
Výskyt periferního primitivního neuroektodermálního nádoru v průběhu spinálního kořene – kazuistika
Occurence of Peripheral Primitive Neuroectodermal Tumor within Spinal Nerve Root – a Case Report
Peripheral primitive neuroectodermal tumor (pPNET) is a highly malignant soft tissue sarcoma probably originating from embryonic neural crest cells. For some common cytological, immunohistochemical and cytogenetic features with bone Ewing’s sarcoma, pPNET is also sometimes inaccurately referred to as Ewing’s sarcoma of soft tissues. pPNET probably is a different morphological expression of the same tumor type. pPNET mainly occurs in younger patients in various locations, always in close proximity to a peripheral nerve. However, its occurrence within a spinal nerve root is very rare and, so far, only a few cases have been described in the literature. In our report, we present a case of 35-year-old woman with pPNET in extraforaminal projection of L5 root. As a result of early diagnosis and treatment, including radical surgery combined with aggressive chemotherapy and irradiation of the tumor bed, no recurrence has been reported so far. The follow-up period is more than five years.
Key words:
peripheral primitive neuroectodermal tumor – spinal nerve root – neurosurgery – chemotherapy – radiotherapy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
P. Buchvald 1; P. Barsa 1; A. Blüml 2; R. Lukáš 3
Působiště autorů:
Neurocentrum, Neurochirurgické oddělení, Krajská nemocnice Liberec, a. s.
1; Oddělení patologie, Krajská nemocnice Liberec, a. s.
2; Traumatologicko-ortopedické centrum, Krajská nemocnice Liberec, a. s.
3
Vyšlo v časopise:
Cesk Slov Neurol N 2015; 78/111(3): 344-347
Kategorie:
Kazuistika
Souhrn
Periferní primitivní neuroektodermální tumor (pPNET) je vysoce maligní sarkom měkkých tkání vycházející pravděpodobně z primitivních buněk zárodečné neurální lišty. Pro některé společné cytologické, imunohistochemické a cytogenetické rysy s kostním Ewingovým sarkomem bývá také někdy nepřesně označován jako Ewingův sarkom měkkých tkání, kdy se může jednat o různou morfologickou expresi stejného nádorového typu. Objevuje se převážně u mladých nemocných v různých lokalizacích, vždy v intimním spojení s periferním nervem. Jeho výskyt v průběhu spinálního kořene je velmi vzácný a literární výčet je v tomto případě omezen pouze na několik kazuistických sdělení. V naší práci představujeme případ 35leté pacientky, u které byl odhalen pPNET v oblasti extraforminálního výstupu kořene L5. Díky časné diagnostice a léčbě zahrnující radikální resekci v kombinaci s agresivní chemoterapií a ozářením lůžka nádoru nebyla dosud zaznamenána recidiva nádorového růstu, přičemž doba sledování činí více než pět let.
Klíčová slova:
periferní primitivní neuroektodermální tumor – nervový kořen – neurochirurgie – chemoterapie – radioterapie
Úvod
Periferní primitivní neuroektodermální tumor (pPNET) je druhým nejčastějším typem sarkomu dětí a mladých dospělých. Pochází nejspíše z maligního bujení perzistujících buněk primitivní neurální lišty a často bývá pro četné společné fenotypické a genotypické rysy s kostním Ewingovým sarkomem ne zcela přesně označován jako Ewingův sarkom měkkých tkání. Jeho lokalizace je možná v různých oblastech těla, ovšem jeho výskyt v míše či oblasti kaudy je velmi vzácný a růst v oblasti kořene spinálního nervu je zcela raritní. V následujícím sdělení popisujeme řešení pPNET rostoucího v extraforaminální oblasti kořene L5 u mladé ženy.
Kazuistika
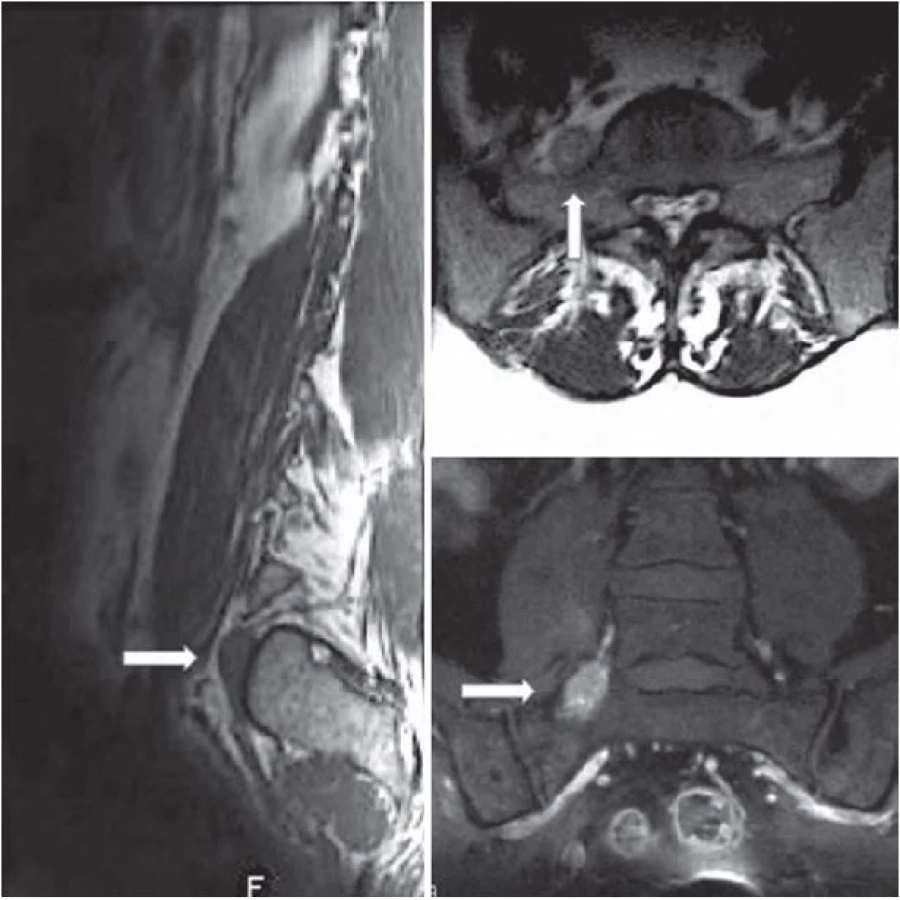
V našem sdělení představujeme případ 35leté ženy na mateřské dovolené, která se dlouhodobě léčila pouze pro hypotyreózu. K lékaři ji přivedly několikatýdenní postupně narůstající bolesti směřující z bederní krajiny do zevní strany pravé dolní končetiny. V rámci objektivního neurologického vyšetření u ní nebyl zjištěn hybný deficit, pouze snížené čití v kořenovém dermatomu L5 vpravo. Vyšetření magnetickou rezonancí (MR) odhalilo extraforaminálně uloženou ohraničenou expanzi velikosti 26 × 12 × 10 mm v kontinuitě s pravostranným kořenem L5 připomínající schwannom (obr. 1). V T1 váženém obraze byl nádor izointenzní se svaly s nepravidelným hyperintenzním sycením po podání kontrastní látky. V T2 váženém obraze byl tumor hyperintenzní v porovnání se svaly a izointenzní v porovnání s tukovou tkání. Pro tento nález byla pacientka indikována k operačnímu řešení. V celkové anestezii jsme ze zadního pravostranného intermuskulárního přístupu k extraforaminální oblasti L5/ S1 provedli selektivní exstirpaci ohraničeného měkkého tumoru uloženého v pochvě kořene L5 s uchováním nádorem nepoškozených nervových fasciklů. Peroperační monitoring jehlové elektromyografie a motorických evokovaných potenciálů nevykazoval po celou dobu výkonu významnou změnu normální aktivity odpovědí ve vyšetřovaných svalech (m. tibialis anterior, m. peroneus longus) oproti předoperačnímu vyšetření. Po operaci byla pacientka zcela bez neurologického postižení, původní radikulární bolesti odezněly a byla velmi brzy vertikalizována a dimitována. Odebraný materiál byl podroben histologickému vyšetření, které překvapivě odhalilo, že se jedná o nádor z malých kulatých buněk s hyperchromatickými jádry, jež vykazují intenzivní membranózní MIC‑ 2 (CD99) pozitivitu. Na základě dalšího imunohistochemického a cytogenetického vyšetření byl nádor klasifikován jako vysoce maligní periferní primitivní neuroektodermální tumor. Nezbytné následné PET vyšetření nepotvrdilo možnou diseminaci procesu. Pacientka byla ihned zařazena do neuroonkologického programu a byla u ní zahájena chemoterapie podle standardizovaného protokolu EuroEWING 99. Po dvou měsících, kdy pacientka absolvovala šest cyklů léčby, byla provedena kontrolní vyšetření MR a PET, která rovněž nenaznačovala recidivu či diseminaci nádoru. Při další kontrole PET šest měsíců po operaci byla odhalena lokální recidiva nádorového růstu v průměru 16 mm bez další blízké či vzdálené propagace. S ohledem na tuto skutečnost bylo nezbytné přistoupit k radikálnější resekci, realizovatelné pouze ze dvou nezávislých přístupů. Nejprve jsme ze zadního klasického interlaminárního přístupu provedli amputaci kořene L5 od durálního vaku v páteřním kanálu. Konec odpojeného kořene jsme označili tantalovou svorkou, protáhli jej do extraforaminální oblasti a deliberovali spolu s částí obsahující nádor presakrálně, až kam bylo možné z daného přístupu bez rizika dostoupit. Ve druhé době, tři dny po první operaci, jsme z předního transperitoneálního přístupu (výhodnější extraperitoneální přístup nebylo pro pevné adheze mezi fascií a peritoneem bohužel možné provést) odstranili zbytek nádoru do zdravé tkáně, přičemž v hledání amputovaného kořene L5 nám významně pomohlo označení výše zmíněným kovovým klipem detekovatelným pod C ramenem. Pacientka, ač byla předem upozorněna na možný pooperační deficit v podobě peroneální plegie, nebyla překvapivě po operaci motoricky postižena. Pravděpodobně došlo během vývoje k inervační redistribuci kořenových funkcí. Jedinou klinickou odezvou byla subjektivní lehká dysestezie na hřbetu nohy, která během několika týdnů zcela vymizela. Patologem byla z odebrané tkáně potvrzena diagnóza výše popsaného nádoru a rovněž byla prokázána radikální exstirpace do zdravé tkáně. Pacientka byla dále podrobena dalším osmi cyklům chemoterapie a bylo provedeno rovněž konkomitantní ozáření nádorového lůžka (celková dávka 45 Gy). Měsíc a následně šest měsíců po druhé operaci dle MR a PET nebyly patrné známky lokální recidivy nádoru či jeho vzdáleného šíření. Tak tomu bylo i na dalších půlročních kontrolách (obr. 2). Celková doba sledování pacientky od druhé operace dnes činí pět let.

Diskuze
Primitivní neuroektodermální tumor je agresivně rostoucí maligní nádor složený z malých buněk pocházejících nejspíše z embryonálně založené primitivní neurální lišty. Nádorové buňky mohou být přitom zastoupeny v různém stupni neurální diferenciace. Výše zmíněný název byl iniciálně použit pro skupinu embryonálních nádorů vyskytujících se v centrálním nervovém systému, dnes označovanou jako centrální PNET, kam řadíme meduloblastom, pineoblastom, centrální neuroblastom, ependymoblastom, meduloepiteliom, primární rabdomyosarkom a atypický teratom [1]. Později byl koncept PNET rozšířen přiřazením velmi histologicky podobných periferně lokalizovaných nádorů, které začaly být označovány jako periferní PNET (pPNET) [2]. K popsání pPNET byla použita v literatuře také celá řada synonym (periferní neuroepitheliom, periferní neuroblastom, Askinův tumor aj.) [3]. Vzhledem k četným shodným histologickým, cytogenetickým a imunohistochemickým rysům bývá také pPNET společně s kostním Ewingovým sarkomem a měkkotkáňovým extraoseálním Ewingovým sarkomem (EES) řazen do skupiny označované jako Ewing Sarcoma Family of Tumours (ESFT) [4]. Horowitz et al v roce 1993 analyzovali publikované případy 1 505 pacientů z USA, Evropy a Japonska, u kterých bylo možné resekovaný nádor zařadit do této skupiny, přičemž 87 % nádorů patřilo typické kostní formě, 8 % bylo zařazeno jako extraoseální Ewingův sarkom a 5 % jako pPNET [5]. Zatímco Ewingův sarkom je druhou nejčastější kostní malignitou dětského věku, pPNET a EES tvoří 4– 17 % všech dětských měkkotkáňových nádorů. Ovšem pPNET může být diagnostikován v jakémkoliv věku. Hlavní incidence je statisticky udávána v období dospívání a u mladých dospělých do 30 let bez sexuální predilekce. Tato forma se může vyskytovat v různých lokalizacích, především v hrudníku, pánvi, břiše a na končetinách. Angervall a Enzinger v roce 1975 a posléze i další autoři popsali možný vznik pPNET také v epidurální či paravertebrální lokalizaci [4,6– 9]. Jejich přítomnost intramedulárně či v oblasti kaudy je extrémně vzácná [10– 14] a pPNET rostoucí z nervového kořene v lumbální extraforaminální oblasti byl dle naší literární analýzy dosud popsán pouze v několika málo případech [15,16]. Tyto nádory obecně vykazují známky rychlého a invazivního růstu a jejich agresivní povaha je dokumentována i tím, že v době stanovení diagnózy má již 9– 20 % pacientů přítomny vzdálené metastázy, především do regionálních uzlin, do plic a kostí [17]. Doba přežití takových nemocných je i při intenzívní léčbě udávána mezi šesti měsíci až třemi roky. Z klinického hlediska nebývají příznaky pro určení nádoru nijak specifické. Většinou se jedná o různé typy bolestí, případně o neurologická postižení podle lokalizace nádoru. Z grafického hlediska při vyšetření na sonografii, CT a MR se nádor jeví jako nehomogenní měkkotkáňová solidní masa různé velikosti a různého stupně invazivity, spíše s malým sycením po podání kontrastní látky. Někdy bývají přítomny zóny prokrvácení či nekrózy, naopak kalcifikace nádor zpravidla neobsahuje [14– 16]. Vyloženě specifické zobrazovací rysy definovány nejsou. Nezbytné je doplnění komplexního vyšetření o PET, které odhalí přítomnost vzdálených metastáz a je rovněž důležitým kontrolním ukazatelem k hodnocení účinnosti zavedené léčby. Diagnostika PNET je založena na histopatologickém, imunohistochemickém a cytogenetickém vyšetření, které odliší tyto nádory od jiných kulatobuněčných nádorů (non‑Hodgkinův lymfom, rabdomyosarkom aj.). V elektronovém mikroskopu je možné v buňkách pPNET odhalit neurosekreční granule s mikrotubuly a mikrofilamenty a krátké dendritické výstupky ležící mezi buňkami (ty u Ewingova sarkomu chybí). Cytogenetická analýza pPNET ukazuje na vztah nádoru k ESFT pro shodnou translokaci genu 22q12 (EWS gen) a 11q24 (FLI‑ 1 gen), která je přítomna až v 85 %. V 5– 10 % bývá translokace nalezena mezi EWS genem a ERG (21q22). Imunohistochemický profil nádoru pak umožňuje odlišit pPNET a další ESFT od jiných malokulatobuněčných nádorů. Vyšetření je zaměřeno jednak na průkaz protilátek FLI‑ 1 genu, jednak tyto nádory také typicky vykazují imunoreaktivitu na povrchový antigen MIC2 (glykoprotein CD99), který je exprimován až v 97 % případů [5]. Tento nález neodliší pPNET od jiných ESFT, ale umožní odlišit pPNET od centrálního PNET (zde není přítomen). Zvýšený průkaz nespecifických neuronálních a gliálních ukazatelů, jakými jsou S‑ 100, neuron‑ specifická enoláza, GFAP, synaptofyzin aj., poukazuje na vyšší stupeň neurální diferenciace nádorových buněk, a tedy zařazení nádoru spíše k pPNET než k jiným ESFT, pro které jsou příznačné spíše velmi málo diferencované buňky. To je také společně s nálezem neurosekrečních granul na elektronovém mikroskopu jeden z mála odlišných faktorů mezi pPNET a jinými ESFT. V rámci ESFT má pPNET horší prognózu proti EES.
Jedinou účinnou léčbou je radikální exstirpace nádoru doplněná chemoterapií a ozářením lůžka nádoru, která je možná pouze, pokud je nádor odhalen časně, výrazně neinvaduje a není generalizován. V terapii je dnes uplatňováno léčebné schéma podle protokolu EuroEWING 99 spočívající v indukční chemoterapii (šest cyklů – kombinace vinkristinu, doxorubicinu, ifosfamidu a etoposidu) následované radikální resekcí do zdravé tkáně u lokalizované choroby a posléze navazuje konsolidační chemoterapie (kombinace vinkristinu, aktinomicinu D s cyklofosfamidem nebo ifosfamidem) doprovázená konkomitantním ozářením lůžka nádoru (celková dávka 45– 55 Gy) [18]. Pětileté přežití je pak možné očekávat u 55– 65 % pacientů. U generalizované či reziduální formy, pokud je možné uplatnit vysokodávkovanou chemoterapii a aktinoterapii, má naději na tříleté přežití asi 30 % léčených. Při pouhé konvenční systémové léčbě přežívá jeden až dva roky pouhých 10– 20 % pacientů.
Závěr
Výskyt pPNET v průběhu spinálního nervu v páteřním kanálu či paravertebrálně je velmi vzácný. Přesto je zcela nezbytné zvažovat tuto možnost v rámci diferenciální diagnostiky měkkotkáňových tumorů, a to především u mladých nemocných. Jedině časné odhalení tohoto vysoce maligního nádoru a jeho radikální léčba v podobě makroradikální resekce, adjuvantní chemoterapie a aktinoterapie dává naději na příznivý průběh onemocnění.
Přijato k recenzi: 14. 11. 2014
Přijato do tisku: 10. 3. 2015
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Pavel Buchvald
Neurocentrum
Neurochirurgické oddělení
Krajská nemocnice Liberec, a.s.
Husova 10
460 63 Liberec
e-mail: pavel.buchvald@nemlib.cz
Zdroje
1. Hart MN, Earle KM. Primitive neuroectodermal tumours of the brain in children. Cancer 1973; 32(4): 890– 897.
2. Kielhuer P, Bueger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol 1993; 3(3): 255– 268.
3. Barbara J. Primitive neuroectodermal tumours. In: Wilkins RH, Rengachary SS (eds). Neurosurgery. New York: McGraw Hill 1996: 1707– 1713.
4. Angervall L, Enzinger FM. Extraskeletal neoplasm resembling Ewing’s sarcoma. Cancer 1975; 36(1): 240– 251.
5. Horowitz ME, Malawer MM, DeLaney TF, Tsokos MG. Ewing’s sarcoma family of tumors: Ewing’s sarcoma of bone and soft tissue and the peripheral primitive neuroectodermal tumors. Principles and practice of pediatric oncology. 2nd ed. Philadelphia: Lippincott Williams & Wilkins 1993: 795– 821.
6. Fink LH, Meriwether MW. Primary epidural Ewing’s sarcoma presenting as a lumbar disc protrusion. J Neurosurg 1979; 51(1): 120– 123.
7. Hadfield MG, Quezado MM, Williams RL, Luo VY. Ewing’s family of tumors involving structures related to the central nervous system: a review. Pediatr Dev Pathol 2000; 3(3): 203– 210.
8. Benesch M, Urban C, Lackner H, Kerbl R, Schwinger W, Beham‑ Schmid C et al. Atypical extraosseous Ewing’s sarcoma of the spinal canal with bone marrow involvement in a two‑month‑ old boy. Med Pediatr Oncol 1999; 32(6): 471– 473.
9. Spaziante R, de Divitiis E, Giamundo A, Gambardella A, Di Prisco B. Ewing’s sarcoma arising primarily in the spinal eoidural space: fifth case report. Neurosurgery 1983; 12(3): 337– 341.
10. Hisaoka M, Hashimoto H, Murao T. Peripheral primitive neuroectodermal tumor with ganglioneuroma‑like areas arising in the cauda equina. Virchows Arch 1997; 431(5): 365– 369.
11. McDermott VG, el‑ Jabbour JN, Sellar RJ, Bell J. Primitive neuroectodermal tumor of the cauda equina. Neuroradiology 1994; 36(3): 228– 230.
12. Isotalo PA, Agbi C, Davidson B. Primary primitive neuroectodermal tumor of the cauda equina. Human Pathol 2000; 31(8): 999– 1001.
13. Weil RJ, Zhuang Z, Pack S, Kumar S, Helman L, Fuller BG et al. Intramedullary Ewing sarcoma of the spinal cord: consequences of molecular diagnosis. J Neurosurg (Spine) 2001; 95 (Suppl 2): 270– 275.
14. Mawrin C, Synowitz HJ, Kirches E, Kutz E, Dietzmann K, Weis S. Primary primitive neuroectodermal tumor of the spinal cord: case report and review of the literature. Clin Neurol Neurosurg 2002; 104(1): 36– 40.
15. Ishikawa S, Ohshima Y, Suzuki T, Oboshi S. Primitive neuroectodermal tumor of spinal nerve root. Acta Pathol Jpn 1979; 29(2): 289– 301.
16. Liu H, Yang WC, Garcia RL, Noh JM, Malhotra V, Leeds NE. Intraspinal primitive neuroectodermal tumor arising from the sacral spinal nerve root. J Comp Tomography 1987; 11(4): 350– 354.
17. Schmidt D, Herrmann C, Jürgens H, Harms D. Malignant peripheral neuroectodermal tumor and its necessary distinction from Ewing’s sarcoma. Cancer 1991; 68(10): 2251– 2259.
18. Lewis I, Jürgens H, Oberlin O, Gadner H, Judson I, Paulussen M. EuroEWING 99. Amended protocol version 2006: 1– 122.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2015 Číslo 3
Nejčtenější v tomto čísle
- Addenbrookský kognitivní test – orientační normy pro českou populaci
- Míšní šok – od patofyziologie ke klinickým projevům
- Diagnostika epileptických záchvatů
- Vzduchová embolie mozku – kazuistika