S, Parisi F et al. Heart transplantation in a child with LGMD2I presenting as isolated dilated cardiomyopathy. Neuromuscul Disord 2008; 18 (2): 153–155. doi: 10.1016/j.nmd.2007.09.013.
72. Nikhanj A, Yogasundaram H, Nichols BM et al. Cardiac intervention improves heart disease and clinical outcomes in patients with muscular dystrophy in a multidisciplinary care setting. J Am Heart Assoc 2020; 9 (2): e014004. doi: 10.1161/jaha.119.014004.
73. Simonds AK. Recent advances in respiratory care for neuromuscular disease. Chest 2006; 130 (6): 1879–1886. doi: 10.1378/chest.130.6.1879.
74. Bartoli M, Roudaut C, Martin S et al. Safety and efficacy of AAV-mediated calpain 3 gene transfer in a mouse model of limb-girdle muscular dystrophy type 2A. Mol Ther 2006; 13 (2): 250–259. doi: 10.1016/j.ymthe.2005.09.017.
75. Griffin DA, Pozsgai ER, Heller KN et al. Preclinical syste-mic delivery of adeno-associated a-sarcoglycan gene transfer for limb-girdle muscular dystrophy. Hum Gene Ther 2021; 32 (7–8): 390–404. doi: 10.1089/hum.2019.199.
76. Israeli D, Cosette J, Corre G et al. An AAV-SGCG dose-response study in a g-sarcoglycanopathy mouse model in the context of mechanical stress. Mol Ther Methods Clin Dev 2019; 13: 494–502. doi: 10.1016/ j.omtm.2019.04.007.
77. Lostal W, Bartoli M, Bourg N et al. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum Mol Genet 2010; 19 (10): 1897–1907. doi: 10.1093/hmg/ddq065.
78. Xu L, Lu PJ, Wang C-H et al. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of a-dystroglycan and improves muscle functions. Mol Ther 2013; 21 (10): 1832–1840. doi: 10.1038/mt.2013.156.
79. Vannoy CH, Leroy V, Lu QL. Dose-dependent effects of FKRP gene-replacement therapy on functional rescue and longevity in dystrophic mice. Mol Ther Methods Clin Dev 2018; 11: 106–120. doi: 10.1016/j.omtm.2018.10.004.
80. Birnkrant DJ, Bushby K, Bann CM et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 2018; 17 (3): 251–267. doi: 10.1016/s1474-4422 (18) 30024-3.
81. Wong-Kisiel LC, Kuntz NL. Two siblings with limb-girdle muscular dystrophy type 2E responsive to deflazacort. Neuromuscular Disord 2010; 20 (2): 122–124. doi: 10.1016/j.nmd.2009.11.005.
82. Bogdanovich S, McNally EM, Khurana TS. Myostatin blockade improves function but not histopathology in a murine model of limb‑girdle muscular dystrophy 2C. Muscle Nerve 2008; 37 (3): 308–316. doi: 10.1002/mus.20920.
83. Mariot V, Joubert R, Hourdé C et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat Commun 2017; 8 (1): 1859. doi: 10.1038/s41467-017-01486-4.
84. Piñol-Jurado P, Suárez-Calvet X, Fernández-Simón E et al. Nintedanib decreases muscle fibrosis and improves muscle function in a murine model of dystrophinopathy. Cell Death Dis 2018; 9 (7): 776. doi: 10.1038/s41419-018-0792-6.
85. REaDY. [online]. Dostupné z URL: https: //ready.registry.cz/.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2022 Číslo 6
Nejčtenější v tomto čísle
- Doporučení pro vývojovou dysfázii – verze 2022
- Validační studie a představení nového testu porozumění větám TEPO pro děti ve věku 3–8 let
- Nové farmakologické možnosti v léčbě Alzheimerovy nemoci
- Pletencové svalové dystrofie