
Nodo-paranodopatie s protilátkami IgG4 proti neurofascinu-155
Autoři:
K. Revendová 1,2; O. Volný 1,2; MUDr. Jana Junkerová 1,2; I. Woznicová 2; J. Slonková 1,2; R. Mazanec 3; M. Bar 1,2
Působiště autorů:
Katedra a centrum klinických neurověd, LF OU, Ostrava
1; Neurologická klinika FN Ostrava
2; Neuromuskulární centrum, 2. LF UK a FN Motol, Praha
3
Vyšlo v časopise:
Cesk Slov Neurol N 2021; 84(6): 567-569
Kategorie:
Dopisy redakci
doi:
https://doi.org/10.48095/cccsnn2021567
Vážená redakce,
poprvé v české odborné literatuře prezentujeme kazuistiku pacienta s nodo-paranodopatií s potvrzenou pozitivitou protilátek IgG4 proti neurofascinu-155 (NF155). Původně byla tato skupina považovaná za variantu chronické zánětlivé demyelinizační polyradikuloneuropatie (chronic inflammatory demyelinating polyradiculoneuropathy; CIDP) [1]. Zatímco CIDP je definována progredujícím nebo relabujícím průběhem delším než 2 měsíce, symetrickou svalovou slabostí s hypo - či areflexií šlachookosticových reflexů a senzitivní poruchou všech končetin s možným postižením hlavových nervů, pro nodo-paranodopatie mohou svědčit rychle progredující chabá kvadruparéza, kraniální neuropatie, senzitivní ataxie, tremor, neuropatická bolest, zejména rezistence na léčbu intravenózními imunoglobuliny (IVIG) a jen parciální efekt terapeutické plazmaferézy (TPE) či kortikosteroidů [1–3].
V posledních letech byly objeveny protilátky proti paranodálním a nodálním proteinům asociované s rozdílným klinickým fenotypem u pacientů, naplňující klinická a elektrofyziologická kritéria pro diagnózu CIDP [2]. Protilátky proti contactinu-1 (CNTN1) byly prokázány u 2,2–8,7 % převážně starších pacientů s CIDP [4]. Tito pacienti mají akutní nebo subakutní nástup nemoci, predominantně motorické postižení a agresivní průběh se špatnou odpovědí na léčbu IVIG [2]. Protilátky proti proteinu-1 asociovaném s contactinem (CASPR1) byly prokázány u 2 z 37 pacientů dle studie Dopplera et al, jeden pacient naplňoval kritéria Guillain-Barrého syndromu a druhý CIDP. Oba měli silnou neuropatickou bolest, distální svalovou slabost a nedostatečnou terapeutickou odpověď na IVIG [2]. Protilátky proti neurofascinu 140/186 (NF140/186) byly přítomny u méně než 2 % pacientů s CIDP [4]. Většina pacientů má přidružené autoimunitní onemocnění se subakutním rozvojem nemoci, senzitivní ataxií a obecně dobrou odpovědí na terapii IVIG a kortikosteroidy [4]. Protilátky proti NF155 se vyskytly u 4–18 % převážně mladších pacientů (průměrný věk 25,2 ± 10,7 let) s CIDP se subakutním nebo chronickým průběhem nemoci [2,5]. Klinicky mají distální svalovou slabost s vyšší frekvencí akrální parézy (tzv. foot drop), poruchou chůze, senzitivní ataxií a v některých případech tremorem [2,5,6]. Typicky bývá prokázána výrazná hyperproteinorachie (průměr 3,17 ± 1,41 g/l) a EMG svědčí pro získaný symetrický demyelinizační typ neuropatie s prodloužením distálních motorických latencí (DML) a latence F vln [5–7]. Vzácně se mohou rozvíjet demyelinizační léze centrálního nervového systému dle MR mozku [5]. Na terapii IVIG odpovídá pouhých 20 až 30 % pacientů, 50 až 60 % pacientů může odpovídat na léčbu kortikosteroidy [6–8]. Kortikosteroidy kombinované s IVIG jsou efektivnější než IVIG samostatně [7]. Dlouhodobá imunosuprese denními perorálními kortikoidy a/nebo imunosupresivy je efektivnější než opakování IVIG [5]. Rituximab se jeví efektivní u pacientů s anti-NF155 a anti-CNTN1 pozitivitou při selhání klasické imunoterapie, nicméně se jednalo pouze o prezentaci série kazuistických případů [4,6]. K ověření účinnosti rituximabu jsou nutné výsledky randomizovaných, placebem kontrolovaných klinických studií [2,4]. V současné době probíhá dvojitě zaslepená, randomizovaná, placebem kontrolovaná studie RECIPE, která je zaměřena na zhodnocení efektu rituximabu u refrakterních pacientů se stanovenou diagnózou CIDP s průkazem nodálních/paranodálních protilátek oproti skupině pacientů s CIDP, u kterých tyto protilátky nebyly prokázány [9].
Šestačtyřicetiletý muž bez přidružených onemocnění v předchorobí byl hospitalizován pro 10 dní progredující chabou kvadruparézu, pravostrannou periferní lézi nervus facialis, silnou neuropatickou bolest a dysestézie v rukavicové a ponožkové distribuci. Při přijetí byla provedena lumbální punkce s obrazem proteinocytologické disociace (proteinorachie 1,32 g/l [referenční rozmezí 0,2–0,4 g/l], 2 elementy/mm3 [referenční rozmezí 0–5 elementů/mm3]). První EMG, provedená 11. den od vzniku potíží, vykazovala lehké a nespecifické projevy demyelinizační polyneuropatie s převahou postižení motorických vláken (prodloužení DML a nevýbavnost F vln v motorických neurogramech, dobrá výbavnost senzitivních neurogramů, nepřítomnost patologické spontánní aktivity při vyšetření jehlovou elektrodou) (tab. 1 A, B). Vyslovili jsme pracovní diagnózu akutní zánětlivé demyelinizační polyradikuloneuritidy (acute inflammatory demyelinating polyradiculoneuropathy; AIDP) a pacient byl následně léčen sérií pěti TPE. Při překladu k ústavní rehabilitaci kompletně regredovalo oslabení horních končetin a zlepšila se mimika obličeje vpravo.
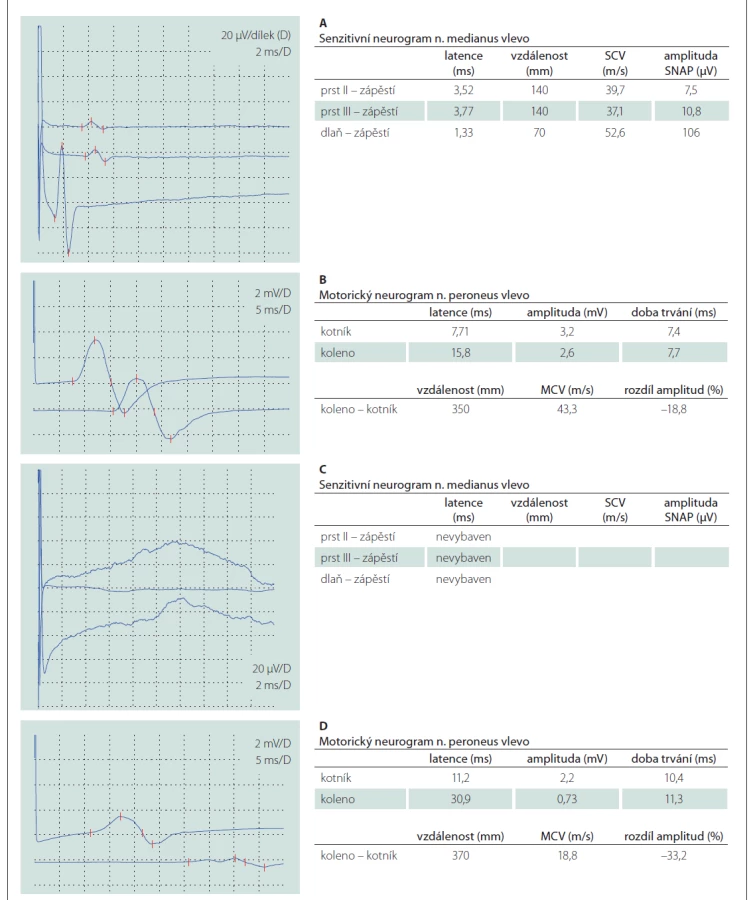
Po 3 měsících došlo k relapsu nemoci, který se projevil rozvojem faciální diparézy a chabé kvadruparézy. Kontrolní EMG potvrdila progresi nálezu, senzitivní neurogramy nebyly výbavné, v motorických neurogramech bylo výrazné prodloužení DML, byly přítomny proximální bloky vedení a nízké amplitudy, všechny F vlny byly nevýbavné (tab. 1 C, D). V jehlové EMG byla přítomná patologická spontánní aktivita (fibrilace, pozitivní ostré vlny a fascikulace) a reinervační změny motorických jednotek s nejvyšší amplitudou kolem 3,5 mV. Pacient byl léčen IVIG v plné dávce, tj. 2 g/kg během 5 dní, nicméně nedošlo ke zlepšení klinického stavu a pacient byl následně přeložen k intenzivní ústavní rehabilitaci.
Ve 4. měsíci nemoci došlo k druhému relapsu a progresi do chabé kvadruparézy těžkého stupně, vč. prohloubení předchozí oboustranné léze lícního nervu s akcentací vpravo. Pacient byl mobilní pouze na mechanickém invalidním vozíku (MIV). V EMG nálezu přetrvávala těžká difuzní demyelinizační léze s bloky vedení postihující motorická a senzitivní vlákna s patrnou postupující axonopatií. Kontrolní lumbální punkce prokázala prohloubení proteinocytologické disociace (proteinorachie 4,38 g/l, 4 elementy/mm3). Vzhledem k naplnění diagnostických kritérii byla upravena diagnóza na CIDP s akutním začátkem. Pro vstupně pozitivní efekt TPE jsme indikovali její opakování. Pacient byl superkonziliárně vyšetřen na specializovaném pracovišti. Bylo vysloveno podezření na nodo-paranodopatii a bylo doporučeno vyšetření protilátek proti nodálním a paranodálním proteinům v séru. Laboratoř v Nuffield Department of Clinical Neuroscience Oxford University & Oxford University Hospitals NHS Foundation Trust potvrdila pozitivitu protilátek proti NF155, titr > 1 : 800, dominantně ve třídě IgG4 a IgG1, třída IgG2 byla přítomna v menší míře. Pacientovi byla doporučena chronická imunosupresivní terapie perorálními kortikosteroidy, ale pro intoleranci bylo v souladu s dostupnými literárními údaji přistoupeno k imunoterapii monoklonální protilátkou anti-CD20 rituximabem. Zvolili jsme schéma 375 mg/m2 v 21denních cyklech celkem 4x, následně 1 000 mg každých 6 měsíců. Aktuálně přetrvává těžká periferní disabilita, ale pacient je samostatně pohyblivý na MIV. Souhrnný vývoj disability hodnocený škálou Inflammatory Neuropathy Cause and Treatment (INCAT), proteinorachie a typ terapie v čase prezentujeme v tab. 2 [10].

Přestože se jedná o vzácnou podskupinu autoimunitní zánětlivé neuropatie, je třeba na ni myslet u pacientů s podezřením na CIDP s akutním nástupem, kraniální neuropatií, ataxií a literálně i tremorem, zejména pokud jsou refrakterní k terapii IVIG. Včasné zahájení léčby rituximabem dává naději na pozitivnější prognózu pacientů, nicméně terapeutické schéma aplikace rituximabu je u nodo-paranodopatií předmětem dalšího výzkumu.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manu script met the ICMJE “uniform requirements” for biomedical papers.
MUDr. Kamila Revendová
Neurologická klinika
LF OU a FN Ostrava
17. listopadu 1790/5
708 00 Ostrava-Poruba
e-mail: kamila.revendova@fno.cz
Přijato k recenzi: 4. 8. 2021
Přijato do tisku: 16. 11. 2021
Zdroje
1. Van den Bergh PYK, Doorn PA, Hadden RDM et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force – Second revision. J Peripher Nerv Syst 2021; 26 (3): 242–268. doi: 10.1111/jns.12455.
2. Van den Bergh PYK, van Doorn PA, Jacobs BC et al. Boundaries of chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst 2020; 25 (1): 4-8. doi: 10.1111/jns.12364.
3. Van Den Bergh PYK, Hadden RDM, Bouche P et al. European federation of neurological societies/peripheral nerve society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripher. Eur J Neurol 2010; 17 (3): 356–363. doi: 10.1111/j.1468-1331.2009.02 930.x.
4. Lehmann HC, Burke D, Kuwabara S. Chronic inflammatory demyelinating polyneuropathy: update on diagnosis, immunopathogenesis and treatment. J Neurol Neurosurg Psychiatry 2019; 90 (9): 981–987. doi: 10.1136/jnnp-2019-320314.
5. Kira JI, Yamasaki R, Ogata H. Anti-neurofascin autoantibody and demyelination. Neurochem Int 2019; 130 : 104360. doi: 10.1016/j.neuint.2018.12.011.
6. Garg N, Park SB, Yiannikas C et al. Neurofascin-155 IGG4 neuropathy: pathophysiological insights, spectrum of clinical severity and response to treatment. Muscle Nerve 2018; 57 (5): 848–851. doi: 10.1002/mus. 26010.
7. Ogata H, Yamasaki R, Hiwatashi A et al. Characterization of IgG4 anti-neurofascin 155 antibody-positive polyneuropathy. Ann Clin Transl Neurol 2015; 2 (10): 960–971. doi: 10.1002/acn3.248.
8. Devaux JJ, Miura Y, Fukami Y et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 2016; 6 (9): 800–807. doi: 10.1212/WNL.0000000000002418.
9. Shimizu S, Iijima M, Fukami Y et al. Efficacy and safety of rituximab in refractory CIDP with or without IgG4 autoantibodies (RECIPE): protocol for a double-blind, randomized, placebo-controlled clinical trial. JMIR Res Protoc 2020; 9 (4): e17117. doi: 10.2196/17117.
10. Breiner A, Barnett C, Bril V. INCAT disability score: a critical analysis of its measurement properties. Muscle Nerve 2014; 50 (2): 164–169. doi: 10.1002/mus.24207.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2021 Číslo 6
Nejčtenější v tomto čísle
- Stiff -person syndrom
- Normotenzní hydrocefalus
- Synukleinopatie a jejich laboratorní biomarkery
- Perorální kladribin v léčbě roztroušené sklerózy – data z celostátního registru ReMuS®