
Fabryho choroba, přehled problematiky a nejčastější neurologické projevy
Fabry disease, an overview and the most common neurological manifestations
Fabry disease is a rare inherited lysosomal storage disorder. Patients with classical multisystemic disease have frequent neurological manifestations. It is of great importance for patients to be diagnosed early and properly since there is specific disease therapy available. This article provides an overview of current knowledge about Fabry disease with emphasis on its neurological manifestations.
Key words:
Fabry disease – small fi ber neuropathy – stroke – enzyme-replacement therapy
P. Reková has been in receipt of travel grant and honoraria for lectures on Fabry disease from Shire HGT and Sanofi Genzyme. K. Sedláková has been in receipt of honoraria for lectures on Fabry disease from Shire HGT. G. Dostálová has been in receipt of travel grants and honoraria for lecures on Fabry disease from Protalix Biotherapeutics, Shire HGT and Sanofi Genzyme. A. Linhart has been in receipt of travel grants and honoraria for lectures on Fabry disease from Shire HGT, Sanofi Genzyme and Amicus Therapeutics.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
P. Reková 1; K. Sedláková 2; G. Dostálová 3; A. Linhart 3
Působiště autorů:
Neurologická klinika a Centrum klinických neurověd 1. LF UK a VFN v Praze
1; Oční klinika 1. LF UK a VFN v Praze
2; II. interní klinika – klinika kardiologie a angiologie 1. LF UK a VFN v Praze
3
Vyšlo v časopise:
Cesk Slov Neurol N 2018; 81(2): 156-163
Kategorie:
Přehledný referát
doi:
https://doi.org/10.14735/amcsnn2018156
Snímky z MR jsou publikovány se svolením Radiologické kliniky 1. LF UK a VFN, za jejich poskytnutí autoři děkují. Za poskytnutí fotografie kožních angiokeratomů patří poděkování MU Dr. Lukáši Lacinovi, Ph.D., z Dermatovenerologické kliniky 1. LF UK a VFN
Souhrn
Fabryho choroba je vzácné dědičné střádavé lyzozomální onemocnění. Nemocní s klasickou multiorgánovou formou nemoci mají časté neurologické projevy. Vzhledem k dostupnosti specifické terapie je důležité pacienty včas a správně diagnostikovat. Článek podává přehled současných znalostí o Fabryho chorobě s důrazem na neurologické projevy choroby.
Klíčová slova:
Fabryho choroba – neuropatie tenkých nervových vláken – cévní mozková příhoda – enzymová substituční terapie
Úvod
Fabryho choroba (FCh; OMIM 301500) je dědičné progresivní metabolické onemocnění vázané na chromozom X. Mutace GLA genu kódujícího enzym α-galaktosidázu A (α-Gal A)vede ke snížené až zcela vymizelé aktivitě enzymu, jejímž důsledkem je akumulace glykosfingolipidů (zejména globotriaosylceramidu – Gb3) v lyzozomech buněk mnoha orgánových systémů, vč. systému nervového [1]. Střádání substrátu iniciuje patofyziologické mechanizmy, jejichž konečným výsledkem jsou klinické projevy onemocnění. Prognosticky nejzávažnější je zejména postižení srdce, ledvin a CNS. Kvalita života je však výrazně ovlivněna rovněž výskytem periferních neurologických komplikací, projevů kožních a postižením smyslových orgánů (oční a vestibulokochleární projevy).
Historické poznámky
FCh byla poprvé popsána na konci 19. století. V roce 1898 dva dermatologové, Angličan William Anderson (1842– 1900) a Němec Johannes Fabry (1860– 1930), nezávisle na sobě publikovali kazuistiky onemocnění u svých pacientů [2,3]. Významnými milníky se staly roky 1950 a 1967. Nejdříve bylo potvrzeno, že střádaný materiál je lipidové povahy [4], a později byl určen enzymatický defekt [5]. Kompletní nukleotidová sekvence genu byla publikována v roce 1989 [6]. Výzkum a vývoj specifických léčebných strategií byl umožněn vytvořením zvířecích modelů (knock out GLA gen u myši) [7]. Specifická léčba pomocí intravenózně aplikované enzymové substituce byla pro pacienty s FCh schválena pro použití v běžné klinické praxi na počátku tohoto tisíciletí a v roce 2015 byl na trh uveden první perorálně podávaný chaperon.
Epidemiologie
FCh je po Gaucherově nemoci druhým nejčastěji se vyskytujícím lyzozomálním střádavým onemocněním. Prevalence FCh ve světě je uváděna v rozmezí 1:40 000– 1:60 000 [8]. Screeningové studie však ukazují na možný vyšší výskyt onemocnění v populaci. Novorozenecký screening provedený italskými autory odhalil výskyt deficitu α-Gal A v poměru 1:3 100 narozených dětí. Frekvence mutací způsobujících FCh byla v této studii 1:4 600 [9]. Screening 110 027 novorozenců na Tchaj-wanu ukázal ještě vyšší zastoupení patologických mutací GLA ve vyšetřené populaci s celkovou prevalencí 1:1 368 narozených chlapců, přičemž známé mutace způsobující onemocnění měly zastoupení 1:1 512. Nález mutace spojené s klasickým fenotypem nemoci odpovídal frekvenci 1:57 000 mužů [10]. Vyšší prevalence FCh byla zaznamenána rovněž ve specifických, pro FCh rizikových, populacích nemocných. Onemocnění se u pacientů s hypertrofií levé komory vyskytuje okolo 1 % (obě pohlaví), u hemodialyzovaných pacientů je zastoupení choroby 0,1 % (ženy) – 0,33 % (muži) [11].Vyšší zastoupení FCh bylo rovněž opakovaně popsáno u mladých nemocných s CMP. V roce 2005 autoři Rolfs et al odhalili mutaci GLA genu u 2,4 % žen a u 4,9 % mužů ve věku 18– 55 let s kryptogenní ischemickou CMP [12]. Belgickými autory byl v kohortě mladých pacientů (18–60 let) s cerebrovaskulárním onemocněním (tranzitorní ischemická ataka [TIA] nebo ischemická CMP, intrakraniální hemoragie, jinak nevysvětlené postižení bílé hmoty, vertebrobazilární dolichoektázie) zjištěn výskyt GLA mutace kolem 1 % [13]. Metaanalýza osmi studií (celkem 8 302 pacientů) zabývajících se výskytem FCh u pacientů s CMP vedla k závěru, že FCh může být příčinou asi u 1 % všech CMP u mladých lidí, vč. 3– 5 % kryptogenních CMP [14].
V ČR je v současné době v Centru pro FCh sledováno 140 pacientů s tímto onemocněním (data autorů). Počet obyvatel ČR k 30. 9. 2017 byl 10 597 473 [15]. Prevalenci FCh v ČR bychom na základě těchto údajů mohli odhadnout na 1:76 000. Vzhledem k heterogenní klinické manifestaci tohoto onemocnění však předpokládáme, že rovněž v ČR je prevalence FCh podhodnocena a výskyt pacientů s tímto onemocněním může odpovídat literárním údajům. Také v ČR v minulosti proběhly studie vyhledávající pacienty s FCh u rizikových skupin nemocných. V roce 2006 byla publikovaná práce, ve které autoři prezentovali výsledky screeningu FCh u dialyzovaných pacientů v ČR. Onemocnění bylo diagnostikováno u 0,26 % mužů a u 0,05 % žen ve sledované populaci [16]. V roce 2014 publikovali čeští autoři výsledky vyhledávání FCh u mužů s nevysvětlitelnou hypertrofií levé komory. V této kohortě byla zjištěna 4% prevalence FCh [17]. V současné době probíhá projekt pod záštitou Cerebrovaskulární sekce České neurologické společnosti Jana Evangelisty Purkyně, jehož primárním cílem je identifikovat pacienty s dosud nepoznanou FCh mezi nemocnými s CMP.
Biologický základ nemoci a klinické projevy
Příčinou FCh je mutace genu GLA pro lyzozomální enzym α-Gal A. GLA gen je lokalizován na dlouhém raménku chromozomu X, v oblasti Xq22.1. Tento gen je tvořen 7 exony. V současné době jsou známy stovky různých mutací tohoto genu [18]. Genovým produktem je polypeptid, enzym α-Gal A. Tento peptid je tvořen 429 aminokyselinami a funkčně je hydrolázou štěpící galaktosylové části v pozici α z glykolipidů, glykoproteinů a oligosacharidů [19]. Důsledkem snížené či vymizelé aktivity enzymu je hromadění substrátu, v případě FCh zejména globotriaosylceramidu, v buňkách různých tkání a orgánů s následným širokým spektrem klinických projevů. Významnou patofyziologickou roli může hrát deacylovaná forma Gb3, globotriaosylsfingosin neboli lyzo-Gb3. Kromě souvislosti hladin lyzo-Gb3 s tíží onemocnění byly popsány i přímé toxické dopady této substance na tkáně [20].
Při klasickém (typickém) průběhu se onemocnění projevuje již v dětství nebo v období dospívání, nejčastěji pálivými bolestmi rukou a nohou, poruchami pocení (hypo- až anhidróza, vzácněji hyperhidróza [21]), přítomností kožních angiokeratomů či některých očních nálezů (cornea verticillata, vinuté retinální cévy) [22]. V pozdějších fázích života se objevují hlavní orgánové komplikace v podobě postižení ledvin, srdce a CNS [1].
Fenotypická manifestace FCh však může být i atypická. U atypických variant (varianty s pozdním začátkem, late-onset) FCh mohou některé z klasických příznaků onemocnění zcela chybět. Tyto varianty mívají pozdní začátek (4.– 6. dekáda) a často mono- či oligosymptomatický průběh. Byla popsána kardiální nebo renální varianta onemocnění [1]. U renální varianty panují pochybnosti o její reálné existenci, naproti tomu nemocní s postižením omezeným na srdce představují velkou skupinu pacientů. První manifestací FCh může být i CMP [23], což potvrzují i zkušenosti autorů. Onemocnění s pozdní manifestací bývá spojeno s reziduální aktivitou enzymu α-Gal A [1].
Dříve se předpokládalo, že ženy s mutací GLA jsou klinicky asymptomatické a o onemocnění se hovořilo jako o X-recesivně vázaném. Ukázalo se však, že také heterozygotní ženy mohou mít klinické projevy nemoci. Tíže postižení u žen má výraznou klinickou variabilitu od asymptomatického průběhu až po těžké orgánové projevy nemoci srovnatelné s klasickým průběhem onemocnění u mužů. Za jeden z možných důvodů rozmanitosti klinických projevů FCh u žen je označován proces inaktivace chromozomu X v časných fázích embryogeneze a jeho případné zešikmení (skewed X-inactivation) upřednostňující inaktivaci mutované alely [24].
Neurologická manifestace nemoci
Neurologické projevy FCh jsou časté a postihují všechny části nervového systému – periferní, autonomní i centrální.
Projevy postižení periferního nervového systému
Patologicko-anatomické poznámky
Akumulace patologického materiálu byla prokázána v různých částech nervového systému. V periferním nervovém systému bývají glykolipidová depozita nalézána zejména ve spinálních gangliích, perineuriu, axonech myelinizovaných i nemyelinizovaných vláken, méně často ve Schwannových buňkách. Z patofyziologického hlediska je významné rovněž střádání v endotelu a buňkách hladké svaloviny epineurálních a endoneurálních cév periferního nervového systému [25,26]. Patologicko-anatomické nálezy u pacientů trpících FCh prokazují ztrátu nemyelinizovaných vláken (A δ, C) periferního nervového systému [25].
Klinické projevy
Nejčastějším a také mnohdy nejčasnějším projevem FCh jsou (převážně) akrální bolesti [26]. Přesný patofyziologický mechanizmus vzniku bolesti u FCh není znám. Předpokládá se, že bolest je převážně důsledkem postižení tenkých nervových vláken (neuropatická bolest), avšak nociceptivní komponenta je rovněž přítomna. Bolest bývá nejčastěji lokalizována do oblasti dlaní, prstů a plant či lýtek, mohou být však postiženy také jiné části těla. Například bolesti kloubů jsou referovány až v 27,2 % mužů a 24,1 % žen s FCh [27– 30]. Pacienti charakterizují bolest nejčastěji jako pálivou, bodavou, bolestivý chlad. Epizodické bolestivé ataky či krátkodobé zhoršení chronické bolesti je popisováno u 70 % mužů a 52 % žen, chronická bolest pak obtěžuje asi 50 % mužů a 33 % žen s FCh. Specifická situace, charakteristická pro pacienty s klasickým průběhem FCh, jsou tzv. krize. Jde o hodiny až dny trvající bolestivé záchvaty, které začínají většinou akrálně a rozšiřují se do celého těla. Krize pacienty velmi vyčerpávají, bolesti jsou kruté, nesnesitelné, agonizující. Ataky bolestí či zhoršení chronických obtíží jsou často spouštěny interkurentním onemocněním, stresem, změnou venkovní teploty, tělesnou aktivitou [29,30]. Bolesti výrazně snižují kvalitu života pacientů s FCh [31,32]. Literární údaje, které potvrzují i zkušenosti autorů, ukazují, že u některých nemocných dochází v dospělosti spontánně ke snížení intenzity bolesti, u části pacientů dokonce obtíže zcela mizí. Na druhé straně v pozdějších stadiích nemoci, zejména u pacientů s poklesem renálních funkcí, se mohou objevit známky postižení silných nervových vláken [27].
Projevy postižení autonomního nervového systému
Patologicko-anatomické poznámky
Glykolipidová depozita byla popsána také v různých částech autonomního nervového systému, v autonomních centrech (např. nucleus supraopticus, nucleus paraventricularis, gyrus parahipocampalis, nucleus n. vagi), autonomních gangliích či v intestinálních autonomních plexech. Inkluze glykolipidů jsou však přítomny také v samotných eferentních orgánech, např. buňkách potních žláz či v buňkách hladké svaloviny střeva [1,26].
Klinická manifestace
Nejčastějšími klinickými projevy, na kterých se spolupodílí postižení autonomního nervstva, jsou gastrointestinální projevy a poruchy pocení. Bolestí břicha trpí až 41 % mužů a 32 % žen s FCh [28]. Dalšími gastrointestinálními projevy jsou nadýmaní, průjmy, nauzea. Hypohidróza či anhidróza jsou jiným příkladem spojeným s postižením autonomního nervového systému. V důsledku snížené schopnosti pocení pacienti špatně tolerují vyšší teploty či fyzickou zátěž. V literatuře byly popsány i další projevy autonomní dysregulace, např. poruchy pupilární konstrikce, snížení tvorby slz, slin, dysregulace krevního tlaku vedoucí k ortostatickým hypotenzím [33,34]. Autonomní dysfunkce se může podílet i na častém sklonu k bradykardii a zhoršovat dysfunkci srdečního sinusového uzlu [35]. Postižení cerebrovaskulární reaktivity se může spolupodílet na zvýšené četnosti mozkových příhod u pacientů s FCh [36].
Postižení CNS
Patologicko-anatomické poznámky
Glykolipidová depozita byla popsána rovněž v různých částech CNS, příkladem mohou být hypothalamická jádra, amygdala, pontinní retikulární formace, substancia nigra, míšní neurony [37]. Důležitou roli v patofyziologii projevů z CNS hraje akumulace Gb3 ve strukturách cév zásobujících mozek [38]. Prokázáno bylo též leptomeningeální střádání [39].
Klinické projevy
Hlavní klinické projevy z oblasti CNS ukazuje tab. 1. Podrobný popis neuropsychiatrických a otoneurologických obtíží přesahuje rámec tohoto sdělení. Dále budou popsány cerebrovaskulární manifestace a typické nálezy při vyšetření CNS pomocí MR.
![Hlavní projevy Fabryho choroby z oblasti CNS [1].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/d8c1953a84b618fd31887c49fe6bc721.jpg)
CMP a tranzitorní ischemické ataky jsou nejčastějším projevem cerebrovaskulárního postižení u pacientů s FCh [12]. Retrospektivní studie čítající menší počty pacientů s FCh uvádí frekvenci výskytu iktů, ať už klinicky manifestních příhod či nálezů němých ischemií při zobrazení CNS, v širokém rozmezí (7– 48 %) [40,41]. Vzhledem k malému počtu pacientů trpících FCh jsou anonymní data z celého světa shromažďována v registrech (Fabry Registry a Fabry Outcome Survey), které jsou zdrojem informací nejen o přirozeném průběhu nemoci a klinických projevech, ale také o účinnosti léčby u pacientů. Údaje z registrů potvrzují častý výskyt CMP u nemocných s FCh [23]. Data z Fabry Registry ukazují, že většina pacientů prodělala první iktus ve věku mezi 20 a 50 lety, přičemž 22 % pacientů v době první příhody bylo mladších 30 let. Navíc CMP se objevila u velkého procenta pacientů (50 % mužů a 38 % žen) ještě před stanovením diagnózy FCh [23].
Při vyšetření MR lze u pacientů s FCh identifikovat některé častěji se vyskytující abnormality. Nejčastěji se u pacientů setkáváme s postižením velkých tepen, malých tepen, výskytu lézí v oblasti bílé hmoty a tzv. pulvinar sign.
Makroangiopatické změny zahrnují dilatace, elongace a tortuozity tepen jak v karotické, tak vertebrobazilární oblasti [38]. Rozšíření bazilární tepny může upozornit na možnou FCh [42].
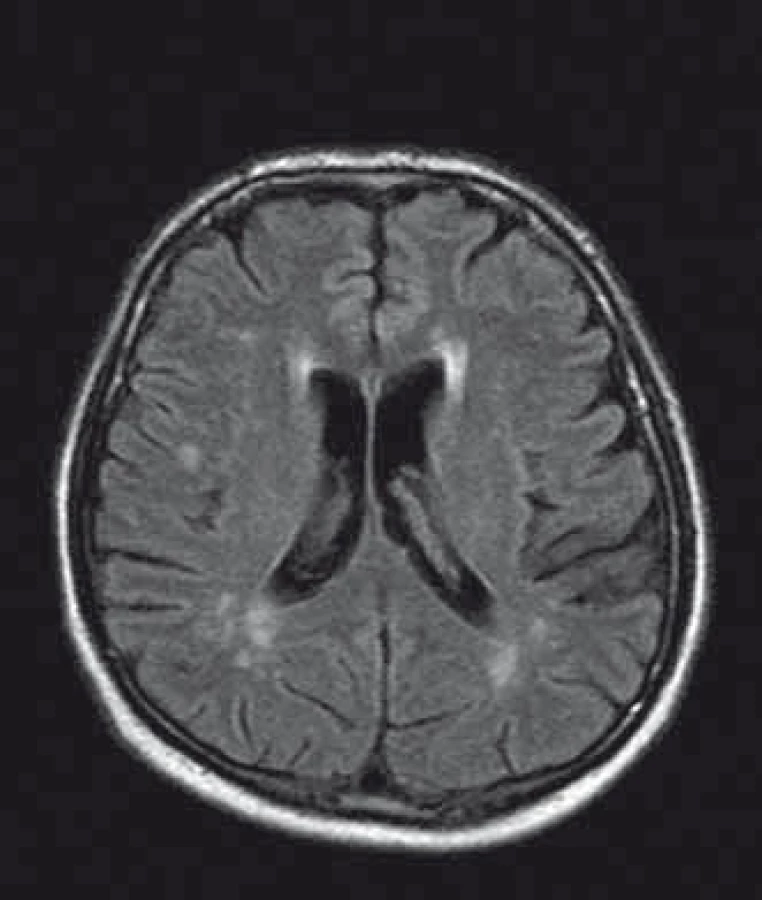
U pacientů často nacházíme lakunární infarkty odrážející postižení malých tepen (obr. 1). Chronické změny v oblasti bílé hmoty mozkové (white matter lesions; WMLs), na MR nejlépe viditelné v podobě hyperintenzit v T2 vážených obrazech či ve FLAIR sekvencích, jsou rovněž časté (obr. 2). Postihují subkortikální, hluboké i periventrikulární oblasti bílé hmoty. Mohou být tečkovité, vícečetné i splývající. Léze v bílé hmotě se vyskytují již v mladém věku, byly popsány dokonce i u dětí [43]. Mají progresivní charakter. Tíže postižení u mužů a žen je srovnatelná. Patogeneze WMLs není zcela objasněna. Roli hrají vaskulární změny. Přepokládá se však, že na jejich vzniku se podílejí ještě jiné patofyziologické mechanizmy než pouze vaskulárně-ischemické [44]. Léze v oblasti bílé hmoty mohou někdy napodobovat nález u RS. Na rozdíl od RS se však WMLs jen zřídka vyskytují v oblasti corpus callosum [45], nemívají enhancement, a pokud je autorům známo, také spinální lokalizace nebyla dosud popsána.

Tzv. pulvinar sign, hyperintenzity v zadní thalamické oblasti na T1 vážených obrazech, byl poprvé popsán v roce 2003 jako charakteristický projev u mužů s přirozeným průběhem FCh (data pocházejí z éry před enzymovou substituční terapií) s četností výskytu kolem 20 % [46]. Častěji se vyskytuje oboustranně, nicméně jednostranný výskyt byl rovněž popsán [47]. Nedávno publikovaná práce však naznačuje, že frekvence jeho výskytu může být výrazně nižší, než se původně předpokládalo. V kohortě 133 pacientů s FCh byl tento projev identifikován pouze u 3 % z nich [48].
Postižení jiných orgánových systémů
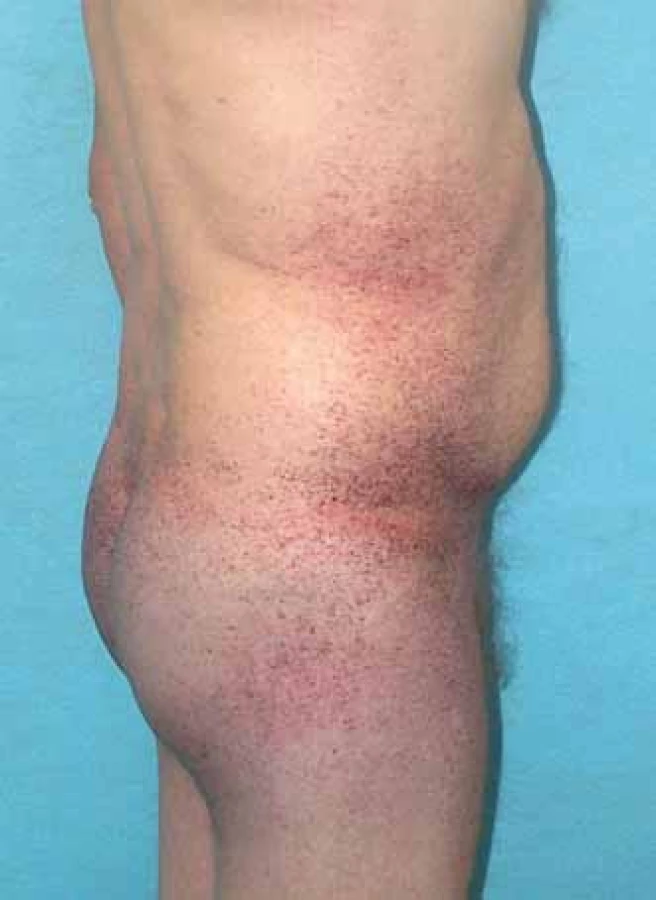
Mezi hlavní projevy mimo nervový systém patří postižení srdce, ledvin, kůže, očí, vestibulokochleárního systému. Pro kožní postižení jsou typické angiokeratomy (obr. 3). Nacházejí se zejména v oblasti mezi pupkem a koleny s maximem výskytu periumbilikálně a v oblasti genitálu. U pacientů se objevují již v dětství a jejich počet s věkem často narůstá.
U klasického průběhu onemocnění dochází u mužů ve věku kolem 20– 30 let, u žen asi o 10 let později, k postupnému zhoršování funkce ledvin. Prvním projevem je mikroalbuminurie, poté malá až středně velká proteinurie a současně klesá glomerulární filtrace. Pokud pacienti nejsou léčeni, pak v případě renálního postižení bývá asi od 3.– 4. dekády života nutná dialyzační léčba a event. transplantace ledvin.
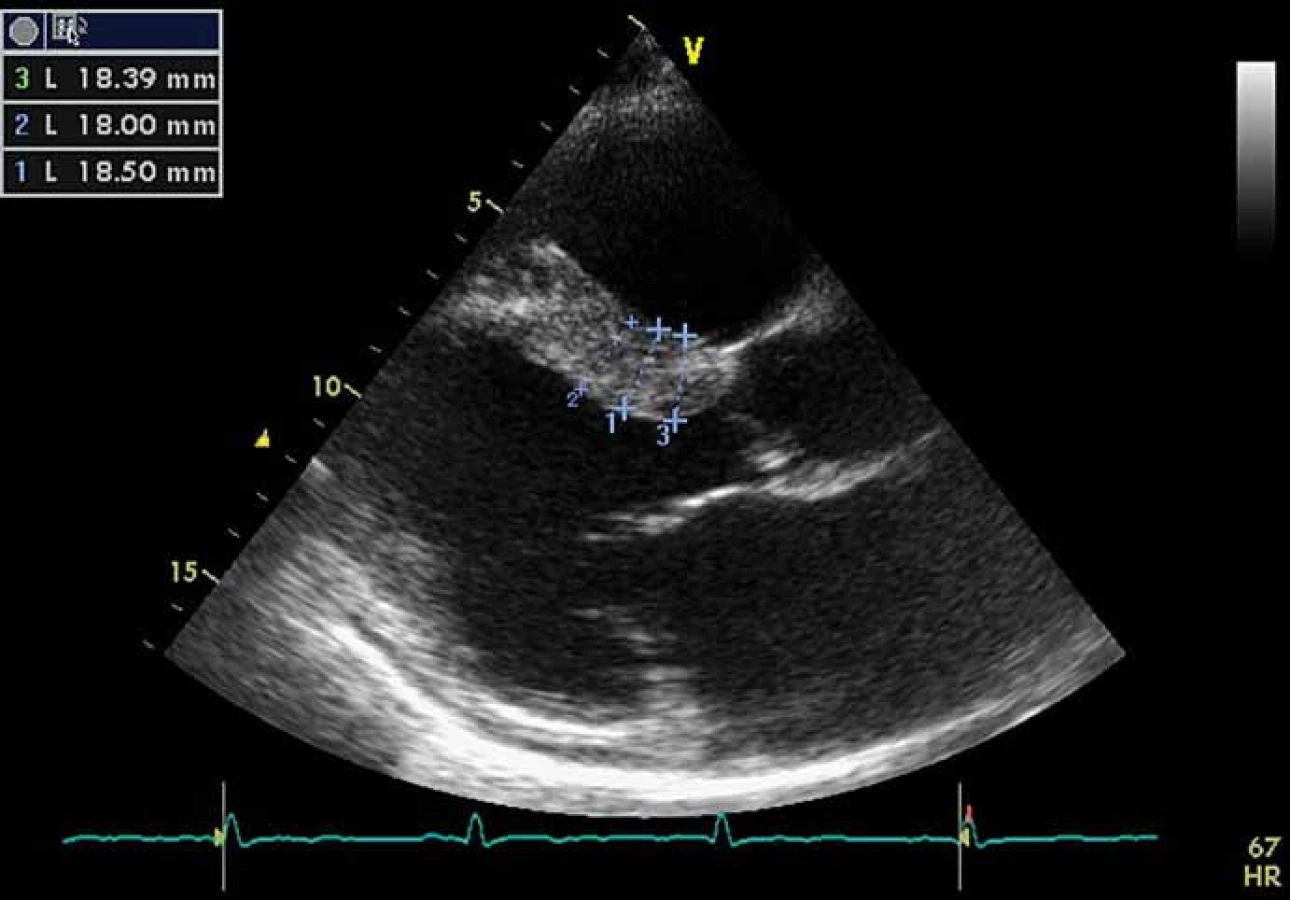
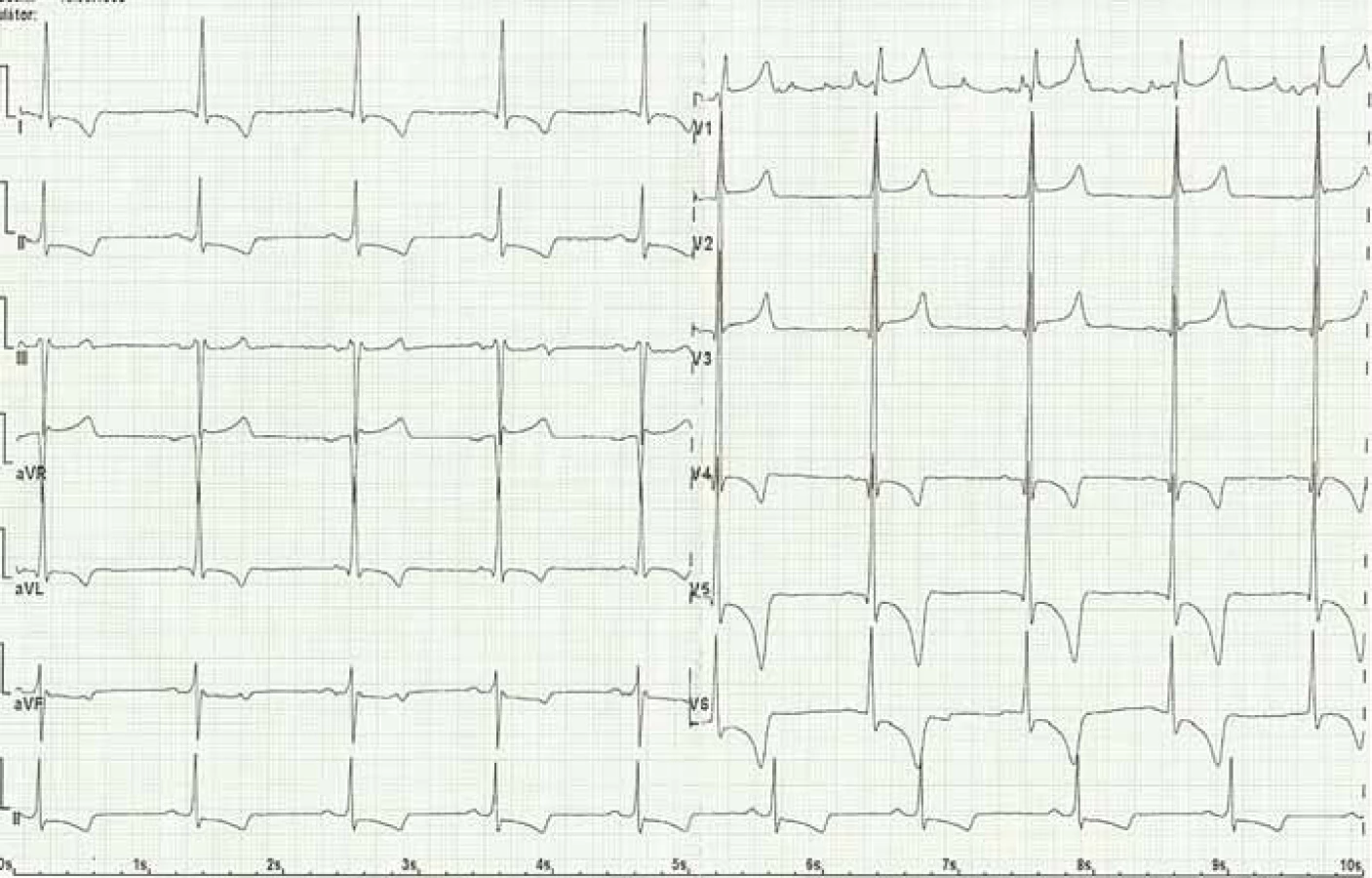
Kardiální postižení je u pacientů s FCh velmi časté. Pro postižení srdce je charakteristická hypertrofie myokardu. Může jít o hypertrofii obou komor, ale též pouze o hypertrofii septa (obr. 4). Již elektrokardiografie (EKG) může upozornit na FCh. Nález na EKG křivce s výrazně navýšenou voltáží a v další fázi i změnami ST úseku (obr. 5) by měl být indikací k echokardiografickému vyšetření. Kromě vysoké voltáže odrážející hypertrofii myokardu je u části nemocných přítomno zkrácení PQ intervalu (pod 120 ms). Postupně však dochází ke zhoršování atrioventrikulárního vedení s rozvojem převodních poruch s nutností implantace kardiostimulátoru. U řady nemocných se rozvíjí fibróza myokardu lokalizovaná typicky posterolaterálně. Nemocní trpí projevy srdečního selhání, poruchami rytmu a kardiovaskulární úmrtí jsou velice častá [49].
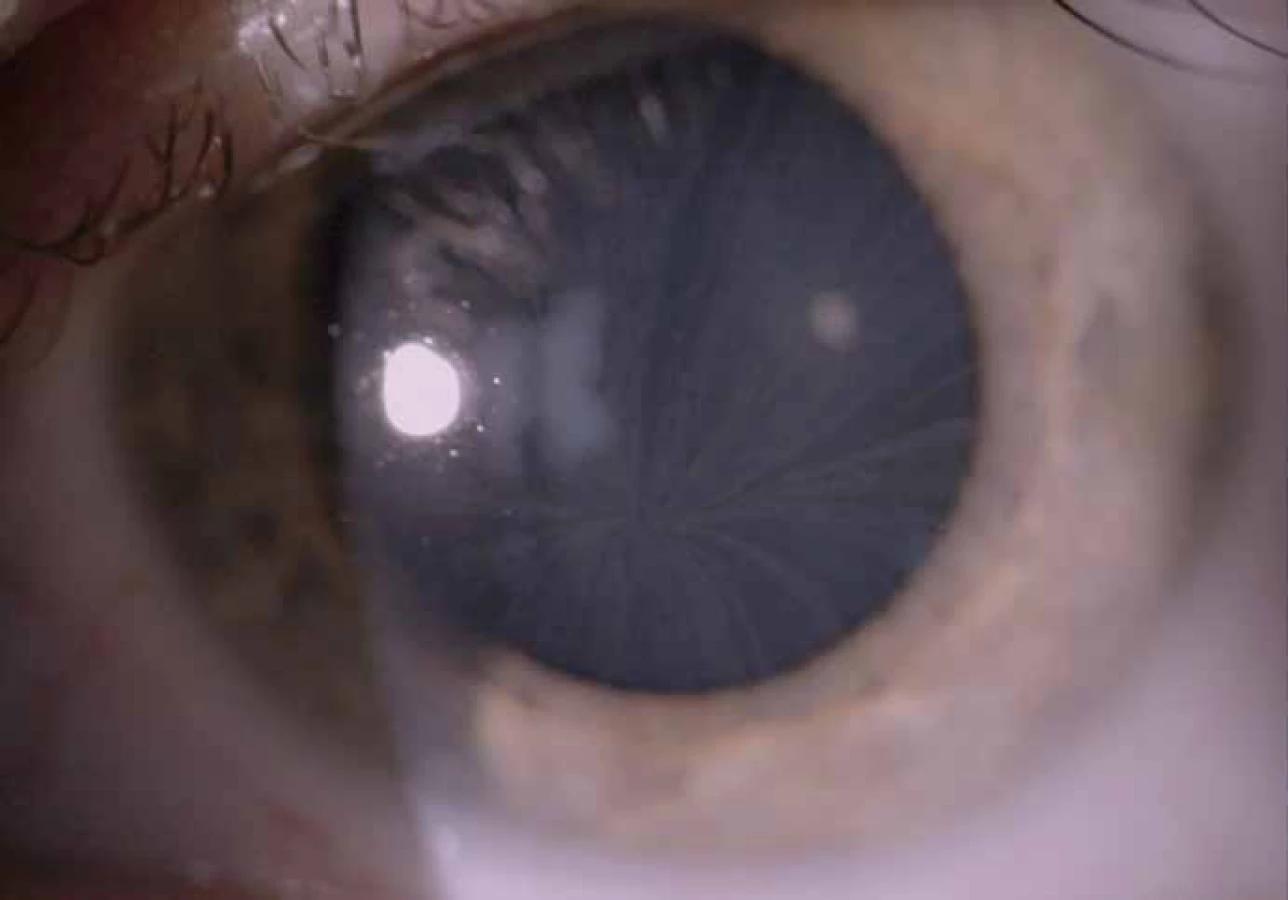
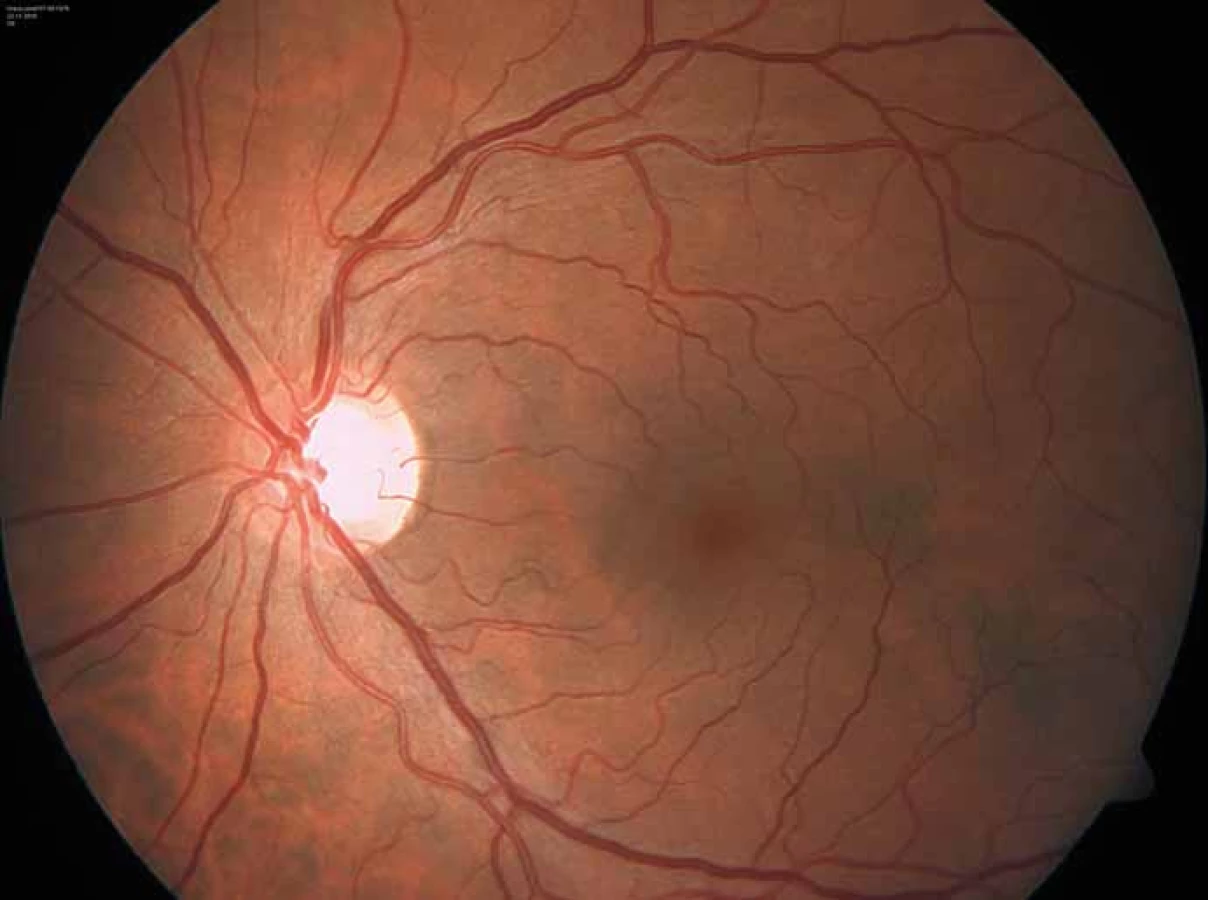
Zajímavým, častým a poměrně specifickým projevem jsou oční manifestace, jejichž přítomnost přispívá nejen k diagnostice, ale slouží i jako marker závažnějšího průběhu onemocnění [50]. Z diagnostického hlediska je nejpřínosnější průkaz přítomnosti cornea verticillata (výskyt u 44– 95 % pacientů s Fch [51]). Jedná se o paprsčitě se větvící opacity v povrchních vrstvách rohovky viditelné při vyšetření na štěrbinové lampě (obr. 6). Cornea verticillata se dále vyskytuje pouze jako poléková (chronická medikace např. amiodaronem, chlorochinem, indometacinem, fenothiazinem, tamoxifenem aj.) [52]. Mezi další oční projevy patří např. Fabryho zadní subkapsulární katarakta nebo přítomnost vinutých retinálních cév, zvláště venul (obr. 7).
Vestibulokochleární postižení je převážně zastoupeno vertigem a postupnou ztrátou sluchu. Také výskyt tinnitu je u pacientů s FCh častý [53].
Diagnostika
FCh je progresivní život zkracující onemocnění. Vzhledem k heterogenním projevům a vzácnému výskytu nemoci je však stanovení diagnózy v obecné populaci obtížné. Uvádí se, že více než 25 % nemocných s FCh je léčeno pod jinou diagnózou. Průměrná doba od prvních projevů do stanovení diagnózy se pohybuje kolem 13,7 roku u mužů a 16,3 roku u žen [6]. Stanovení správné diagnózy a včas zahájená léčba jsou pro osud nemocných nesmírně důležité, neboť mohou zamezit rozvoji nezvratného orgánového poškození. Navíc odhalíme-li nemoc u pacienta, je pravděpodobné, že pomocí genetického vyšetření rodinných příslušníků můžeme diagnostikovat až pět dalších příbuzných trpících FCh [29]. Klinické podezření na FCh vychází z podrobné anamnézy s cíleným zaměřením na známé projevy onemocnění a z fyzikálního vyšetření doplněného zobrazovacími metodami. Definitivní potvrzení diagnózy je možné u mužů pomocí stanovení aktivity enzymu α-Gal Av plazmě, leukocytech či fibroblastech. U žen je nutné molekulárně genetické vyšetření pro častý překryv hodnot aktivity enzymu pacientek s hodnotami vyskytujícími se v běžné populaci [54].
Léčebné možnosti
Léčebné strategie u pacientů s FCh zahrnují postupy specifické i nespecifické (tab. 2) Mezi specifickou terapii řadíme léčbu pomocí enzymové substituce (enzyme replacement therapy; ERT), která je v ČR k dispozici od roku 2004 a terapii pomocí molekulárního chaperonu – migalastatu. ERT spočívá v dodávání uměle vyrobeného enzymu pacientům. K dispozici jsou dva preparáty – agalsidáza α a agalsidáza β. Léky jsou podávány pacientům v infuzi trvající 120– 180 min 1× za 14 dní. Klinické studie prokázaly, že během léčby dochází ke snížení nálože akumulovaného Gb3 ve tkáních a ke zpomalení progrese nemoci. ERT zmírňuje intenzitu bolestí u pacientů s FCh, redukuje nutnost užívání analgetik, zlepšuje percepci tepelných a vibračních podnětů [27]. Bylo potvrzeno zpomalené zhoršování renálních funkcí, zpomalení progrese hypertrofie myokardu, snížení kardiovaskulárních komplikací a zlepšení kvality života [55– 58]. Údaje z registrů rovněž potvrzují efekt ERT [59]. Důležité je včasné zahájení terapie, léčba je účinnější, pokud se zahájí před rozvinutím ireverzibilních orgánových změn.

Nově je v ČR schválen lék migalastat, perorálně podávaný chaperon. Chaperony jsou malé molekuly, které vazbou na endogenní (ale i exogenně dodávaný) enzym jeho strukturu stabilizují a zvyšují jeho účinnost. Migalastat je vhodný jen pro některé nemocné s určitými mutacemi genu, kdy produkovaná endogenní α-Gal A je nestabilní [60]. Specifická terapie pomocí ERT a chaperonů bude možná v brzké době rozšířena o další možnosti. Cílená terapie zaměřená na redukci substrátu je nyní již v prvních fázích klinického zkoušení (např. lucerastat – blokátor glukosylceramid-syntázy) [61]. Rovněž genová terapie FCh vzbuzuje naděje. Je testován přenos genetické informace pomocí lentivirových vektorů s následnou tvorbou enzymu v modifikovaných CD34+ hematopoetických buňkách [62].
Zahájení a případné ukončení cílené specifické léčby FCh se v zásadě řídí doporučením evropské skupiny odborníků. O nasazení léčby rozhoduje typ postižení (mutace), kdy u klasických forem choroby je třeba léčbu indikovat při známkách prvních závažných symptomů a známek orgánového postižení ve vztahu k nemoci (konkrétně u neurologických projevů většinou indikujeme léčbu při výskytu neuropatické bolesti, TIA či CMP; může být rovněž zvážena u postižení bílé hmoty CNS). Naopak léčba není doporučena u pokročilých forem postižení a u nemocných s limitovanou prognózou [63]. Problematická je stále situace u nemocných s pozdními formami, tedy především s izolovanou kardiální manifestací. Léčba je na místě u pacientů se známkami hypertrofie a remodelace levé komory, poruchami rytmu či s projevy srdečního selhání. V současné době ale probíhá řada projektů, které mají za cíl prokázat, že i u těchto nemocných je časné nasazení terapie přínosné.
Významnou roli v terapii pacientů s FCh hrají i nespecifické léčebné postupy. Příkladem může být chorobu ovlivňující léčba (např. použití inhibitorů angiotenzin konvertujícího enzymu a sartanů v terapii proteinurie, primární a sekundární prevence CMP) či léčba symptomatická, která zahrnuje např. medikamentózní ovlivnění neuropatických bolestí. V komplexním terapeutickém přístupu je však velmi důležitá i podpůrná nefarmakologická léčba, jako je úprava stravovacího režimu u pacientů s gastrointestinálními projevy, minimalizace spouštěčů bolestivých atak (rychlá terapie horečky a infekcí, přiměřená fyzická aktivita, používání klimatizace, udržování adekvátní hydratace apod.), a v neposlední řadě psychologická podpora pacientů.
Závěr
Fch je vzácné dědičné nemocnění, jehož častými projevy jsou i projevy neurologické. Na onemocnění bychom měli myslet zejména u pacientů s neuropatií tenkých nervových vláken, pro kterou není jiné vysvětlení, u mladých pacientů s kryptogenní CMP či v rámci širší diferenciální diagnostiky RS. Laboratorní potvrzení diagnózy je dobře dostupné. Fch je onemocnění léčitelné a včasná diagnóza zásadní, protože umožní pacientům léčbu ještě před rozvinutím ireverzibilního orgánového poškození.
P. Reková prohlašuje, že od Shire HGT a Sanofi Genzyme přijala v souvislosti s přednáškovou činností týkající se Fabryho choroby finanční odměnu a cestovní grant. K. Sedláková prohlašuje, že od Shire HGT přijala v souvislosti s přednáškovou činností týkající se Fabryho choroby finanční odměnu. G. Dostálová prohlašuje, že od firmy Protalix Biotherapeutics, Shire HGT a Sanofi Genzyme přijala v souvislosti s přednáškovou činností týkající se Fabryho choroby finanční odměnu a cestovní granty. A. Linhart prohlašuje, že od Shire HGT, Sanofi Genzyme a Amicus Therapeutics přijal v souvislosti s přednáškovou a konzultační činností týkající se Fabryho choroby finanční odměnu a cestovní granty.
Autoři prohlašují, že žádné další finanční či jiné závazky, či komerční zájmy, které by mohly vést ke konfliktu zájmů, nemají.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 22. 1. 2018
Přijato do tisku: 7. 3. 2018
MUDr. Petra Reková
Neurologická klinika a Centrum klinických neurověd 1. LF UK a VFN
Kateřinská 30
128 21 Praha 2
e-mail: petra.rekova@vfn.cz
Zdroje
1. Germain DP. Fabry disease. Orphanet J Rare Dis 2010; 5: 30. doi: 10.1186/ 1750-1172-5-30.
2. Anderson W. A case of angiokeratoma. BR J Dermatol 1898; 10: 113– 117.
3. Fabry J. Ein Beitrag zur Kenntnis der Purpura hae-morhagica nodularis (Purpura papulosa haemorrhagica herbae). Arch Derm Syph 1898; 43: 187– 200.
4. Sweeley CC, Klionsky B. Fabry‘s disease: classification as a sphingolipidosis and partial characterization of a novel glycolipid. J Biol Chem 1963; 238: 3148– 3150.
5. Brady RO, Gal AE, Bradley RM et al. Enzymatic defect in Fabry‘s disease. Ceramidetrihexosidase deficiency. N Engl J Med 1967; 276(21): 1163– 1167. doi: 10.1056/ NEJM196705252762101.
6. Kornreich R, Desnick REJ, Bishop DF. Nucleotide sequence of the human alpha-galactosidase A gene. Nucleic Acids Res 1989; 17(8): 3301– 3302. doi: 10.1093/ nar/ 17.8.3301.
7. Ohshima T, Murray GJ, Nagle JW et al. Structural organization and expression of the mouse gene encoding alpha-galactosidase. Gene 1995; 166(2): 277– 280. doi: 10.1016/ 0378-1119(95)00592-7.
8. Meikle PJ, Hopwood JJ, Clauge AE et al. Prevalence of lysosomal storage disorders. JAMA 1999; 281(3): 249– 254. doi: 10.1001/ jama.281.3.249.
9. Spada M, Pagliardini S, Yasuda M et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet 2006; 79(1): 31– 40. doi: 10.1086/ 504601.
10. Lin HY, Chong KW, Hsu HJ et al. High incidence of the cardiac variant of fabry disease revealed by newborn screening in the taiwan Chinese population. Circ Cardiovasc Genet 2009; 2(5): 450– 456. doi: 10.1161/ CIRCGENETICS.109.862920.
11. Linthorst GE, Bouwman MG, Wijburg FA et al. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet 2010; 47(4): 217– 222. doi: 10.1136/ jmg.2009.072116.
12. Rolfs A, Bottcher T, Zschiesche M et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet 2005; 366(9499): 1794– 1796. doi: 10.1016/ S0140-6736(05)67635-0.
13. Brouns R, Thijs V, Eyskens F et al. Belgian Fabrystudy: prevalence of Fabry disease in a cohort of 1000 young patients with cerebrovascular disease. Stroke 2010; 41(5): 863– 868. doi: 10.1161/ STROKEAHA.110.579409.
14. Shi Q, Chen J, Pongmoragot J et al. Prevalence of Fabry disease in stroke pacients – a systematic review and meta-analysis. J Stroke Cerebrobvasc Dis 2014; 23(5): 985– 992. doi: 10.1016/ j.jstrokecerebrovasdis.2013.08.010.
15. Český statistický úřad: Obyvatelstvo. Česká republika 2017. Dostupné z URL: https: / / www.czso.cz/ csu/ czso/ obyvatelstvo_lide.
16. Merta M, Reiterová J, Ledvinová J et al. A nationwide blood spot screening study for Fabry disease in the Czech Republic haemodialysis patient population. Nephrol Dial Transplant 2007; 22(1): 179– 186. doi: 10.1093/ ndt/ gfl528.
17. Paleček T, Honzíková J, Poupětová H et al. Prevalence of Fabry disease in male patients with unexplained left ventricular hypertrophy in primary cardiology practice: prospective Fabry cardiomyopathy screening study (FACSS). J Inherit Metab Dis 2014; 37(3): 455– 460. doi: 10.1007/ s10545-013-9659-2.
18. Ross MT, Grafham DV, Coffey AJ et al. The DNA sequence of the human X chromozome. Nature 2005; 434(7031): 325– 337. doi: 10.1038/ nature03440.
19. National Center for Biotechnology Information. ClinVar. Bethesda, USA: National Library of Medicine 2018. Dostupné z URL: https:/ / www.ncbi.nlm.nih.gov/ clinvar/ .
20. Aerts JM, Groener JE, Kuiper S et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A 2008; 105(8): 2812– 2817. doi: 10.1073/ pnas.0712309105.
21. Lidove O, Ramaswai U, Jaussaud R et al. Hyperhidrosis: a new and often early symptom in Fabry disease. International experience and data from the Fabry Outcome Survey. Int J Clin Pract 2006; 60(9): 1053– 1059. doi: 10.1111/ j.1742-1241.2006.01061.x
22. Kalkum G, Pitz S, Karabul N et al. Paediatric Fabry disease: prognostic significance of ocular changes for disease severity. BMC Ophthalmol 2016; 16(1): 202. doi: 10.1186/ s12886-016-0374-2.
23. Sims K, Politei J, Banikazemi M et al. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke 2009; 40(3): 788– 794. doi: 10.1161/ STROKEAHA.108.526293.
24. Dobrovolný R, Dvořáková L, Ledvinová J et al. Relationship between X-inactivatin and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. J Mol Med (Berl) 2005; 83(8): 647– 654. doi: 10.1007/ s00109-005-0656-2.
25. Kolodny EH, Pastores GM. Anderson – Fabry disease: extrarenal, neurologic manifestations. J Am Soc Nephrol 2002; 13 (Suppl 2): S150– S153. doi: 10.1097/ 01.ASN.0000015239.57436.18.
26. Kahn P. Anderson – Fabry disease: a histopathological study of three cases with observations on the mechanism of production of pain. J Neurol Neurosurg Psychiatry 1973; 36: 1053– 1062.
27. Politei JM, Bouhassira D, Germain DP et al. Pain in Fabry disease: practical recommendations for diagnosis and treatment. CNS Neurosci Ther 2016; 22(7): 568– 576. doi: 10.1111/ cns.12542.
28. Hoffmann B, Beck M, Sunder-Plassmann G et al. Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy – a retrospective analysis from the Fabry Outcome Survey. Clin J Pain 2007; 23(6): 535– 542. doi: 10.1097/ AJP.0b013e318074c986.
29. Mehta A, Ricci R, Widmer U et al. Fabry disease defined: baseline clinical manifestation of 48366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004; 34(3): 236– 242. doi: 10.1111/ j.1365-2362.2004.01309.x.
30. Ginsberg L. Nervous system manifestations of Fabry disease: data from FOS – the Fabry Outcome Survey. In: Mentha A, Beck M, Sunder-Plassmann G (eds). Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis 2006: 227– 232.
31. Miners AH, Holmes A, Sherr L et al. Assessment of health-related quality of life in males with Anderson Fabry Disease before therapeutic intervention. Qual Life Res 2002; 11(2): 127– 133. doi: 10.1023/ A: 1015009210639.
32. Baehner F, Kampmann C, Whybra C et al. Enzyme replacement therapy in heterozygous females with Fabry disease: results of a phase IIIB study. J Inherit Metab Cis 2003; 26(7): 617– 627. doi: 10.1023/ B: BOLI.0000005658.14563.77.
33. Cable WJ, Kolodny EH, Adams RD. Fabry disease: impaired autonomic function. Neurology 1982; 32(5): 498– 502. doi: 10.1212/ WNL.32.5.498.
34. Mutoh T, Senda Y, Sugimura K. Severe orthostatic hypotension in Female a carrier of Fabry disease. Arch Neurol 1988; 45(4): 468– 472. doi: 10.1001/ archneur.1988.00520280122030.
35. O‘Mahony C, Coats C, Cardona M et al. Incidence and predictors of anti-bradycardia pacing in patients with Anderson-Fabry disease. Europace 2011; 13(12): 1781– 1788. doi: 10.1093/ europace/ eur267.
36. Hilz MJ, Kolodny EH, Brys M et al. Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. J Neurol 2004; 251(5): 564– 570. doi: 10.1007/ s00415-004-0364-9.
37. Kaye EM, Kolodny EH, Logigian EL et al. Nervous system involvement in Fabry’s disease: clinicopathological and biochemical correlation. Ann Neurol 1988; 23(5): 505– 509. doi: 10.1002/ ana.410230513.
38. Moore DF, Kaneski CR, Askari H et al. The cerebral vasculopathy of Fabry disease. J Neurol Sci 2007; 257(1– 2): 258– 263. doi: 10.1016/ j.jns.2007.01.053.
39. Elleder M, Chrostomanou H, Kustermann-Kuhn B et al. Leptomeningeal lipid storage patterns in Fabry disease. Acta Neuropathol 1994; 88(6): 579– 582. doi: 10.1007/ BF00296496.
40. Gupta S, Ries M, Kotsopoulos S et al. The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous woman. Medicine (Baltimore) 2005; 84(5): 261– 268. doi: 10.1097/ 01.md.0000178976.62537.6b.
41. Vedder AC, Linthorst GE, Van Breemen MJ et al. The Dutch Fabry cohort: diversity of clinical manifestations and Gb3 levels. J Inherit Metab Dis 2006; 30(1): 68– 78. doi: 10.1007/ s10545-006-0484-8.
42. Fellgiebel A, Keller I, Martus P et al. Basilar artery diameter is a potential screening tool for Fabry disease in young stroke patients. Cerebrovasc Dis 2011; 31(3): 294– 299. doi: 10.1159/ 000322558.
43. Carbera-Salazar MA, O‘Rourke E, Charia-Ortiz G et al. Radiological evidence of early cerebral microvascular disease in young children with Fabry disease. J Paediatr 2005; 147(1): 102– 105. doi: 10.1016/ j.jpeds.2005.03.004.
44. Rost NS, Cloonan L, Kanakis AS et al. Determinants of white matter hyperintensity burden in patients with Fabry disease. Neurology 2016; 86(20): 1880– 1886. doi: 10.1212/ WNL.0000000000002673.
45. Cocozza S, Olivo G, Riccio E et al. Corpus callosum involvement: a useful clue for differentiating Fabry disease from multiple sclerosis. Neuroradiology 2017; 59(6): 563– 570. doi: 10.1007/ s00234-017-1829-8.
46. Moore DF, Ye F, Schiffmann R et al. Increased signal intensity in the pulvinar on T1-wighted images: A pathognomonic MR imaging sign of Fabry disease. AJNR Am J Neuroradiol 2003; 24(6): 1096– 1101.
47. Gavazzi C, Borsini W, Guerrini L et al. Subcortical damage and cortical functional changes in men and women with Fabry disease: a multifaceted MR study. Radiology 2006; 241(2): 492– 500. doi: 10.1148/ radiool.2412051122.
48. Cocozza S, Russo C, A. Pisani et al. Redefining the pulvinar sign in Fabry disease. AJNR Am J Neuroradiol 2017; 38(12): 2264– 2269. doi: 10.3174/ ajnr.A5420.
49. Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart, 2007; 93(4): 528– 535. doi: 10.1136/ hrt.2005.063818
50. Pitz S, Kalkum G, Arash L et al. Ocular signs correlate well with disease severity and genotype in Fabry disease. PloS One 2015; 10(3): e0120814. doi: 10.1371/ journal.pone.0120814.
51. Sodi A, Ioannidis A, Pitz S. Ophthalmological manifestations of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G (eds). Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis 2006.
52. Raizman MB, Hamrah P, Holland EJ et al. Drug-induced corneal epithelial changes. Surv Ophthalmol 2017; 62(3): 286– 301. doi: 10.1016/ j.survophthal.2016.11.008.
53. Keilmann A, Hegemann S, Conti G et al. Fabry disease and the ear. In: Mehta A, Beck M, Sunder-Plassmann G (eds). Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis 2006: 227-232.
54. Laney DA., Fernhoff PM. J Diagnosis of Fabry disease via analysis of family history. J Genet Couns 2008; 17(1): 79-83. doi: 10.1007/ s10897-007-9128-x.
55. Eng CM, Guffon N, Wilcox WR et al. Safety and efficacy of recombinant human alpha-galactosidase A – replacement therapy in Fabry’s disease. N Engl J Med 2001; 345(1): 9– 16. doi: 10.1056/ NEJM200107053450102.
56. Thurberg BL, Byers HR, Granter SR et al. Monitoring the 3-year efficacy of enzyme replacement therapy in Fabry disease by repeated skin biopsies. J Invest Dermatol 2004; 122(4): 900– 908. doi: 10.1111/ j.0022-202X.2004. 22425.x.
57. Banikazemi M, Bultas J, Waldek S et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 2007; 146(2): 77– 86. doi: 10.7326/ 0003-4819-146-2-200701160-00148.
58. Schiffmann R, Kopp JB, Austin HA 3rd et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001; 285(21): 2743– 2749. doi: 10.1001/ jama.285.21.2743.
59. Ortiz A, Abiose A, Bichet DG et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: data from the Fabry Registry. J Med Genet 2016; 53(7): 495– 502. doi: 10.1136/ jmedgenet-2015-103486.
60. Goláň L. Migalastat v terapii Fabryho choroby. Interní Med 2017; 19(3). 167– 170.
61. Guérard N, Oder D, Nordbeck P et al. Lucerastat, an iminosugar for substrate reduction therapy: tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry Disease on enzyme replacement. Clin Pharmacol Ther 2018; 103(4): 703– 711. doi: 10.1002/ cpt.790.
62. Huang J, Khan A, Au BC et al. Lentivector iterations and pre-clinical scale-up/ toxicity testing: targeting mobilized cd34+ cells for correction of Fabry disease. Mol Ther Methods Clin Dev 2017; 5: 241– 258. doi: 10.1016/ j.omtm.2017.05.003.
63. Biegstraaten M, Arngrímsson R, Barbey F et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphane J Rare Dis 2015; 10: 36. doi: 10.1186/ s13023-015-0253-6.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2018 Číslo 2
Nejčtenější v tomto čísle
- Ataxie
- Biopsie mozku v deseti bodech – co může neurolog očekávat od neurochirurga a neuropatologa?
- Fabryho choroba, přehled problematiky a nejčastější neurologické projevy
- Prof. MUDr. Pavel Haninec, CSc. slaví 60 let