
Anestezie a nervosvalová onemocnění
Anaesthesia and neuromuscular disorders
fragile balance can lead to postponing of necessary interventions, but on the other hand, it can lead to serious risks with improper approaches. Myasthenia is a disease where there is no risk of malignant hyperthermia, and there is an altered sensitivity to peripheral myorelaxants. We prefer to avoid benzodiazepines as a premedication, and after the procedure we always place the patient on a monitored bed with the possibility of artificial ventilation. We do not give suxamethonium to muscular dystrophy and myotonic dystrophy patients and do not use volatile gases due to risk of rhabdomyolysis. There is also no risk of malignant hyperthermia in this group. Malignant hyperthermia is a pharmacogenetic disorder manifested by abnormal hypermetabolic response when exposed to halogenated inhalation anesthetics (halothane, isoflurane, desflurane, sevoflurane) or peripheral muscle relaxants of the depolarizing type (suxamethonium). It can rarely occur even after physical, excessive or heat stress. It is associated with electromechanical coupling and most often occurs with mutations in the ryanodine receptor, but rarely with some other mutations in the genes that are related to calcium metabolism.
Key words:
anaesthesia – myasthenia gravis – muscular dystrophy – myotonic dystrophy – neuromuscular blockade – malignant hyperthermia
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
S. Voháňka 1; P. Štourač 2; M. Klincová 2
Působiště autorů:
Neuromuskulární centrum, Neurologická klinika LF MU a FN Brno
1; Klinika dětské anesteziologie a resuscitace, LF MU a FN Brno
2
Vyšlo v časopise:
Cesk Slov Neurol N 2018; 81(5): 501-514
Kategorie:
Minimonografie
doi:
https://doi.org/10.14735/amcsnn2018501
Souhrn
Anestezie představuje složitou situaci v léčbě pacientů s nervosvalovými chorobami. Obavy z prolomení křehké rovnováhy mohou vést k odkládání potřebných zákroků na jedné straně a závažným rizikům při nesprávném postupu na straně druhé. Myastenie je onemocnění, kde nehrozí riziko maligní hypertermie, je zde změněná citlivost k periferním myorelaxanciím. V premedikaci se raději vyhýbáme benzodiazepinům a po zákroku vždy pacienta umístíme na monitorované lůžko s možností umělé ventilace. U svalových dystrofií a myotonické dystrofie nepodáváme suxamethonium a nepoužíváme volatilní plyny pro riziko rabdomyolýzy. Taktéž u této skupiny není riziko maligní hypertermie. Maligní hypertermie je farmakogenetická porucha, která se manifestuje abnormální hypermetabolickou odpovědí při expozici halogenovanými inhalačními anestetiky (halothan, isofluran, desflurane, sevofluran) nebo periferními svalovými relaxanciemi depolarizačního typu (suxamethonium). Vzácně může vzniknout i po fyzické, nadměrné stresové nebo tepelné zátěži. Souvisí s elektromechanickým spřažením a nejčastěji vzniká při mutacích v ryanodinovém receptoru, vzácně při některých jiných mutacích v genech, které souvisejí s metabolizmem kalcia.
Klíčová slova:
anestezie – myastenie – svalová dystrofie – myotonická dystrofie – nervosvalová blokáda – maligní hypertermie
Obecné poznámky
Neuromuskulární choroby představují širokou skupinu onemocnění, která vyžadují při plánování operačního výkonu a anesteziologického postupu řadu opatření. Jde o přímý (svalová relaxancia) či nepřímý (inhalační či některá intravenózní anestetika) zásah do nervosvalového přenosu (myastenie, Lambert Eatonův myastenický syndrom a kongenitální myastenie), dále o riziko rabdomyolýzy a maligní hypertermie (MH) u některých svalových dystrofií a kanálopatií, respirační a kardiální postižení či multisystémová postižení (myotonické dystrofie). Pozornost vyžaduje i dlouhodobá terapie kortikoidy (myastenie, autimunitní myopatie a neuropatie, Duchennova svalová dystrofie) nebo imunosupresivy. Velmi často jde v případě operačního výkonu o riziko prolomení nastavené křehké rovnovány. Vlastní indikaci operačního výkonu by tedy měla být věnována mimořádná pozornost, a to celého perioperačního týmu. Kromě anesteziologů se na těchto rozhodnutích a péči musí podílet ošetřující neurologové, kardiologové, pneumologové, fyzioterapeuti a podle situace i odborníci dalších oborů [1].
Lze také narazit na situaci, kdy není jasné, zda se jedná o nervosvalové onemocnění nebo pacient neví, že tímto onemocněním trpí. Jde o choroby, které se často vyvíjejí mnoho roků a incipientní stadia mohou být nenápadná. Každopádně by k úvaze o dosud nediagnostikovaném nervosvalovém onemocnění měla vést zvýšená hladina kreatinkinázy, nejasná slabost kořenových svalů, atrofie nebo naopak svalové hypertrofie (zvláště lýtek) či skolióza. Další postup potom závisí na tom, zda se jedná o urgentní či elektivní výkon a na možnosti alespoň rychlé rámcové diagnostiky a klasifikace choroby.
Obecně lze říci, že předoperační stratifikaci rizika je třeba založit na zhodnocení parametrů uvedených v následujících odstavcích.
Respirační kapacita
Při usilovné vitální kapacitě (forced vital capacity; FVC) 20 ml/ kg tělesné hmotnosti a méně lze očekávat problémy s odpojením od umělé plicní ventilace [2]. Ventilační porucha má převážně restrikční charakter, ale vzhledem k často opakovaně prodělaným infekčním komplikacím bývá problematický i stav vlastního plicního parenchymu. Poruchy polykacího aktu zvyšují riziko aspirace. Problémy s toaletou dýchacích cest pak následně zvyšují riziko vzniku pneumonie.
Skolióza
Jde o významný faktor, který přispívá k zhoršení respiračních funkcí a může vytvářet morfologickou překážku či ztížit aplikaci subarachnoidální anestezie. Těžká skolióza se často vyskytuje u spinální muskulární atrofie, u svalových dystrofií odpovídá tíži trupového postižení.
Kardiální postižení
Kardiomyopatie je pro některé svalové dystrofie obligátní (Duchennova svalová dystrofie) nebo dominantní příznak (Emeryho--Dreifussova svalová dystrofie), u jiných se prakticky nevyskytuje (facioskapulohumerální svalová dystrofie). U některých chorob jde o dilatační kardiomyopatie (Duchennova svalová dystrofie), jindy o zvýšené riziko závažných arytmií (myotonická dystrofie), charakteristickou poruchu převodního systému (síňové zástavy a riziko fibrilace komor u Emeryho-Dreifussovy svalové dystrofie, long QT u některých svalových kanálopatií).
U hereditárních chorob je tedy při preoperačním plánování důležitá přesná znalost nozologické jednotky a s ní spojené patofyziologie, která nás nasměruje k potenciálním specifickým rizikům. U autoimunitních onemocnění (myastenie, zánětlivé neuropatie či myopatie) je nutná detailní znalost medikamentózní terapie, kterou jsou pacienti léčeni.
V pooperačním období je u řady nemocných nutné počítat s obtížným odpojením od umělé plicní ventilace a větším rizikem především respiračních komplikací, které vyplývá jak z nízké respirační kapacity, tak z obtížné toalety dýchacích cest (aspirační pneumonie). Je třeba mít k dispozici neinvazivní podporu dechu, respirační fyzioterapii, asistent kašle apod. Prakticky vždy je tedy po zákroku nutné umístění na intenzívní lůžko s možností monitorace. Pokud mají nemocní výrazné omezení hybnosti, tak je na místě důsledná prevence tromboembolické nemoci.
V perioperačním období čelíme u pacientů s neuromuskulárními chorobami nejčastěji následujícím komplikacím:
- rabdomyolýza;
- kardiální komplikace;
- respirační problémy;
- akcentace myotonie;
- hypertermie či hypotermie;
- autonomní dysfunkce.
Moderní trendy v anestezii ve vztahu k nervosvalovým onemocněním
V případě nervosvalových onemocnění je třeba průběh celého perioperačního období velice pozorně dopředu naplánovat. Anesteziologické aspekty hodné zvláštního zřetele jsou u jednotlivých onemocnění popsány přímo v příslušných odstavcích. Obecně lze říct, že kde je možné použít lokální, neuroaxiální či regionální formu anestezie, měli bychom ji preferovat.
Anesteziologickou péči můžeme rozdělit na předoperační anesteziologické zhodnocení pacienta, které má vyústit v rozhodnutí o optimálním způsobu anestezie, dále na vlastní průběh anestezie a pooperační péči. V rámci předoperačního anesteziologického vyšetření se stanovuje míra perioperačního rizika, nejčastěji dle American Society of Anesthesiologists (ASA) skóre (tab. 1). Součástí předoperačního doporučení mohou být další vyšetření směřující ke zjištění aktuálního stavu pacienta a doporučení premedikace, která má za cíl snížit anxietu z výkonu, např. benzodiazepiny, a/ či zmírnit vegetativní reakci (parasympatolytika). Předpokládaná míra operační zátěže se dá stratifikovat do tří skupin dle rozsahu a náročnosti výkonu (tab. 2). U pacientů s nervosvalovými onemocněními nebývá premedikace často podávána. Bezprostřední perioperační anesteziologická péče pak začíná podáním anestezie, pokračuje udržovací fází a končí zotavením z jejích účinků. Pozornost je třeba věnovat všem fázím anestezie, protože přinášejí svá specifická rizika. V rámci úvodu do celkové anestezie je to především riziko obtížné intubace. V průběhu anestezie u některých nervosvalových onemocnění riziko arytmií a na konci anestezie, zejména při použití svalových relaxans, je to riziko přetrvávající nervosvalové blokády a s ním spojená rizika dechové nedostatečnosti či aspirace. Těmto rizikům se snaží předcházet některé nové anesteziologické techniky. Součástí moderní pooperační péče je také akcentace léčby pooperační bolesti a pacienti s nervosvalovými onemocněními netvoří v tomto směru výjimku. Míru předpokládané pooperační bolesti lze predikovat např. dle tab. 3 [3].



Monitorace pacienta v průběhu anestezie
Moderní přístrojová technika vybavená komplexní monitorací životních funkcí pacienta zvýšila v několika posledních dekádách bezpečnost anestezie na úroveň vyšší než „six sigma“ (úroveň řízení procesu, kdy na jeden milión příležitostí připadá max. 3,4 chyby). Neinvazivní sledování hladiny kyslíku v arteriální krvi (SpO2), monitorace hladiny CO2 na konci výdech (etCO2), sledování vdechovaných a vydechovaných plynů vč. volatilních anestetik a rutinní možnost měření neinvazivního i invazivního tlaku krve či sledování EKG patří ke standardům monitoringu v průběhu anestezie či monitorované anesteziologické péče (analgosedace, anesteziologický dohled u rizikového výkonu apod.). Nejnovější směřování sledování pacienta v průběhu výkonu vede k co nejmenší invazivitě vyšetření, co nejrychlejšímu získání aktuálních laboratorních dat o pacientovi (bed-side monitoring) a k získání informací nutných k optimálnímu vedení anestezie (bispektrální index, relaxometrie, hemodynamický monitoring) [3].
Měření hloubky anestezie
Na probuzení či bdělost pacienta v celkové anestezii nás historicky v průběhu výkonu upozorňovaly jen nepřímé klinické známky (tachykardie, tachypnoe, arteriální hypertenze, nadměrné pocení). V horším případě tuto skutečnost demaskoval po výkonu sám pacient. Tradiční Guedelovo schéma stadií hloubky anestezie ztrácí s příchodem moderních anestetik na významu a není vhodné k užití v běžné klinické praxi. V současnosti existuje několik dostupných metod (bispektrální index, entropie), které pracují na principu analýzy EEG záznamu Fourierovou analýzou. Výsledná kvantifikace stanovuje hloubku uměle navozeného bezvědomí a může optimalizovat dávkování a časování podání anestetik. Například u metody bispektrální index jsou to hodnoty: 0 – silentní EEG záznam, 40– 60 stadium chirurgické anestezie, 100 plné vědomí [4]. Pro měření hloubky analgezie lze také využít pupilární algeziometrii, která hodnotí úroveň analgezie dle velikosti zornic a jejich reaktivity [5,6].
Měření hloubky nervosvalové blokády
Blokáda nervosvalového přenosu dočasně oslabí všechny příčně pruhované svaly, což je velice výhodné pro operatéra (klidné operační pole, přehledná dutina břišní atd.), ale klade velké nároky na vedení anestezie. Anesteziolog musí neustále sledovat průběh operace a udržovat svaly paralyzované, aby usnadnil chirurgovi operační přístup. Na druhou stranu musí při probouzení pacienta zajistit plný návrat svalové síly. Tento problém z velké části řeší sledování svalové síly v průběhu operace pomocí relaxometrie, která se stala součástí doporučení odborné anesteziologické společnosti. Zatímco dříve se anesteziolog orientoval pouze podle nepřímých známek (počínající dechové pohyby, hlášení operatéra) a znalosti farmakokinetiky daného relaxans, dnes umožňuje měření svalové síly anesteziologovi velmi precizně dávkovat svalová relaxancia. Pacient je tak dostatečně relaxován až do konce operačního výkonu (uzavření dutiny břišní) a nehrozí předávkování, které pak může výrazně prodloužit probouzení pacienta, příp. se podílet na některých časných pooperačních komplikacích. Velký význam má objektivizace hloubky nervosvalové blokády právě u pacientů s nervosvalovým onemocněním, protože jiným vyšetřením nelze přetrvávající nervosvalový blok identifikovat [3].
Zajištění dýchacích cest
Obávanou noční můrou anesteziologa je scénář „Nemohu intubovat, nemohu ventilovat (pacienta)“. Jedním z anesteziologických evergreenů je otázka identifikace pacienta, u kterého lze předpokládat obtížnou intubaci. Žádná ze současných v praxi užívaných klasifikací nedosahuje ani vzdáleně hodnot 100% pozitivní predikce. Každoročně výrobci uvádí na trh mnoho novinek v oblasti obtížného zajištění dýchacích cest. Velkou revoluci v algoritmech obtížného zajištění dýchacích cest se stalo rutinní zavedení laryngeální masky, zejména intubační (umožňuje přes vlastní lumen intubaci), do algoritmů obtížného zajištění dýchacích cest. Pro její zavedení není třeba pacienta relaxovat, což je u pacientů s nervosvalovou poruchou výhodné. Další revolucí je rozvoj tzv. videolaryngoskopů (jsou opatřeny vizualizačním zařízením na rukojeti nebo na samostatné obrazovce, které získává obraz ze špice laryngoskopu). Zlepšení vizualizace vstupu do hlasivkové štěrbiny je pak významné. V emergentních situacích a při selhání výše zmíněných postupů jsou v široké míře dostupné sety umožňující koniopunkci. Žádná pomůcka, zejména v neškolených rukou, nemá absolutní účinnost, a proto nácvik dovedností na modelech a následně při běžné praxi je nezbytný [7].
Total Intravenous Anesthesia/ Target Controlled Infusion
Ekonomická i faktická dostupnost moderních léčiv užívaných při celkové anestezii, konkrétně ultrakrátce působícího nitrožilního anestetika propofol a opioidu remifentanil, umožnila rozvoj anesteziologických technik totální intravenózní anestezie (Total Intravenous Anesthesia; TIVA) s užitím moderních pump s programem na udržení plazmatické hladiny léčiva (Target Controlled Infusion; TCI). Techniky vycházejí z farmakokinetiky léčiv a existence matematických modelů popisujících vliv podání léku na jeho plazmatickou hladinu, které jsou implementovány do programovatelné infuzní pumpy. Výhodou metody je dobrá řiditelnost takto podané anestezie, limitací pak skutečnost, že metoda je založena jen na matematickém modelu. U mnoha pacientů s nervosvalovými poruchami je tato metoda nejen výhodná, ale dokonce metodou volby (pacienti v riziku MH apod.) [8].
Sugammadex
Situaci v oblasti svalových relaxancií značně mění i nově uvedený lék, který neutralizuje účinek steroidních myorelaxancií, konkrétně rokuronia a vekuronia. Jedná se o sugammadex, v ČR dostupný jako BridionTM (Merck Sharp & Dohme Limited, Hertfordshire, Velká Británie), který patří do velké skupiny látek zvaných cyklodextriny. Cyklodextriny se již delší dobu používají i v jiných odvětvích než ve farmacii, např. v potravinářství. Jejich zvláštností je velká molekula schopná „polapit“ jinou molekulu do svého nitra. Konkrétně sugammadex je tvořen osmi cukernými zbytky spojenými do kruhu a vzniklá dutina uvnitř kruhu svojí velikostí přesně odpovídá velikosti steroidních relaxancií rokuroniu a vekuroniu. Rokuronium zachycené uvnitř sugammadexu potom nemůže účinkovat na nervosvalové ploténce a způsobit paralýzu příčně pruhovaných svalů. Navíc sugammadex s navázaným rokuroniem se velmi snadno a rychle vylučuje ledvinami, což je rozdíl oproti samotnému rokuroniu, které podléhá především jaternímu metabolizmu. Celý tento mechanizmus neutralizace účinku léku je v anestezii naprosto revoluční, protože zatím většina postupů zrušení účinku léků použitých při anestezii byla založena na vytěsnění jedné molekuly z místa účinku jinou molekulou. Až dosud používaná dekurarizace (zrušení účinku svalového relaxancia) navíc přinášela řadu nežádoucích účinků, byla použitelná pouze u doznívajícího svalového bloku a u mnoha pacientů s nervosvalovými onemocněními byla přímo kontraindikována (myotonia congenita apod.). Sugammadex naproti tomu je použitelný v kterékoliv fázi svalového bloku a s minimem nežádoucích účinků. Masovému rozšíření zatím brání pouze vyšší cena. Jeho vyšší ekonomická náročnost je však právě u pacientů s nervosvalovými onemocněními ospravedlnitelná. V kombinaci s objektivní monitorací míry zotavení z nervosvalové blokády maximalizuje bezpečnost anestezovaných pacientů a v případě nutnosti použít svalovou relaxaci v průběhu výkonu tvoří kombinace rokuronium, sugammadex a objektivizace hloubky nervosvalové blokády preferovaný způsob vedení svalové relaxace u těchto pacientů [9,10].
Speciální část
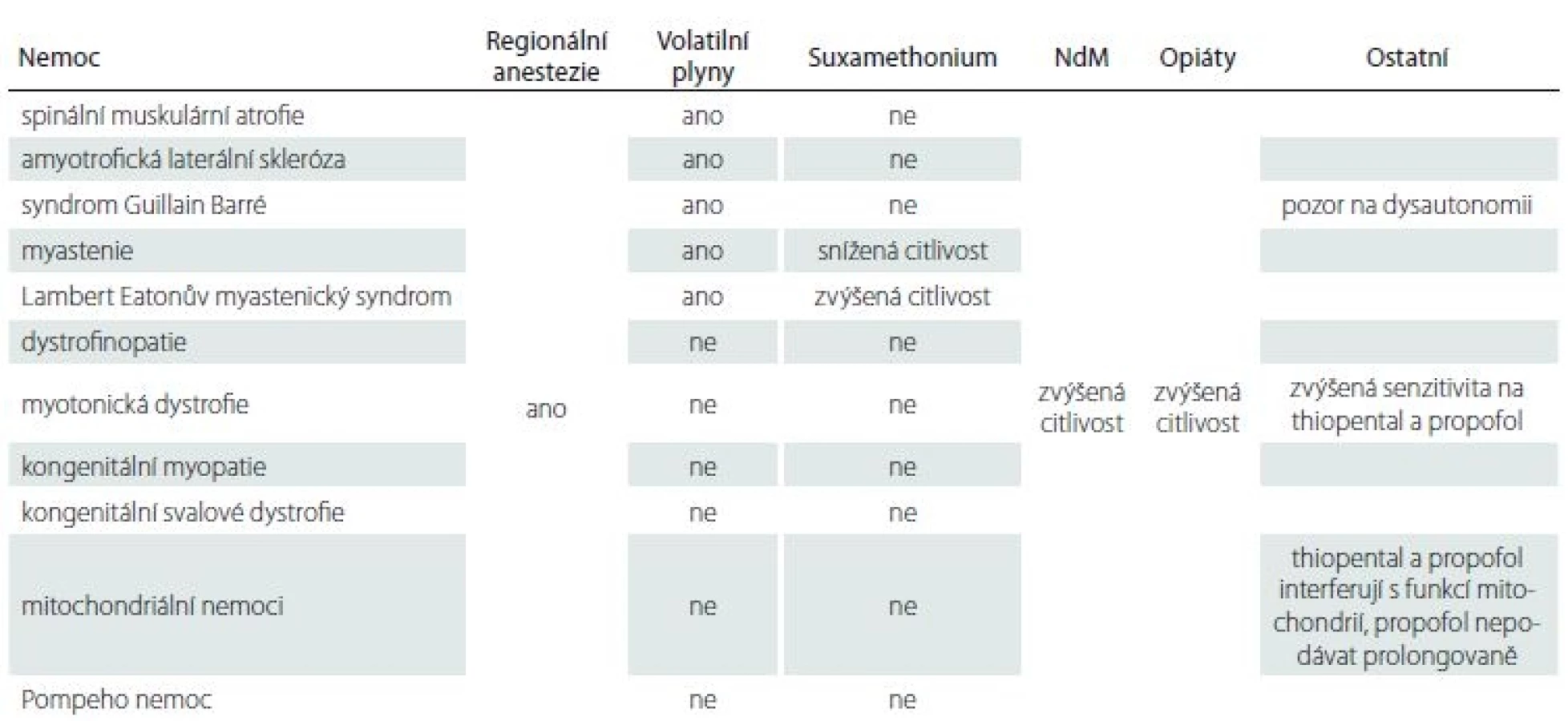
Základní přehled o jednotlivých chorobách a rizikových medikamentech podávají tab. 4, 5 [11].

Myastenie
Myastenie je autoimunitní onemocnění charakterizované svalovou unavitelností. Příčinou je autoimunitní děj, jehož cílem je postsynaptická část nervosvalové ploténky. Dominuje okulobulbární predilekce, respirační svaly jsou postiženy přibližně ve 20 % případů. Choroba je asi v 10 % asociována s jinými autoimunitními chorobami a až v 15 % je přítomen thymom. Chorobu charakterizuje kolísavá unavitelnost s cirkadiálním kolísáním (akcentace zpravidla večer) a ponámahovým horšením. Terapie je obvykle vedena kombinací podávání inhibitorů cholinesterázy, imunomodulační terapie (kortikoidy, imunosupresiva, monoklonální protilátky, imunoglobulin, plazmaferéza) a thymektomie. Řada léků vykazuje významnou interakci s léčivy užívanými při podávání anestezie. Lze konstatovat, že samotná choroba představuje zvýšené perioperační riziko. Elektivní chirurgický výkon by měl být plánován do období navozené remise. Pokud je nutné operaci provést bezodkladně navzdory nedostatečně kompenzovanému stavu, potom se snažíme navodit remisi rychle pomocí intravenózních imunoglobulinů, event. plazmaferézy [12]. Typickými operačními výkony u pacientů s myastenií gravis jsou thymektomie a tracheostomie [13].
V oblasti anestezie a perioperační péče u nemocných s myastenií panuje řada obav, které mají částečně historický charakter, ale na druhé straně odráží obtížné možnosti získávání relevantních poznatků v této oblasti. Většina publikací jsou kazuistiky, řada prací je starších než 15 let. Snižování mortality a zlepšování kvality života nemocných s myastenií však nepřímo ukazují na nesporný pokrok i v této oblasti. Nakolik se ovšem jedná o celkové zlepšení kompenzace myastenie a nakolik o nové anesteziologické techniky, nelze rozhodnout. Je nepochybné, že obě odbornosti ke zvýšení bezpečnosti pacientů přispívají. V letech 2013 a 2014 byly v časopise Anesteziologie a intenzivní medicína představeny nové postupy k anestezii u pacientů s myastenia gravis, a to v oblasti volby myorelaxans [14] a volby inhalačního anestetika [15]. V roce 2015 bylo publikováno doporučení evropského týmu Orphan Anaesthesia pro vedení anestezie u pacientů s myastenií [13]. V témže roce byl českými autory otištěn největší soubor pacientů s myastenií, jež podstoupili thymektomii či cholecystektomii [16].
Předoperační rozvaha musí vždy zahrnovat zhodnocení respiračních funkcí [17], vč. FVC a bulbárních funkcí – tedy schopnosti toalety dýchacích cest (kašlání, polykání). Důležité je také posoudit, zda tyto funkce jsou zachovány v horizontální poloze (např. léze bránice se projevuje právě zhoršením ventilace v horizontální poloze). U nemocných s rozsáhlejším nádorem thymu mohou nastat problémy s intubací (deviace trachey a kvůli kompresi velkých cév i kardiovaskulární problémy) [18]. Jinak ale myastenie není na rozdíl od řady jiných svalových onemocnění choroba postihující myokard a kromě obecných doporučení nejsou v tomto smyslu třeba žádná specifická opatření.
Umožňuje-li to charakter operačního výkonu, preferujeme při rozhodování o typu anestezie lokální, regionální či neuroaxiální techniky (epidurální či subarachnoidální).
Analgosedace (kombinace podání benzodiazepinu s opioidem) není bezpečná technika pro obtížnou regulovatelnost míry sedace a možnost rozvoje dechové deprese a zhoršení myastenické symptomatologie.
Pro celkovou anestezii s řízenou ventilací není jednoznačné doporučení. Volíme však co nejlépe řiditelná anestetika s min. ovlivněním nervosvalového přenosu, je-li to vzhledem k charakteru výkonu možné. Je třeba poznamenat, že v řadě případů u plně kompenzovaných pacientů jsou pro vedení anestezie důležitější další komorbidity než vlastní myastenie.
Při plánování perioperační péče je také třeba zvažovat podání řady dalších léků, které mohou vést k exacerbaci myastenie (především aminoglykosidová a makrolidová antibiotika, některá antiarytmika apod.) [13,19] (tab. 6).
![Léčiva s potencionálním ovlivněním průběhu anestezie u myastenie [13].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/c894ca80a9cc7f220d809e7d3926d30e.jpeg)
Význam stávající medikace
Inhibitory cholinesterázy
Inhibitory cholinesterázy (ICHE) mají různou délku působení a mohou interferovat s léky, které se používají při anestezii (tab. 7). Pokud není možné provést zákrok mezi jednotlivými dávkami inhibitorů, nahrazujeme je parenterálním podáváním neostigminu v odpovídající dávce. Jde o léky, které významně zvyšují svalovou sílu a používají se k symptomatické úlevě. Někteří pacienti jsou na ICHE závislí psychicky a obávají se jakéhokoli vynechání dávky. Na místě je podrobné vysvětlení a ujištění o správném postupu [12]. Hlavní nežádoucí účinky vyplývají z cholinergního efektu: akumulace acetylcholinu na muskarinových receptorech hladkých svalů a žláz se zevní sekrecí (průjmy, nauzea, zvracení, slinění, slzení, bronchospazmus). Dále dochází k aktivaci svalových nikotinových receptorů (svalové křeče, fascikulace). Gastrointestinální potíže lze potlačit loperamidem nebo antimuskarinovými látkami (propantelin, atropin).

Kortikoidy
Jde o léky významné z řady důvodů: zvláště ve vyšších dávkách zhoršují hojení ran a zvyšují riziko infekcí, dále zvyšují glykemii a riziko stresového vředu. Je třeba mít na paměti, že výrazné i krátkodobé zvýšení dávky kortikoidů může vést k zhoršení myastenie či vzniku myastenické krize [20]. Mírné zvýšení dávky (do 20 %) odpovídající zvýšené stresové zátěži je bezpečné. Při nutnosti parenterálního podávání máme dostatečnou škálu galenických forem.
Imunosupresiva
Imunosupresivní účinek obvykle podávaných látek je po navození remise dlouhodobý, a tak není problém je na krátké perioperační období několika dnů vynechat. Účinek nejčastěji používaného azatioprinu trvá řadu měsíců, u cyklosporinu A se jedná o týdny. Pokud není pacient po delší období po operaci schopen perorálního příjmu, je třeba vždy zajistit do sondy nebo gastrostomie odpovídající galenickou formu léčivé látky.
Premedikace
V premedikaci se nedoporučuje podávání benzodiazepinů a opioidů. Ani jedna skupina nemá přímý vliv na nervosvalovou ploténku, předpokládá se ale určitá sumace účinků. U benzodiazepinů jde o myorelaxaci a vliv na svalovou sílu, v případě opioidů o depresivní efekt na ventilaci. U plně kompenzovaných pacientů s dobrou respirační rezervou nebo u okulárních forem lze i tyto lékové skupiny s jistou opatrností použít [12].
Peroperační průběh
Lokální, regionální a neuroaxiální techniky
Při volbě lokálního anestetika, především u regionálních technik, volíme preferenčně amidová oproti esterovým. Současně se vyvarujeme překračování max. dávek, protože vysoká plazmatická hladina každého lokálního anestetika může vyvolat svalovou slabost. Snížení hladiny lokálního anestetika v plazmě může být dosaženo přidáním adjuvans (adrenalin).
Celková anestezie
Úvod do anestezie
Indukce anestezie všemi intravenózními krátce působícími anestetiky je považována u pacientů s myastenií gravis za bezpečnou (propofol, thiopental, etomidát nebo ketamin).
Vedení anestezie
Opioidní analgetika neinterferují s nervosvalovým přenosem. Lze tedy použít všechna komerčně dostupná. Přesto umožňuje-li to charakter výkonu, je vhodné pro peroperační analgezii použít remifentanil, ultrakrátce působící opioid s farmakokinetikou nultého řádu. Pooperační analgezii je poté nutno zajistit buď neopioidními analgetiky nebo jiným opioidem s delším poločasem účinku.
V případě použití remifentanilu je velmi dobrou strategií podat TIVA kombinací s kontinuálně aplikovaným propofolem. Nejmodernější přístup počítá s aplikací v režimu TCI, který zohledňuje farmakokinetické modely, a je tak dobře řiditelný prostřednictvím ovlivnění efektivní či plazmatické hladiny léčiva v organizmu nemocného.
V případě použití inhalační anestezie je nutno konstatovat, že všechna aktuálně dostupná anestetika z řady halogenidovaných éterů jsou dobře řiditelná a přes určitou míru schopnosti navodit svalovou relaxaci nelze jednoznačně říct, že je třeba se jich u pacientů s myastenií striktně vyvarovat. Senzitivita nemocných s myastenií se vůči jednotlivým volatilním plynům liší (Kadosaki et al) [12]. Nejlépe řiditelným je v této oblasti desfluran, nejpoužívanější sevofluran.
Periferní myorelaxancia se v anesteziologii používají k usnadnění úvodního zajištění dýchacích cest intubací, udržování svalové relaxace při řízené ventilaci a k usnadnění zejména nitrobřišních a nitrohrudních výkonů. V anesteziologii se používají dva typy periferních myorelaxancií: nedepolarizující a depolarizující. U pacientů s myastenií mají tyto látky určitý nepředvídatelný efekt: změněnou citlivost na obvyklou dávku a prodloužený interval neuromuskulární blokády. Dále je přítomen vliv současně podávaných inhibitorů cholinesterázy. Adekvátní dávka myorelaxans se u myasteniků pohybuje v širokém rozmezí mezi 5 a 90 % dávky obvyklé. Zejména z těchto důvodů je striktně doporučeno u pacientů s myastenií gravis při použití svalové relaxace objektivizovat hloubku nervosvalové blokády, a to nejen peroperačně, ale v celém perioperačním období. Zlatý standard monitorace hloubky nervosvalové blokády je akcelerometrická metoda. Je-li to možné, je vhodné objektivizovat hodnotu train of four (TOF) ratio – poměr intenzity 4. a 1. záškubu vyvolaného přístrojem již předoperačně při vědomí. Hodnota by měla být 1,0 (nepřítomnost tzv. fadingu v průběhu stimulace). U pacienta s myastenií gravis může být již iniciálně hodnota nižší než 0,9, což značí únavnost (fading) a tato skutečnost by měla rezultovat ve snížení dávky myorelaxans. Na konci operačního výkonu by mělo být dosaženo předoperačních hodnot.
Obecně však lze říct, že není-li to nezbytné, je spíše tendence se svalovým relaxans při anestezii nemocných s myastenií vyhýbat [21]. V literatuře jsou také popsány případy, kdy po použití periferních myorelaxancií dojde k manifestaci dosud latentní myastenie [21,22]. Vhodnou alternativou pro zajištění dýchacích cest intubací bez nutnosti podání myorelaxans je podání kombinace propofolu a remifentanilu, které však není vhodné v případě nutnosti použití tzv. bleskového úvodu do celkové anestezie.
Depolarizující myorelaxancia (succinylcholin, suxamethonium [SCH]) působí naopak jako agonisté a k myorelaxaci dojde abnormální aktivitou ploténky a depolarizačním blokem. Látka je rozkládána acetylcholinesterázou a její účinek není zrušitelný neostigminem. Při použití depolarizujících myorelaxancií je nutné počítat s určitou deplecí postsynaptických receptorů a tím nutností vyšší dávky. Eisenkraft prokázal, že efektivní dávka pro 50 % populace (ED50) a efektivní dávka pro 95 % populace (ED95) je 2×, resp. 2,6× vyšší u nemocných s myastenií [23]. Dávka SCH používaná obvykle pro úvod do anestezie (1– 1,5 mg/ kg) však ED95 překračuje, takže stačí mírné navýšení (1,5– 2 mg/ kg) k dosažení adekvátní rychlé relaxace. SCH je po podání během minut v krevním řečišti metabolizováno pseudocholinesterázou. Inhibitory cholinesterázy (kterými jsou pacienti často léčeni) snižují její efekt a může tak dojít k prolongaci myorelaxačního efektu [9,24]. Děje se tak i u mivacuria ze skupiny nedepolarizujících myorelaxans.
Nedepolarizující (atracurium, cis-atracurium, mivacurium, vecuronium, pancuronium, rocuronium) působí tak, že kompetitivně obsazují acetylcholinové receptory. Jednotlivé druhy se od sebe liší délkou trvání účinku a dalšími farmakologickými vlastnostmi. Jsou odvozeny od kurare, šípového jedu rostlinného původu, používaného americkými indiány. Při jejich podání (na rozdíl od depolarizujících) jsou nemocní s myastenií výrazně citlivější než běžná populace. Je to dáno menším množstvím receptorů a motorických plotének, které jsou obsazovány. Je nutná redukce dávky a je třeba počítat s delší dobou účinku. Z tohoto důvodu se nedoporučuje používání relaxans s dlouhým účinkem (např. pancuronium), krátkodobé lze s opatrností použít. Pro urychlení zotavení z účinků těchto léků na konci anestezie se obvykle používají inhibitory cholinesterázy (např. neostigmin). U pacientů s myastenia gravis se však aktivní reverzi nervosvalové blokády inhibitory cholinesterázy snažíme vyhnout pro možnost vyvolání cholinergní krize. Lze je alespoň podávat titračně. Po podání vysoké dávky neostigminu se může vyskytnout paradoxní svalová slabost při již téměř úplném zotavení neuromuskulárních funkcí. Navíc hluboký neuromuskulární blok nemůže neostigmin antagonizovat ani ve vysoké dávce, protože již nemůže dále zvýšit dostupnost acetylcholinu.
Jednou z možností vyvarování se podání inhibitorů cholinesterázy je vyvést pacienta z nervosvalové blokády při řízené ventilaci tzv. spontánním zotavením, což však při známé nepředvídatelné prolongaci blokády u těchto pacientů může být otázka desítek min až jednotek h. V průběhu zotavení je nezbytné monitorovat hloubku nervosvalové blokády a extubovat pacienta až při dosažení předoperačních hodnot TOF ratio či hodnoty 1,0.
Změnu v přístupu k aktivní reverzi nervosvalové blokády přináší zavedení léčivé látky sugammadex (Bridion). Jde o modifikovaný γ-cyklodextrin, který ireverzibilně (chelací, enkapsulací) váže myorelaxancia aminosteroidního typu (rokuronium, vecuronium) a ruší jejich účinek. Jedná se o zcela jinou koncepci antagonizace svalových relaxancií než s dosud běžně používanými inhibitory cholinesterázy (neostigmin), čímž se stává použití těchto myorelaxancií u rizikových pacientů výrazně bezpečnějším [23,25]. Při mělké blokádě (je patrný záškub v režimu TOF či změřitelný TOF ratio) je indikována dávka 2 mg/ kg, při hluboké blokádě (není patrný záškub v režimu TOF, ale jsou patrné záškuby v režimu PostTetanicCount) se podává dávka 4 mg/ kg. Pro bezprostřední antagonizaci vysokých dávek, zejména intubačních, rokuronia se doporučuje 16 mg/ kg sugammadexu. Výhodou je možnost udržování hlubokého neuromuskulárního bloku až do konce chirurgického výkonu a možnost aktivně revertovat jakoukoli hloubku nervosvalové blokády. Rychlost antagonizace je mnohonásobně vyšší než při použití neostigminu, v publikované práci Vymazala et al došlo k plnému zotavení v průměru do 2 min od podání sugammadexu [16].
Rokuronium je aktuálně svalové relaxans volby v případě nutnosti zahájit anestezii u myasteniků tzv. bleskovým úvodem (rapid sequence induction) při známém vyšším riziku aspirace či rychlé desaturace pacienta v průběhu úvodu do anestezie. Sugammadex, pro svoji schopnost zvrátit i velmi hlubokou nervosvalovou blokádu, je nezbytnou součástí bezpečného postupu v tomto případě.
Pooperační péče
Protože prolongovanou nebo rekurentní hypoventilaci nelze u pacientů s myastenia gravis nikdy vyloučit, je nezbytné pacienta umístit až do plného zotavení z účinků všech složek anestezie na monitorované lůžko s možností rychlého zavedení respirační podpory [10,26– 28]. V případě, že byla použita nedepolarizující svalová relaxans, je vhodná objektivní monitorace zotavení z nervosvalové blokády akcelerometricky v režimu TOF ratio až do dosažení předoperačních hodnot či hodnoty 1,0. Pacient má obvykle v časném pooperačním období menší svalovou zátěž a tomu je třeba přizpůsobit dávky inhibitorů cholinesterázy [29]. Předejdeme tak zbytečným nežádoucím cholinergním účinkům.
V pooperačním období je také vhodné věnovat pozornost dostatečné analgetizaci, protože silná bolest může zhoršit průběh onemocnění. Například u thymektomie, s předpokládanou střední až vysokou mírou pooperační bolesti, je vhodné využít hrudní epidurální analgezii k tlumení pooperační bolesti. Lze-li u ostatních výkonů dosáhnout dobré kontroly bolesti (na vizuální analogové škále pod 4) bez použití opioidů, není jejich použití nutné a je možné kombinovat nesteroidní analgetika, paracetamol a metamizol.
Ambulantní anestezie
Je třeba konstatovat, že operační výkon i anestezie u pacientů s myastenia gravis v ambulantních podmínkách nejsou bezpečné a nemůžeme je doporučit.
Maligní hypertermie
Jde o farmakogenetickou poruchu, která se manifestuje abnormální hypermetabolickou odpovědí při expozici halogenovanými inhalačními anestetiky (halotan, isofluran, desflurane, sevofluran) nebo periferními svalovými relaxanciemi depolarizačního typu (SCH). Vzácně může vzniknout i po fyzické, nadměrné stresové nebo tepelné zátěži [30]. Tato porucha je známa také ve veterinární medicíně, nejčastější je tzv. stresový syndrom prasat (porcine stress syndrome), ale vyskytuje se i u koní či myší a jiných zvířat.
Klíčovým prvkem v patogenezi je zhroucení metabolizmu kalcia. Mutovaný ryanidový nebo dihydropyridínový receptor reaguje při vystavení spouštěčům MH inadekvátně, tj. dochází ke spuštění nekontrolovaného úniku kalcia ze sarkoplazmatického retikula, což následně vede k trvalé svalové kontrakci, nastává deplece adenosintrifosfátu (ATP). Dochází k dramatickému zvýšení spotřeby kyslíku a nadměrné tvorbě CO2 a tepla. Deplece ATP jako zásobníku energie vede k dezintegraci sarkolemy a úniku draslíku, kreatinkinázy a myoglobinu do cirkulace, což má za následek sled přímo život ohrožujících dějů.

Epidemiologie
Incidence MH je odhadována na 1 případ na 10– 250 tisíc anestezií [30,31]. Vzhledem k dědičnosti se její výskyt může výrazně lišit oblast od oblasti, např. v USA je nejvyšší ve státu Wisconsin. Epizoda může, ale nemusí nastat již při první expozici, v průměru se uvádí tři předchozí expozice bez rozvoje MH. Osoby mužského pohlaví jsou náchylnější v poměru 2 : 1 [32– 34]. Průměrný věk manifestace je 18 let. Rozmezí popsaných případů je 6 měsíců až 78 let. Přesnou incidenci asi nikdy nezjistíme, protože řada nemocných s citlivostí s MH se nikdy v životě nedostane do rizikové situace [34– 36]. Prevalence genetických abnormalit, které se podílejí na citlivosti k MH, bývá odhadována na 1 : 3 000 až 1 : 8 500. Recentní data získaná celoexomovým sekvenováním však ukazují na podstatně vyšší kumulativní prevalenci nejčastějších genetických poruch asociovaných s MH, tedy mutace v genu ryanodinového receptoru (RYR1) a dihydropyridinového receptoru (CACNA1S), 1 : 400 [37].
Farmakologické spouštěče
Všechna inhalační anestetika (kromě oxidu dusného) a depolarizující myorelaxans SCH mohou spouštět MH. Žádné ostatní látky používané k vedení anestezie (propofol, ketamin, tiopental, benzodiazepiny, opioidy, nedepolarizující myorelaxancia [NDMR], katecholaminy atd.) nemají potenciál ke spuštění MH.
U pacientů s tendencí k MH může dojít k spuštění krize i výraznou fyzickou zátěží, expozicí teplu nebo za situace zvýšeného stresu, aniž je podáno některé z výše uvedených farmak, a existuje domněnka, že riziko MH je větší, pokud měl pacient před zákrokem větší fyzickou zátěž [29,37,38].
Onemocnění spojená s MH
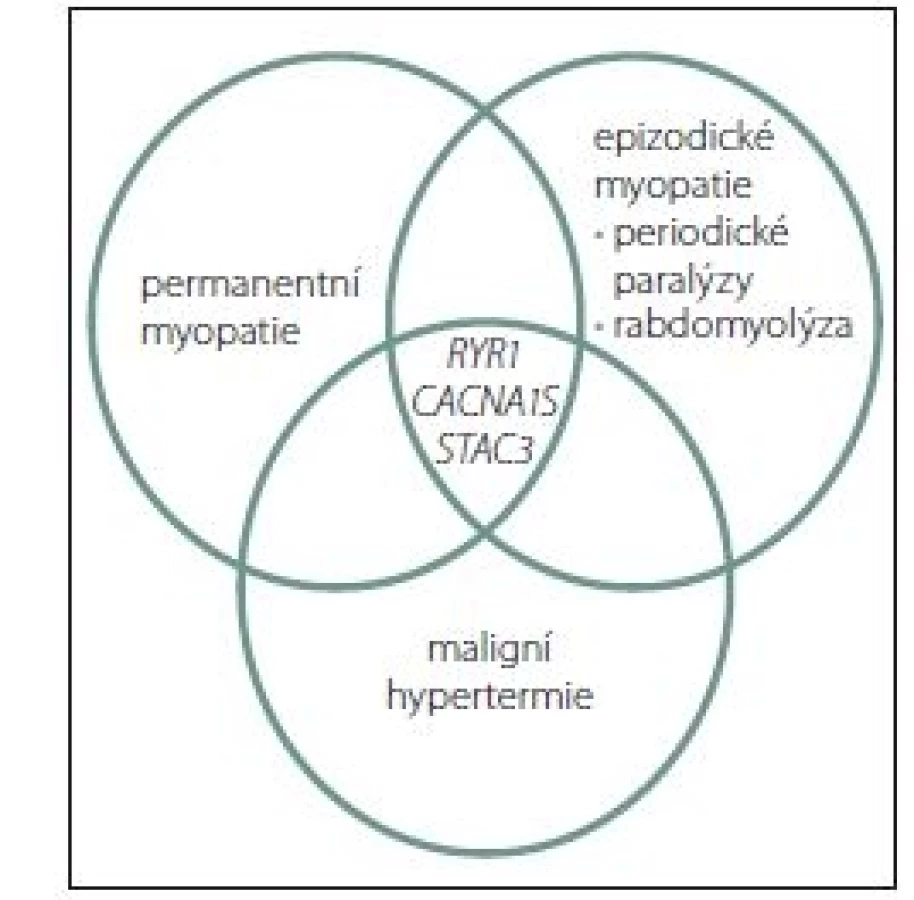
Dosud byly identifikovány tři genové poruchy související s MH. Všechny souvisí s metabolizmem kalcia, resp. s tzv. elektromechanickým spřažením (excitation contraction coupling; ECC). Zatím je prokázána řada kauzálných mutací v genu RYR1 [37– 39], dvě mutace (c.3257G>A / p.Arg1086His, exon 26 a c.520C>T / p.Arg174Trp, exon 4 [40,41]) v genu membránového napětím řízeného kalciového kanálu, resp. jeho alfa podjednotce CACNA1S a nově mutace v genu STAC3 (jde o jednu rodinu původních obyvatel Ameriky se vzácnou autozomálně recesívní myopatií – AR NAM). Klinické potíže a obrazy však vytvářejí alespoň částečně se překrývající množiny (obr. 1). Většina případů (75 %) MH je spojena s mutacemi v RYR1 [42]. Mutace v CACNA1S se na celkovém počtu MH podílí 1 % [41]. Kromě těchto tří genů bylo identifikováno dalších min. pět genů a lokusů se vztahem k MH, většinou s AD dědičností (CACNL2A/ 7q21, SCNA4/ 17q11, CPT2/ 1p32 a dva lokusy 5p a 3q13) [43].
Fig. 1. Clinical manifestations of gene mutations
in muscular diseases that contribute
to the sensitivity to malignant
hyperthermia.
RYR1 asociovaná kongenitální myopatie
Pravděpodobně se jedná o nejčastější kongenitální myopatii. Podstatou je patologická mutace v genu RYR1, který je odpovědný za uvolňování kalcia ze sarkoplazmatického retikula. Izoforma RYR2 je exprimována v myokardu a RYR3 v mozku. Gen je lokalizován na dlouhém raménku 19. chromozomu (19q13.2) a obsahuje 104 exonů – výsledný protein je složen z 5037 aminokyselin. Poruchy jsou převážně autozomálně dominantní, některé autozomálně recesivní. Histologicky jde nejčastěji o central core obraz, dále se může histologicky jednat o typovou dysproporci svalových vláken (congenital fibre type dysproportion), centronukleární obraz či obraz tyčinkové myopatie.
Klinické příznaky jsou extrémně variabilní. Většina se manifestuje již v kojeneckém věku hypotonií, svalovou slabostí, opožděním motorického vývoje a ortopedickými deformitami. Slabost je převážně axiální, více je postižen pánevní pletenec, může být i ptóza. Průběh je většinou lehký a neprogreduje [44]. Ne všechny mutace v RYR1 vedou k MH – dosud jich bylo popsáno asi 200 [42], mutací v RYR1 je ale celkem > 300.
U složených heterozygotů (různé mutace na obou alelách) s příznaky kongenitální myopatie byla popsána i neanestetická MH, která bývá spuštěna mimořádnou fyzickou zátěží, zvláště v horku nebo během horečky při infekci. Léčebně se podává dantrolen a intenzivní chlazení [45].



Histologické a morfologické varianty svalových chorob spojované s MH [17].
Central core disease
Central core disease (CCD) je autozomálně dominantní (ale i autozomálně recesivní) onemocnění spojené s mutací v ryanodinovém receptoru RYR1 (svalová izoforma). Gen je lokalizován na dlouhém raménku 13. chromozomu (13q13.2) Klinická manifestace: 1. kongenitální myopatie s projevy neonatální hypotonie, opožděným motorickým vývojem, skoliózou, deformitami nohou a svalovou slabostí; 2. syndrom King Denborough – skeletální abnormality, dysmorfizmus, malý vzrůst, charakteristická facies; 3. některé mutace mohou ale způsobovat pouze MH bez klinického postižení svalového systému [46,47]; 4. obraz pletencové svalové dystrofie (limb girdle muscular dystrophy; LGMD).
Multiminicore disease
Autozomálně recesivní myopatie s axiálním, respiračním, bulbárním a extraokulárním postižením. Neprogresívní onemocnění, které není spojeno s vnímavostí k MH [46,47].
Nemalinové tyčinkové myopatie
Klinicky i geneticky heterogenní onemocnění. Je známo nejméně 12 genů s dominantní či recesivním typem dědičnosti, které mohou způsobit tento histologický obraz. Jedním z nich může být vzácně i mutace v RYR1 [48].
Centronukleární myopatie
Také tato histologická klasifikace má za podklad celou řadu genových defektů, přičemž jedním z nich může být recesivní mutace v RYR1 (nejčastěji typem je X-linked myotubular myopathy, která ale nesouvisí s MH) [49].
Klinický obraz
Maligní hypertermie může vzniknout kdykoliv během anestezie i v časném pooperačním období, ale nejpozději do 1 h po ukončení expozice volatilním anestetikům. Časnými příznaky jsou tachykardie, arteriální hypertenze a především vzestup koncentrace CO2 ve vydechovaném vzduchu (etCO2) navzdory vzestupu minutové ventilace. Dále následuje svalová rigidita (zvl. při podání SCH). Vzestup teploty je velmi rychlý: 1– 2 °C každých 5 min, ale samotná hypertermie je až pozdním příznakem. Může dojít až k vzestupu nad 44 °C, což vede k extrémní produkci CO2, rozsáhlé orgánové dysfunkci a diseminované intravaskulární koagulopatii a následně k smrti. Nekontrolovaný hypermetabolizmus vede k respirační a ve většině případů i k metabolické acidóze. Pokud není stav léčen, dochází k rozsáhlé rabdomyolýze vedoucí k hyperkalemii a selhání ledvin při masivní myoglobinurii. Dalšími komplikacemi je městnavé srdeční selhání, ischemie střev a kompartment syndrom způsobený otoky na končetinách. Přesáhne-li teplota 41 °C, je nejčastější příčinou smrti diseminovaná intravaskulární koagulace [30]. Je-li šokový stav a nástup acidózy rychlý, nemusí výjimečně k vzestupu teploty vůbec dojít [50].
Diskutuje se, zda je samotná rabdomyolýza, vzniklá během anestezie nebo do 24 h po expozici, známkou náchylnosti k MH či nikoli [51]. Současný názor je spíše takový, že MH je způsobena přesně definovanými genovými defekty a rabdomyolýza je obecná komplikace u řady svalových chorob, ale může přispět k podezření a následné diagnostice MH.
Někdy může být úvodním příznakem SCH indukovaná svalová rigidita masseterů vyskytující se asi u jednoho dítěte ze 100 dětí, které byly uvedeny do anestezie volatilním plynem a jimž bylo podáno uvedené myorelaxans. Asi v 15 % se následně rozvíjejí příznaky MH.
Pro včasnou diagnostiku MH je nejdůležitější tuto dianózu mít na paměti. Rozhodující je monitorace vitálních funkcí pacienta, hodnoty vydechovaného etCO2 a centrální tělesné teploty během zákroku. Údaje publikované z amerického registru (North American Malignant Hyperthermia Registry; NAMHR) ukazují, že riziko úmrtí na MH bylo 14× větší u nemocných, kde nebyla monitorována centrální teplota a téměř 10× vyšší, byla-li sledována pouze teplota kožní [52].
Diagnostika
Donedávna byl zlatým standardem v diagnostice halothanem a kofeinem indukovaný in vitro kontrakční test (IVCT) [30]. Evropský protokol má senzitivitu 99 % a specifitu 94 %, americká varianta 97 % senzitivitu a 78 % specificitu [53,54]. Pro tuto diagnostiku je však třeba poměrně velký úsek viabilní svalové tkáně, zpravidla m. quadriceps (2,5– 5 cm) s provedením testu velmi krátce po odběru [36]. V dnešní době je tato metoda proto stále více nahrazován přímou DNA diagnostikou kandidátních genů. Pokud se u pacienta detekuje jedna z kauzálních mutací, považuje se pacient za MH suspektního a toto vyšetření má stejnou váhu jako IVCT, aniž musí být proveden. Seznam kauzálních mutací se neustále rozšiřuje a je k nalezení na webových stránkách Evropské společnosti pro MH [55]. V případě nedetekovaní kauzální mutace se o sklonu k MH nedá přesvědčivě rozhodnout a pacient se k její vyloučení nebo potvrzení musí podrobit IVCT.
Jsou známy farmakologické spouštěče a kandidátní geny: tato množina podstatně zužuje skupinu kandidátů. V praxi tedy stojíme před otázkou¸ u kterých pacientů je třeba anestezii modifikovat a jak. Odpověď je snadná – u všech nemocných s dědičným (genově podmíněným) svalovým onemocněním, kteří nemají jasně stanovený genový podklad nebo u nemocných s naléhavým podezřením na svalové onemocnění (hyperCKemie, kořenová svalová slabost).
Terapie
Kritickým elementem v terapii MH je bezprostřední podání dantrolenu. Jde o derivát difenylhydantoinu inhibující uvolňování kalcia ze sarkoplazmatického retikula (před jeho zavedením dosahovala mortalita při MH 60– 80 %). Při podezření na MH je třeba zastavit podávání spouštěcí látky, použít 100% oxygenaci a co nejrychleji ukončit chirurgický zákrok za použití nevyvolávajících anestetik. Na to, zda je třeba i vyměnit součásti okruhu a absorbér jsou kontroverzní názory, ale v běžné klinické praxi je toto opatření doporučeno [50,56]. Dantrolen se podá v iniciální dávce 2,5 mg/ kg i.v., co nejdříve. Podle reakce se stoupá až do dávky 10 mg/ kg. Je dobré si uvědomit, že v jedné lahvičce dantrolenu je 20 mg účinné látky a toto množství se ředí 60 ml solvens, tudíž pro průměrného 80kilového dospělého pacienta je v iniciální dávce nutno naředit a podat 10 lahviček dantrolenu. Proto je důležité přivolat na pomoc další personál. Dále je nutno pacienta aktivně chladit a korigovat acidózu, hypoxemii, arytmie a forsírovat diurézu furosemidem k ochraně renálních funkcí (riziko akutní tubulární nekrózy způsobené precipitací myoglobinu). Diuréza by měla být udržována na hodnotě alespoň 2 ml/ kg/ hod. Pokud je stav zvládnut, je nutné pacienta umístit na jednotku intenzivní péče na dobu nejméně 24 h. Mortalita je při použití dantrolenu 3– 5 %. U kompartment syndromu jsou někdy nutné operační fasciotomie.
Po zvládnutí stavu dochází k postupné úpravě svalového metabolizmu, což můžeme sledovat poklesem hodnoty kreatinkinázy, která trvá až 2 týdny, symptomy mohou přetrvávat déle [43].
Svalové dystrofie
Jde o heterogenní skupinu dědičně podmíněných svalových onemocnění s manifestací od raného dětství do pozdní dospělosti. Nejčastější jsou dystrofinopatie, myotonické dystrofie, facioskapulohumerální svalová dystrofie a heterogenní skupina tzv. pletencových myopatií (LGMD). Při anestezii je u těchto pacientů obecně kontraindikováno podávání SCH pro riziko rabdomyolýzy, hyperkalemie a srdeční zástavy. Také použití volatilních anestetik je spojeno s rizikem tzv. anestezií indukované rabdomyolýzy (AIR), při které současně dochází k hyperkalemii a tím ke zvýšenému riziku srdeční zástavy [56– 60]. Tato AIR je často nesprávně zaměňována za MH. Ta se přitom týká jen úzké skupiny geneticky podmíněných onemocnění spojených s přenosem kalcia a tzv. elektromechanickým spřažením.
Dystrofinopatie
U Duchennovy svalové dystrofie se musíme u klinicky pokročilých fází onemocnění vyrovnat nejen s pokročilou restrikční ventilační poruchou, ale i se závažnou dilatační kardiomyopatií. Snížení FVC na ≤ 30 % výrazně zvyšuje riziko respiračních komplikací [1]. Respirační poměry a zajištění dýchacích cest je navíc zhoršováno častou skoliózou. Respirační funkce mají přímý vliv na stratifikaci operačního rizika. Pacienti s Beckerovou variantou, kde je svalové postižení lehčího stupně, se dožívají podstatně vyššího věku a do popředí potíží, které limitují kvalitu a délku života, se dostává dilatační kardiomyopatie. Anestezie tedy musí být vedena především s ohledem na tento aspekt.
Je popisován opožděný nástup nedepolarizujících myorelaxancií, depolarizující jsou pak kontraindikována. Při použití rokuronia či vekuronia ze skupiny nedepolarizujících myorelaxans lze využít sugammadex k rychlému zotavení, ideálně za současné objektivní monitorace hloubky nervosvalové blokády [61].
Preferovanou metodou celkové anestezie je TIVA, sedace a regionální anestezie jsou zpravidla dobře tolerovány [61– 64]. Vyhýbáme se lékům s arytmogenním a negativně inotropním potenciálem.
Myotonické dystrofie
Jde o onemocnění charakterizovaná základní triádou příznaků: svalová slabost, která většinou nemá vliv na respiraci, katarakta a myotonie, tedy porucha relaxace po volní kontrakci. K tomu je nutné přidat, především u myotonické dystrofie 1. typu (MD1), řadu závažných multisystémových postižení. Obě choroby spojuje molekulárně genetický podklad a patogeneze (expanze trinukleotidů, resp. tetranukleotidů a rozsáhlé ovlivnění intracelulárních procesů sekvestrovanou RNA [65]). Druhý typ myotonické dystrofie (MD2) vzniká až v dospělosti, nemá tedy kongenitální a dětské formy a postihuje dominantně kořenové svalstvo [66].
Názor na možnost použití halogenovaných plynů je kontroverzní, spíše se nedoporučuje je používat [11]. Také depolarizující myorelaxancia se nedoporučují pro akcentaci myotonie a riziko rabdomyolýzy [67]. Byla popsána zvýšená senzitivita pacientů s MD1 na thiopental a propofol [66,67]. Thiopental je relativně kontraindikován pro riziko prolongované respirační deprese, propofol je možné použít při pečlivé titraci dávky jak pro úvod, tak pro udržování anestezie [11]. Pro riziko závažné arytmie a náhlé smrti je během anestezie nezbytná pečlivá monitorace kardiálních funkcí a omezení medikace s arytmogenním účinkem [67– 71]. Významný je tepelný management: podchlazení pacienta a vznik třesavky vede k akcentaci myotonie [67]. Dalšími riziky v peroperačním období jsou dysfagie, gastroezofageální reflux a riziko hyperglykemie [68]. Retrospektivní studie provedená na souboru 219 dospělých v regionu Saguenay (Quebec), kde je globálně nejvyšší výskyt MD1, nalezla periprocedurální komplikace u 8 % pacientů. Nejčastěji šlo o respirační postižení: atelektázy, respirační selhání a pneumonie. Větší riziko bylo u nemocných po břišních operacích a pacientů s těžším stupněm svalové slabosti. Je třeba ale dodat, že u 73 % nebyla v době zákroku diagnóza známa, anestezie byla tedy vedena běžným způsobem. U diagnostikovaných pacientů bylo procento komplikací podstatně nižší [72]. Podobně australská studie ukázala u dětí a mladistvých větší množství komplikací v případě závažnějšího postižení a dále při podávání myorelaxancií pro intubaci bez následné reverze [73]. Stran zajištění dýchacích cest během zákroku ukazují některé práce na menší množství pooperačních komplikací při použití laryngeální masky ve srovnání se zajištěním dýchacích cest intubací [73].
V pooperačním období je nutná pečlivá monitorace respiračních funkcí, srdečního rytmu a polykání (riziko aspirace). Neinvazívní ventilační podporu je nutné použít za pečlivé monitorace – zvýšené riziko aspirace [67]. Při použití svalové relaxace je vhodné monitorovat objektivně míru zotavení relaxometrií.
Myotonická dystrofie 2. typu má zpravidla podstatně lehčí klinický průběh a Weingarten et al [74] nenalezli při retrospektivní analýze žádné závažné komplikace vedení anestezie běžným způsobem. Asi v 15 % nicméně pacienti referovali po celkové anestezii snížení svalové síly, zhoršení svalových bolestí nebo křeče.
Wahbi uvádí, že nemocní s MD1 mají až 10× vyšší riziko hluboké žilní trombózy a závažného tromboembolizmu ve srovnání s běžnou populací [75]. Důsledná prevence je tedy jedním z hlavních opatření, na které myslíme.
Svalové kanálopatie
Nejčastěji se můžeme setkat s poruchou v napětím řízeném chloridovém kanálu, tedy s myotonia congenita, a to autozomálně recesivní tzv. Beckerovou variantou. Na rozdíl od myotonických dystrofií zde není žádné kardiální ani multisystémové postižení ani svalová slabost. Riziko představuje akcentace myotonie při podání depolarizujících myorelaxancií. Podchlazení pacienta zvyšuje riziko myotonie – tepelné zajištění je tedy důležitou součástí managementu. Pro natriovou kanálopatii (paramyotonia congenita) platí podobné doporučení. Navíc je třeba se vyhnout riziku iontové dysbalance – zvýšená hladina K+ může stejně jako chlad a fyzická zátěž provokovat periodické paralýzy. V případě hypokalemické periodické paralýzy, tedy mutace v napětím řízeném vápníkovém kanálu (CACNA1S), hrozí riziko MH pouze u 2 mutací. Periodické paralýzy vyvolává také hladovění [9,43].
Mitochondriální myopatie
Jde o heterogenní skupinu nemocných, kdy mutace v mitochondriální DNA vede ke svalovému postižení, laktátové acidóze a různému stupni postižení periferního a centrálního nervového systému.
Propofol, halogenované plyny midazolam a thiopental mohou indukovat laktátovou acidózu. Na druhou stranu je třeba poznamenat, že každý z těchto preparátů byl úspěšně použit pro anestezii takto postižených pacientů [76,77]. Neexistuje tedy žádné jasně vyloučené anestetikum [11].
Pompeho nemoc
U dětí je třeba zohlednit hypertrofickou kardiomyopatii, která vede k poruše diastolického plnění a může vyústit v náhlou srdeční zástavu [78]. V kohortě dětských pacientů je kardiální problematika klíčový problém pro úspěšné vedení anestezie. Za rizikový se považuje propofol i thiopental pro riziko snížení preloadu a diastolického tlaku a riziko myokardiální ischemie. Jako indukční agens jsou za bezpečné považovány ketamin a etomidát [79].
Dospělí pacienti nemají srdce postižené, mají však časté postižení trupového svalstva vedoucí k zhoršení respirační kapacity. Ventilace je u řady pacientů limitujícím faktorem kvality a délky života [80]. V perioperační péči je nutná aktivní monitorace metabolických funkcí a respirace, důležitá je prevence hypotermie [25].
Amyotrofická laterální skleróza
V pokročilejších stadiích nemoci s postižením respiračních svalů a polykání je vždy třeba počítat s rizikem respiračních komplikací, problémy s odpojením od umělé plicní ventilace a toaletou dýchacích cest [2]. Pacient před elektivní operací by měl mít vyjasněný „living-will“. Nedoporučuje se aplikace SCH pro vyšší výskyt fascikulací, křečí a vyšší citlivost pacientů k hyperkalemii. Na NDMR je naopak vyšší senzitivita [25].
Spinální muskulární atrofie
Vzhledem k významnému postižení respiračního svalstva lze očekávat pooperační problémy s odpojením od umělé plicní ventilace a toaletou dýchacích cest. Pokročilá kyfoskolióza může vést k zásadním problémům při spinální anestezii. Pro použití myorelaxancií platí stejné doporučení jako u amyotrofické laterální sklerózy.
Polyradikuloneuritida Guillain-Barré
Asi 20– 30 % nemocných s touto chorobou má vegetativní dysautonomii [17,81], nejčastěji (70 %) se jedná o sinusovou tachykardii, může ale být přítomna i závažná bradykardie nebo změny krevního tlaku, zvláště posturální hypotenze. Pacienti také mohou mít hyponatremii způsobenou syndromem nepřiměřené sekrece antidiuretického hormonu nebo paralytický ileus. Během výkonu je doporučována intenzivní monitorace vitálních funkcí (i s použitím semi- či invazivních metod monitorace hemodynamiky), dostatečná hydratace, bilance iontů. Protože denervace vede ke zvýšenému množství extrajunkčních acetylcholinových receptorů, je třeba se vyhnout SCH pro riziko hyperkalemické srdeční zástavy. NDMR lze použít s vědomím vyšší citlivosti těchto pacientů k jejich účinku [25]. Jedná se nicméně o fenomén závislý na fázi onemocnění. Zvýšená citlivost je ve fázi reinervace, naopak v denervační fázi může být přítomna i určitá rezistence na NDMR [17]. Při riziku vegetativní dysautonomie se u těchto pacientů nedoporučuje subarachnoidální anestezie pro riziko prohloubení dysautonomie blokádou bederního sympatiku a tím způsobenou oběhovou nestabilitu [2].
Hereditární neuropatie Charcot-Marie-Tooth
Byla popsána zvýšená senzitivita na thiopental. Tento fenomén je sice zmiňován v odborné literatuře, ale byl popsán pouze v jedné práci [82]. Jinak lze použít bezpečně celkovou anestezii jak vyváženou (halogenované preparáty), tak nitrožilní (propofol). Neuroaxiální bloky jsou také bezpečné. Web orphananaesthesia.eu nedoporučuje podávat SCH a uvádí také, že NDMR mohou mít nepředvídatelný efekt [83]. Důležité je pečlivé polohování pro zvýšené riziko vzniku otlakových paréz.
Seznam zkratek
AIR - anestezií indukovaná rabdomyolýza
ATP - adenosintrifosfát
ED50 - efektivní dávka pro 50 % populace
ED95 - efektivní dávka pro 95 % populace
FVC - usilovná vitální kapacita (forced vital capacity)
ICHE - inhibitory cholinesterázy
IVCT - in vitro kontrakční test
LGMD - pletencové svalové dystrofie
MD1 - myotonická dystrofie 1. typu
MD2 - myotonická dystrofie 2. typu
NDMR - nedepolarizující myorelaxancia
MH - maligní hypertermie
SCH - succinylcholin, suxamethonium
TCI - target controlled infusion TIVA totální intravenózní anestezie
TOF - train of four
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Stanislav Voháňka, CSc., MBA
Neurologická klinika LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: vohanka.stanislav@fnbrno.cz
Přijato k recenzi: 26. 3. 2018
Přijato do tisku: 29. 8. 2018
MUDr. Stanislav Voháňka, CSc., MBA

Dr. Voháňka promoval na Masarykově univerzitě v roce 1982. Neurologickou erudici zahájil na Klinice dětské neurologie tehdejší Fakultní dětské nemocnice v Brně. Od roku 1989 pracuje na Neurologické klinice MU a FN Brno, kde je od roku 1993 zástupcem přednosty pro LPP. Dlouhodobě se zabývá problematikou nervosvalových onemocnění. V 90. letech se intenzivně věnoval elektrofyziologii a tyto zkušenosti využil při budování elektromyografické laboratoře a pracoviště evokovaných potenciálů v nově vzniklé Fakultní nemocnici Brno. V roce 1993 obhájil kandidátskou dizertační práci, jejímž těžištěm byly elektrofyziologické nálezy u vybraných vertebrogenních onemocnění. Vhled do této problematiky získal při pobytu na spinální jednotce Schulthessovy kliniky v Zurichu pod vedením prof. Dvořáka (1991–1992). Je zakládajícím členem Neuromuskulární sekce České neurologické společnosti a od roku 2005 jejím předsedou. Prezentoval více než 350 sdělení a je autorem nebo spoluautorem 250 publikací převážně z oblasti neuromuskulárních chorob. Je členem TREAT-NMD Alliance a kurátorem 3 národních neuromuskulárních registrů. V roce 2016 se pod jeho vedením Neuromuskulární centrum FN Brno stalo součástí evropské sítě neuromuskulárních center (European Reference Network).
Zdroje
1. Katz JA, Murphy GS. Anesthetic consideration for neuromuscular diseases. Curr Opin Anaesthesiol 2017; 30(3): 435– 440. doi: 10.1097/ ACO.0000000000000466.
2. Klingler W, Lehmann-Horn F, Jurkat-Rott K. Complications of anaesthesia in neuromuscular disorders. Neuromuscul Disord 2005; 15(3): 195– 206. doi: 10.1016/ j.nmd.2004.10.017.
3. Adamus M, Cvachovec K, Černý V et al. Doporučený postup: zásady bezpečné anesteziologické péče. Anest Intenziv Med 2018; 29(2): 107– 110.
4. Tschiedel E, Müller O, Schara U et al. Sedation monitoring during open muscle biopsy in children by Comfort Score and Bispectral Index - a prospective analysis. Paediatr Anaesth 2015; 25(3): 265– 271. doi: 10.1111/ pan.12547.
5. Isnardon S, Vinclair M, Genty C et al. Pupillometry to detect pain response during general anaesthesia following unilateral popliteal sciatic nerve block: a prospective, observational study. Eur J Anaesthesiol 2013; 30(7): 429– 434. doi: 10.1097/ EJA.0b013e32835f0030.
6. Guglielminotti J, Grillot N, Paule M et al. Prediction of movement to surgical stimulation by the pupillary dilatation reflex amplitude evoked by a standardized noxious test. Anesthesiology 2015; 122(5): 985– 993. doi: 10.1097/ ALN.0000000000000624.
7. Mukaihara K, Godai K, Yamada T et al. Successful airway management using a MultiViewScope handle with a stylet scope in a patient with Schwartz-Jampel syndrome. JA Clin Rep 2016; 2(1): 36. doi: 10.1186/ s40981-016-0062-5.
8. Gritti P, Carrara B, Khotcholava M et al. The use of desflurane or propofol in combination with remifentanil in myasthenic patients undergoing a video-assisted thoracoscopic-extended thymectomy. Acta Anaesthesiol Scand 2009; 53(3): 380– 389. doi: 10.1111/ j.1399-6576.2008.01853.x.
9. Stourac P, Krikava I, Seidlova J et al. Sugammadex in a parturient with myotonic dystrophy. Br J Anaesth 2013; 110(4): 657– 658. doi: 10.1093/ bja/ aet037.
10. Kosinova M, Stourac P, Harazim H et al. Anaesthesia and orphan disease: rocuronium and sugammadex in the anaesthetic management of a parturient with Becker’s myotonia congenita. Eur J Anaesthesiol 2016; 33(7): 545– 547. doi: 10.1097/ EJA.0000000000000442.
11. Racca TM. Recommendations for anesthesia and perioperative management of patients with neuromuscular disorders. Minerva Anestesiol 2013; 79(4): 419– 433.
12. Abel M, Eisenkraft JB. Anesthetic implications of myasthenia gravis. Mt Sinai J Med 2002; 69(1– 2): 31– 37.
13. Fischer T. Anaesthesia recommendations for patients suffering from Myasthenia gravis. Anästh Intensivmed 2015; 56: S654– S661.
14. Vymazal T, Horáček M, Bicek V et al. Myasthenia gravis a anestezie – nový bezpečnější postup. Anest Intenziv Med 2014; 25(1): 21– 24.
15. Bicek V, Vymazal T. Desfluran jako vhodné inhalační anestetikum u pacientky s těžkou formou myasthenia gravis. Anest Intenziv Med 2013; 24(3): 160– 162.
16. Vymazal T, Krecmerova M, Bicek V et al. Feasibility of full and rapid neuromuscular blockade recovery with sugammadex in myasthenia gravis patients undergoing surgery – a series of 117 cases. Ther Clin Risk Manag 2015; 11: 1593– 1596. doi: 10.2147/ TCRM.S93009.
17. Romero A, Joshi GP. Neuromuscular disease and anesthesia. Muscle Nerve 2013; 48(3): 451– 460. doi: 10.1002/ mus.23817.
18. Brambrink AM, Kirsch JR. Perioperative care of patients with neuromuscular disease and dysfunction. Anesthesiol Clin 2007; 25(3): 483– 509, viii– ix. doi: 10.1016/ j.anclin.2007.05.005.
19. Voháňka S. Léky a nervosvalový přenos. Neurol Praxi 2017; 18(1): 11– 14.
20. Bae JS, Go SM, Kim BJ. Clinical predictors of steroid-induced exacerbation in myasthenia gravis. J Clin Neurosci Off J Neurosurg Soc Australas 2006; 13: 1006– 1010. doi: 10.1016/ j.jocn.2005.12.041.
21. Rangasamy V, Kumar K, Rai A et al. Sevoflurane and thoracic epidural anesthesia for trans-sternal thymectomy in a child with juvenile myasthenia gravis. J Anaesthesiol Clin Pharmacol 2014; 30(2): 276– 278. doi: 10.4103/ 0970-9185.130088.
22. Dunsire MF, Clarke SG, Stedmon JJ. Undiagnosed myasthenia gravis unmasked by neuromuscular blockade. Br J Anaesth 2001; 86(5): 727– 730.
23. Eisenkraft JB, Book WJ, Mann SM et al. Resistance to succinylcholine in myasthenia gravis: a dose-response study. Anesthesiology 1988; 69(5): 760– 763.
24. Bowie RA. Myasthenia gravis unmasked by neuromuscular blockade. Br J Anaesth 2002; 88(1): 153– 154.
25. Marsh S, Pittard A. Neuromuscular disorders and anaesthesia. Part 2: specific neuromuscular disorders. Contin Educ Anaesth Crit Care Pain 2011; 11(4): 119– 123. doi: 10.1093/ bjaceaccp/ mkr019.
26. Wefki Abdelgawwad Shousha AA, Sanfilippo M, Sabba A et al. Sugammadex and reversal of neuromuscular block in adult patient with duchenne muscular dystrophy. Case Rep Anesthesiol 2014; 2014: e680568. doi: 10.1155/ 2014/ 680568.
27. Batistaki C, Tentes P, Deligiannidi P et al. Residual neuromuscular blockade in a real life clinical setting. Correlation with sugammadex or neostigmine administration. Minerva Anestesiol 2015; 82(5): 550– 558.
28. White MC, Stoddart PA. Anesthesia for thymectomy in children with myasthenia gravis. Paediatr Anaesth 2004; 14(8): 625– 635. doi: 10.1111/ j.1460-9592.2004.01292.x.
29. Racca F, Mongini T, Wolfler A et al. Recommendations for anesthesia and perioperative management of patients with neuromuscular disorders. Minerva Anestesiol 2013; 79(4): 419– 433.
30. Rosenberg H, Pollock N, Schiemann A et al. Malignant hyperthermia: a review. Orphanet J Rare Dis 2015; 10: 93. doi: 10.1186/ s13023-015-0310-1.
31. Halliday NJ. Malignant hyperthermia. J Craniofac Surg 2003; 14(5): 800– 802.
32. Ording H. Incidence of malignant hyperthermia in Denmark. Anesth Analg 1985; 64(7): 700– 704.
33. Riazi S, Larach MG, Hu C et al. Malignant hyperthermia in Canada: characteristics of index anesthetics in 129 malignant hyperthermia susceptible probands. Anesth Analg 2014; 118(2): 381– 387. doi: 10.1213/ ANE.0b013e3182937d8b.
34. Brady JE, Sun LS, Rosenberg H et al. Prevalence of malignant hyperthermia due to anesthesia in New York State, 2001-2005. Anesth Analg 2009; 109(4): 1162– 1166. doi: 10.1213/ ane.0b013e3181ac1548.
35. Brandom BW, Callahan PM. Malignant hyperthermia: an update. Adv Anesth 2015; 33(1): 113– 128. doi: 10.1016/ j.aan.2015.07.007.
36. Campion GH, Hadi AS, Berman AJ et al. Questions regarding the diagnosis of malignant hyperthermia. Anesthesiology 2015; 123(3): 731– 732. doi: 10.1097/ ALN.0000000000000760.
37. Gonsalves SG, Ng D, Johnston JJ et al. Using exome data to identify malignant hyperthermia susceptibility mutations. Anesthesiology 2013; 119(5): 1043– 1053. doi: 10.1097/ ALN.0b013e3182a8a8e7.
38. Groom L, Muldoon SM, Tang ZZ et al. Identical de novo mutation in the type 1 ryanodine receptor gene associated with fatal, stress-induced malignant hyperthermia in two unrelated families. Anesthesiology 2011; 115(5): 938– 945. doi: 10.1097/ ALN.0b013e3182320068.
39. Thomas J, Crowhurst T. Exertional heat stroke, rhabdomyolysis and susceptibility to malignant hyperthermia. Intern Med J 2013; 43(9): 1035– 1038. doi: 10.1111/ imj.12232.
40. Carpenter D, Ringrose C, Leo V et al. The role of CACNA1Sin predisposition to malignant hyperthermia. BMC Med Genet 2009; 10: 104. doi: 10.1186/ 1471-2350-10-104.
41. Stewart SL, Hogan K, Rosenberg H et al. Identification of the Arg1086His mutation in the alpha subunit of the voltage-dependent calcium channel (CACNA1S) in a North American family with malignant hyperthermia. Clin Genet 2001; 59(3): 178– 184.
42. Fiszer D, Shaw M-A, Fisher NA et al. Next-generation Sequencing of RYR1 and CACNA1S in malignant hyperthermia and exertional heat illness. Anesthesiology 2015; 122(5): 1033– 1046. doi: 10.1097/ ALN.00000 00000000610.
43. Jurkat-Rott K, McCarthy T, Lehmann-Horn F. Genetics and pathogenesis of malignant hyperthermia. Muscle Nerve 2000; 23(1): 4– 17.
44. Jungbluth H, Dowling JJ, Ferreiro A et al. 217th ENMC International Workshop: RYR1-related myopathies, Naarden, The Netherlands, 29–31 January 2016. Neuromuscul Disord 2016; 26(9): 624– 633. doi: 10.1016/ j.nmd.2016.06.001.
45. Lehmann-Horn F, Klingler W, Jurkat-Rott K. Nonanesthetic malignant hyperthermia. Anesthesiology 2011; 115(5): 915– 917. doi: 10.1097/ ALN.0b013e318232008f.
46. Davis PJ, Brandom BW. The association of malignant hyperthermia and unusual disease: when you’re hot you’re hot or maybe not. Anesth Analg 2009; 109(4): 1001– 1003. doi: 10.1213/ ane.0b013e3181b493d4.
47. McCarthy TV, Quane KA, Lynch PJ. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat 2000; 15(5): 410– 417. doi: 10.1002/ (SICI)1098-1004(200005)15: 5<410::AID-HUMU2>3.0.CO; 2-D.
48. Sanoudou D, Beggs AH. Clinical and genetic heterogeneity in nemaline myopathy--a disease of skeletal muscle thin filaments. Trends Mol Med 2001; 7(8): 362– 368.
49. Fattori F, Maggi L, Bruno C et al. Centronuclear myopathies: genotype-phenotype correlation and frequency of defined genetic forms in an Italian cohort. J Neurol 2015; 262(7): 1728– 1740. doi: 10.1007/ s00415-015-7757-9.
50. Vymazal T. Maligní hypertermie. Anest Intenziv Med 2016; 27(2): 71– 74.
51. McKenney KA, Holman SJ. Delayed postoperative rhabdomyolysis in a patient subsequently diagnosed as malignant hyperthermia susceptible. Anesthesiology 2002; 96(3): 764– 765.
52. Larach MG, Brandom BW, Allen GC et al. Malignant hyperthermia deaths related to inadequate temperature monitoring, 2007-2012: a report from the North American malignant hyperthermia registry of the malignant hyperthermia association of the United States. Anesth Analg 2014; 119(6): 1359– 1366. doi: 10.1213/ ANE.0000000000000421.
53. Ording H, Brancadoro V, Cozzolino S et al. In vitro contracture test for diagnosis of malignant hyperthermia following the protocol of the European MH Group: results of testing patients surviving fulminant MH and unrelated low-risk subjects. The European Malignant Hyperthermia Group. Acta Anaesthesiol Scand 1997; 41(8): 955– 966.
54. Allen GC, Larach MG, Kunselman AR. The sensitivity and specificity of the caffeine-halothane contracture test: a report from the North American Malignant Hyperthermia Registry. The North American Malignant Hyperthermia Registry of MHAUS. Anesthesiology 1998; 88(3): 579– 588.
55. European malignant hyperthermia group. Diagnostic MH mutations. [online]. Available form URL: https: / / www.emhg.org/ diagnostic-mutations/ .
56. Isaak RS, Stiegler MP. Review of crisis resource management (CRM) principles in the setting of intraoperative malignant hyperthermia. J Anesth 2016; 30(2): 298– 306. doi: 10.1007/ s00540-015-2115-8.
57. Yemen TA, McClain C. Muscular dystrophy, anesthesia and the safety of inhalational agents revisited; again. Paediatr Anaesth 2006; 16(2): 105– 108. doi: 10.1111/ j.1460-9592.2005.01801.x.
58. Girshin M, Mukherjee J, Clowney R et al. The postoperative cardiovascular arrest of a 5-year-old male: an initial presentation of Duchenne’s muscular dystrophy. Paediatr Anaesth 2006; 16(2): 170– 173. doi: 10.1111/ j.1460-9592.2005.01698.x.
59. Hayes J, Veyckemans F, Bissonnette B. Duchenne muscular dystrophy: an old anesthesia problem revisited. Paediatr Anaesth 2008; 18(2): 100– 106. doi: 10.1111/ j.1460-9592.2007.02302.x.
60. Hayes J, Veyckemans F, Bissonnette B. Rhabdomyolysis and anesthesia. Paediatr Anaesth 2008; 18(9): 897– 898. doi: 10.1111/ j.1460-9592.2008.02599.x.
61. de Boer HD, van Esmond J, Booij LH et al. Reversal of rocuronium-induced profound neuromuscular block by sugammadex in Duchenne muscular dystrophy. Paediatr Anaesth 2009; 19(12): 1226– 1228. doi: 10.1111/ j.1460-9592.2009.03178.x.
62. Bang SU, Kim YS, Kwon WJ et al. Peripheral nerve blocks as the sole anesthetic technique in a patient with severe Duchenne muscular dystrophy. J Anesth 2016; 30(2): 320– 323. doi: 10.1007/ s00540-015-2127-4.
63. Muenster T, Mueller C, Forst J et al. Anaesthetic management in patients with Duchenne muscular dystrophy undergoing orthopaedic surgery: a review of 232 cases. Eur J Anaesthesiol 2012; 29(10): 489– 494. doi: 10.1097/ EJA.0b013e3283566789.
64. Miles F, Dare T. Scoliosis repair in a teenager with Duchenne’s muscular dystrophy: who calls the shots? Paediatr Anaesth 2009; 19(10): 1022– 1024. doi: 10.1111/ j.1460-9592.2009.03071.x
65. Meola G, Cardani R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta 2015; 1852(4): 594– 606. doi: 10.1016/ j.bbadis.2014.05.019.
66. Meola G, Cardani R. Myotonic dystrophy type 2: an update on clinical aspects, genetic and pathomolecular mechanism. J Neuromuscul Dis 2015; 2(s2): S59– S71. doi: 10.3233/ JND-150088.
67. Veyckemans F, Scholtes JL. Myotonic dystrophies type 1 and 2: anesthetic care. Paediatr Anaesth 2013; 23(9): 794– 803. doi: 10.1111/ pan.12120.
68. Bisinotto FMB, Fabri DC, Calçado MS et al. Anesthesia for videolaparoscopic cholecystectomy in a patient with Steinert disease. Case report and review of the literature. Rev Bras Anestesiol 2010; 60(2): 181– 191.
69. Schoser BG, Ricker K, Schneider-Gold C et al. Sudden cardiac death in myotonic dystrophy type 2. Neurology 2004; 63(12): 2402– 2404.
70. Sansone VA, Brigonzi E, Schoser B et al. The frequency and severity of cardiac involvement in myotonic dystrophy type 2 (DM2): long-term outcomes. Int J Cardiol 2013; 168(2): 1147– 1153. doi: 10.1016/ j.ijcard.2012.11.076.
71. Groh WJ, Lowe MR, Zipes DP. Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J Cardiovasc 2002; 13(5): 444– 448.
72. Mathieu J, Allard P, Gobeil G et al. Anesthetic and surgical complications in 219 cases of myotonic dystrophy. Neurology 1997; 49(6): 1646– 1650.
73. Sinclair JL, Reed PW. Risk factors for perioperative adverse events in children with myotonic dystrophy. Paediatr Anaesth 2009; 19(8): 740– 747. doi: 10.1111/ j.1460-9592.2009.03079.x.
74. Weingarten TN, Hofer RE, Milone M et al. Anesthesia and myotonic dystrophy type 2: a case series. Can J Anesth 2010; 57(3): 248– 255. doi: 10.1007/ s12630-009-9244-1.
75. Wahbi K, Bellino A, Duboc D et al. Venous thromboembolism in myotonic dystrophy type 1. [online]. Available from URL: https:/ / clinicaltrials.gov/ ct2/ show/ NCT03424460.
76. Veyckemans F. Can inhalation agents be used in the presence of a child with myopathy? Curr Opin Anaesthesiol 2010; 23(3): 348– 355. doi: 10.1097/ ACO.0b013e3283393977.
77. Driessen JJ. Neuromuscular and mitochondrial disorders: what is relevant to the anaesthesiologist? Curr Opin Anaesthesiol 2008; 21(3): 350– 355. doi: 10.1097/ ACO.0b013e3282f82bcc.
78. Bembi B, Cerini E, Danesino C et al. Management and treatment of glycogenosis type II. Neurology 2008; 71 (23 Suppl 2): S12– S36. doi: 10.1212/ WNL.0b013e31818da93f.
79. Kishnani PS, Steiner RD, Bali D et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8(5): 267– 288. doi: 10.1097/ 01.gim.0000218152.87434.f3.
80. Ploeg AT van der. Monitoring of pulmonary function in Pompe disease: a muscle disease with new therapeutic perspectives. Eur Respir J 2005; 26(6): 984– 985. doi: 10.1183/ 09031936.05.00112005.
81. Pfeiffer G, Schiller B, Kruse J et al. Indicators of dysautonomia in severe Guillain-Barré syndrome. J Neurol 1999; 246(11): 1015– 1022.
82. Kotani N, Hirota K, Anzawa N et al. Motor and sensory disability has a strong relationship to induction dose of thiopental in patients with the hypertropic variety of Charcot-Marie-Tooth syndrome. Anesth Analg 1996; 82(1): 182– 186.
83. Orphananesthesia: a project of the German Society of Anesthesiology and Intensive Care Medicine. [online]. Available from URL: https:/ / www.orphananesthesia.eu/ .
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2018 Číslo 5
Nejčtenější v tomto čísle
- Nové poznatky v diagnostice a léčbě amyotrofické laterální sklerózy
- Přehled onemocnění s obrazem restrikce difuze na magnetické rezonanci mozku
- Cervikální vertigo – fikce či realita?
- Anestezie a nervosvalová onemocnění