
Kennedyho choroba v materiáli Centra pre neuromuskulárne ochorenia Bratislava
Kennedy’s Disease in the Neuromuscular Centre in Bratislava
Background:
Kennedy´s spinal and bulbar muscular atrophy is a hereditary disease and the most common form of spinal muscular atrophy in adult age. This disease is caused by a CAG - repeat expansion in androgen receptor gene on the X-chromosome.
Aim:
A comparison of clinical and genetic characteristics of our patients with patients in three large studies available from Japan, USA and Great Britain.
Methodology:
Between 1990 and 2013, we observed 17 patients with genetically verified diagnosis of Kennedy´s disease. We ascertained detailed medical history, all patients had neurological, electrophysiological and laboratory examinations, including genetic testing using PCR methodology.
Results:
The majority of parameters were similar to data from foreign studies – the mean age at the time of data collection was 53.6 ± 9.7 years and 43.1 ± 8.1 years at the time of first symptoms, the mean time from onset of symptoms was 9.2 ± 7.7 years and the mean time from onset of symptoms to diagnosis was 5.2 ± 4.6 years. Initial disease symptoms occurred, similarly to the other studies, in proximal parts of the lower extremities (47% of patients), followed by bulbar region (17%) and the muscles of the upper limb (12%). Compared to other studies, fewer patients (35%) had a positive family history, while the average CAG repeat size was similar (44.4 ± 3.2). We have identified a very strong correlation between the number of CAG repeats and the maximum detected value of creatine kinase and a strong correlation between the duration of the disease and maximal detected value of creatine kinase.
Key words:
Kennedy´s disease – spinal muscular atrophy – myastenia gravis
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
F. Cibulčík 1; I. Martinka 1; A. Hergottová 1; I. Urminská 1; R. Petrovič 2; H. Zelinková 2; P. Špalek 1
Působiště autorů:
Neurologická klinika LF SZU a Centrum pre neuromuskulárne ochorenia, UN Bratislava
1; Ústav lekárskej biológie, genetiky a klinickej genetiky, LF UK a UN Bratislava
2
Vyšlo v časopise:
Cesk Slov Neurol N 2015; 78/111(3): 335-339
Kategorie:
Krátké sdělení
doi:
https://doi.org/10.14735/amcsnn2015335
Souhrn
Úvod:
Kennedyho spinálna a bulbárna svalová atrofia patrí k hereditárnym ochoreniam s dedičnosťou viazanou na X chromozóm. Je najčastejšou formou spinálnej muskulárnej atrofie v dospelom veku a je spôsobená zmnožením trinukleotidových repetícií v géne pre androgénový receptor.
Cieľ:
Porovnanie klinických a genetických charakteristík našich pacientov s pacientmi v troch veľkých súboroch z Japonska, USA a Veľkej Británie.
Súbor a metodika:
V rokoch 1990– 2013 sme na Neurologickej klinike LF SZU a UN Bratislava v jej Centre pre neuromuskulárne ochorenia diagnostikovali a sledovali 17 pacientov s geneticky verifikovanou diagnózou Kennedyho choroby. U všetkých pacientov sme odobrali podrobnú anamnézu, urobili neurologické, laboratórne a elektrofyziologické vyšetrenia včítane genetického vyšetrenia materiálu PCR metodikou.
Výsledky:
Väčšina zistených parametrov bola podobná údajom zo zahraničných prác – priemerný vek pacientov pri vyšetrení bol 53,6 ± 9,7 roka, priemerný vek pri vzniku klinických ťažkostí bol 43,1 ± 8,1 roka, priemerná doba od vzniku ťažkostí po určenie diagnózy bola 5,2 ± 4,6 roka a priemerná doba trvania ochorenia 9,2 ± 7,7 roka. Podobné boli i lokality úvodných príznakov ochorenia – najčastejšie boli pozorované v pletencoch dolných končatín (47 % pacientov), nasledujú bulbárne svaly (17 %) a pletence horných končatín (po 12 %). V našom súbore sme zistili v porovnaní so zahraničnými nižšiu frekvenciu výskytu ochorenia v rodine (35 %), priemerný počet repetícií CAG trinukleotidu bol podobný s ostatnými súbormi (44,4 ± 3,2). Bola zistená veľmi silná korelácia medzi počtom CAG repetícií a najvyššou zistenou hladinou kreatínfosfokinázy a silná korelácia medzi trvaním ochorenia a najvyššou zistenou hladinou kreatínfosfokinázy, iné významné korelácie sme nezistili.
Kľúčové slová:
Kennedyho choroba – spinálna muskulárna atrofia – myasténia gravis
Úvod
Americký neurológ William R. Kennedy v roku 1966 v abstrakte opísal a s kolektívom v roku 1968 publikoval [1] podrobnosti klinického postihnutia 11 mužov z dvoch rodín. Toto postihnutie bolo charakterizované proximálnou svalovou slabosťou so vznikom v 4.– 5. decéniu. Ochorenie nazval progresívna proximálna spinálna a bulbárna svalová atrofia vyššieho veku a popísal jeho na X chromozóm viazaný spôsob prenosu. Po zistení abnormalít pri vyšetrení senzitívnych nervových vlákien v roku 1982 Hardingová použila pre ochorenie názov na X chromozóm viazaná bulbospinálna neuronopatia, do klinického spektra prejavov pribudli neskôr tras horných končatín, periorálne fascikulácie a endokrinologické poruchy s typickou gynekomastiou. V roku 1986 bol genetický defekt lokalizovaný na proximálnej časti dlhého ramienka chromozómu X, neskôr bola popísaná expanzia repetície cytozín‑ adenín‑ guanínového trinukleotidu v prvom exóne génu pre androgénový receptor [2]. V česky a slovensky písanej literatúre prvý kazuistický popis nájdeme v práci Mayera et al už v roku 1995 [3]. V literatúre boli dodnes publikované tri práce venujúce sa popisu charakteristík väčších súborov – u 223 japonských [4], 57 amerických [5] a 61 britských pacientov [6]. V bratislavskom Centre pre neuromuskulárne ochorenia boli diagnostikovaní 17 pacienti s geneticky verifikovanou Kennedyho chorobou. Cieľom našej práce bolo porovnať klinické a genetické charakteristiky našich pacientov s pacientmi žijúcimi v iných geografických a genetických populáciách.
Súbor a metodika
V rokoch 1990– 2013 sme na Neurologickej klinike LF SZU a UN Bratislava v jej Centre pre neuromuskulárne ochorenia diagnostikovali a sledovali 17 pacientov s geneticky verifikovanou diagnózou Kennedyho choroby. U všetkých pacientov sme odobrali podrobnú anamnézu a urobili neurologické vyšetrenie. Pri retrospektívnom vyhodnotení údajov sme sa zamerali na rodinnú anamnézu (prítomnosť ochorenia s klinickými charakteristikami Kennedyho choroby v rodine), vek pacienta, vek pri začiatku príznakov, iniciálne príznaky ochorenia, čas do určenia diagnózy, trvanie ochorenia, prítomnosť gynekomastie a sprievodné ochorenia. Elektromyografické vyšetrenie (EMG) sme robili na prístroji Medtronic Keypoint, vyšetrovali sme motorické vedenie v nervus (n.) medianus, ulnaris a tibialis obojstranne, senzitívne vedenie v n. medianus, ulnaris a suralis obojstranne a urobili sme protokol repetitívnej stimulácie so zameraním na poruchu neuromuskulárneho prevodu v n. radialis (so snímaním z m. anconeus) a n. facialis (so snímaním z m. nasalis) bilaterálne. U EMG vyšetrenia sme hodnotili prítomnosť odchýliek v porovnaní s normatívnymi dátami neurofyziologického laboratória nášho pracoviska. Ďalej sme u všetkých pacientov vyšetrili základné biochemické parametre v krvi – hladinu glukózy, kreatinínu, kreatínfosfokinázy (CK), laktátdehydrogenázy (LDH) a sérových transamináz (AST, ALT, GMT). U všetkých pacientov bol odobratý materiál na genetické vyšetrenie. Toto bolo robené na Oddelení molekulovej a biochemickej genetiky LF UK a UN Bratislava. Pri vyšetrení bola použitá amplifikácia CAG repetícií v 1. exóne génu pre androgénový receptor metódou PCR, elektroforéza a fragmentačná analýza PCR produktu. Za fyziologický sa považoval počet repetícií CAG trinukleotidu v rozmedzí 11– 35, za patologický počet repetícií 38 a viac. Získané dáta boli sumarizované s použitím deskriptívnej štatistiky – výpočtom mediánu, aritmetického priemeru, smerodajnej odchýlky, intervalu, percentuálneho zastúpenia. Na posúdenie korelácie medzi zistenými veličinami bol použitý Spearmanov korelačný koeficient. Na výpočet bol použitý štatistický modul programu SPSS.
Výsledky
Vyšetrili sme 17 vzájomne nepríbuzných pacientov s diagnózou Kennedyho spinobulbárnej atrofie (tab. 1). Priemerný vek pacientov pri vyšetrení bol 53,6 ± 9,7 roka, priemerný vek pri vzniku klinických ťažkostí 43,1 ± 8,1 roka, priemerná doba od vzniku ťažkostí po určenie diagnózy bola 5,2 ± 4,6 roka a priemerná doba trvania ochorenia 9,2 ± 7,7 roka. Pacienti boli pred určením diagnózy Kennedyho choroby vedení pod rôznymi pracovnými diagnózami – motor neuron disease 3×, Kennedyho choroba 3×, polymyozitída 3×, polyneuropatia 2×, po jednom pacientovi boli zastúpené diagnózy myastenia gravis, benígne fascikulácie, hepatopatia, gynekomastia a bulbárny syndróm nejasnej etiológie.
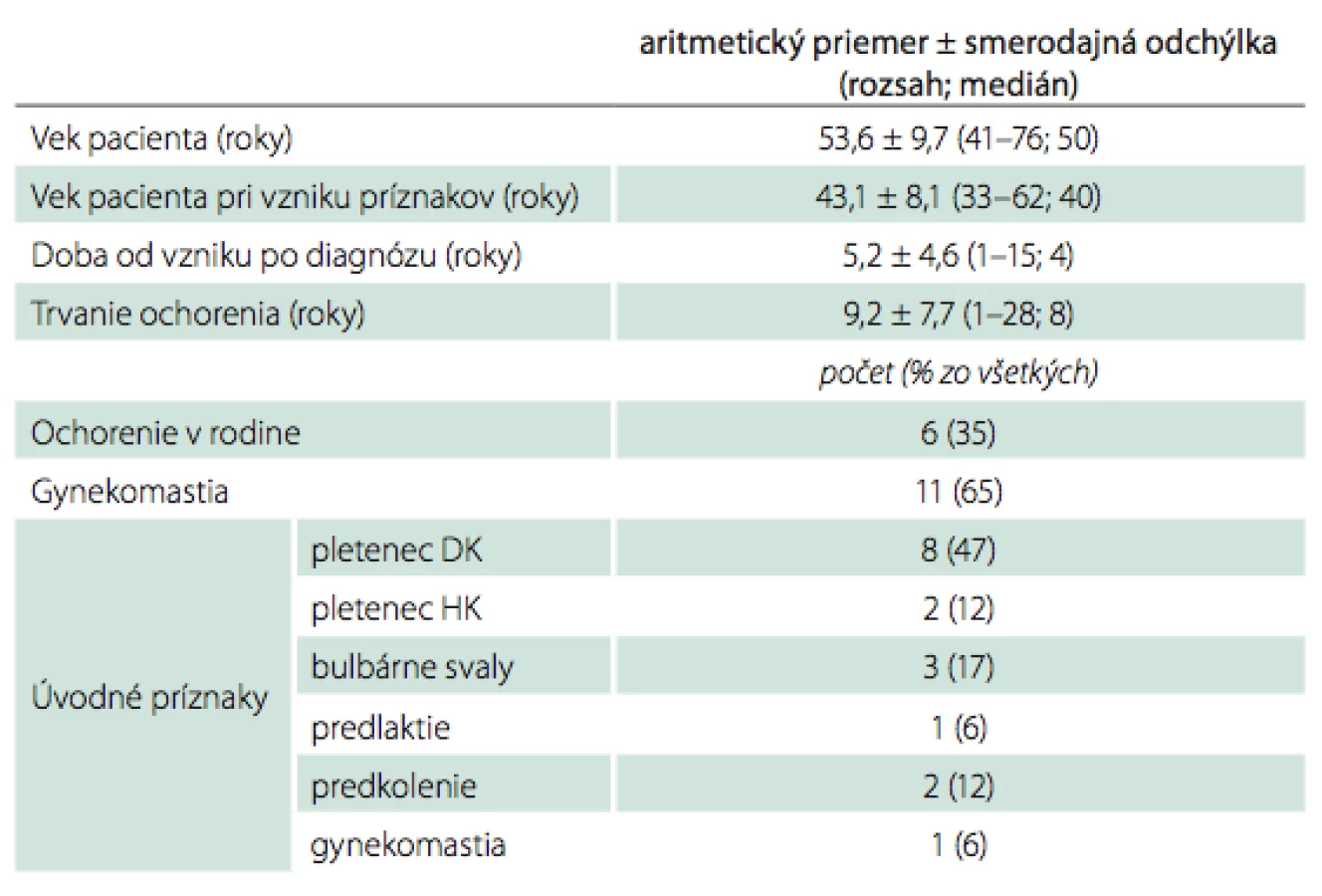
U šiestich pacientov (35 %) sa podarilo zistiť výskyt ochorenia v rodine, u 11 pacientov (65 %) bola zistená gynekomastia. Úvodné príznaky ochorenia – prevažne únavnosť s fascikuláciami a krampami – boli najčastejšie pozorované v pletencoch dolných končatín (47 % pacientov), nasledujú bulbárne svaly (17 %), pletence horných končatín a predkolenia (po 12 %), jeden pacient (6 %) mal ako prvý príznak popísanú gynekomastiu a jeden slabosť svalstva predlaktí. Klinicky významný tras sme v rozpore so skúsenosťami v zahraničí zistili len u čtyroch pacientov a len u jedného dominoval v klinickom obraze vo forme posturálneho trasu. Na rozdiel od pomerne uniformného priebehu ochorenia popisovaného najmä japonskými autormi bola heterogenita príznakov u našich pacientov podstatne väčšia, čo sa týka ich výskytu v čase aj intenzite. V literatúre popisovanú hypertrofiu svalstva lýtok sme nepozorovali, autonómne poruchy vo forme ortostatických ťažkostí sme našli len u pacienta s najdlhším priebehom ochorenia.
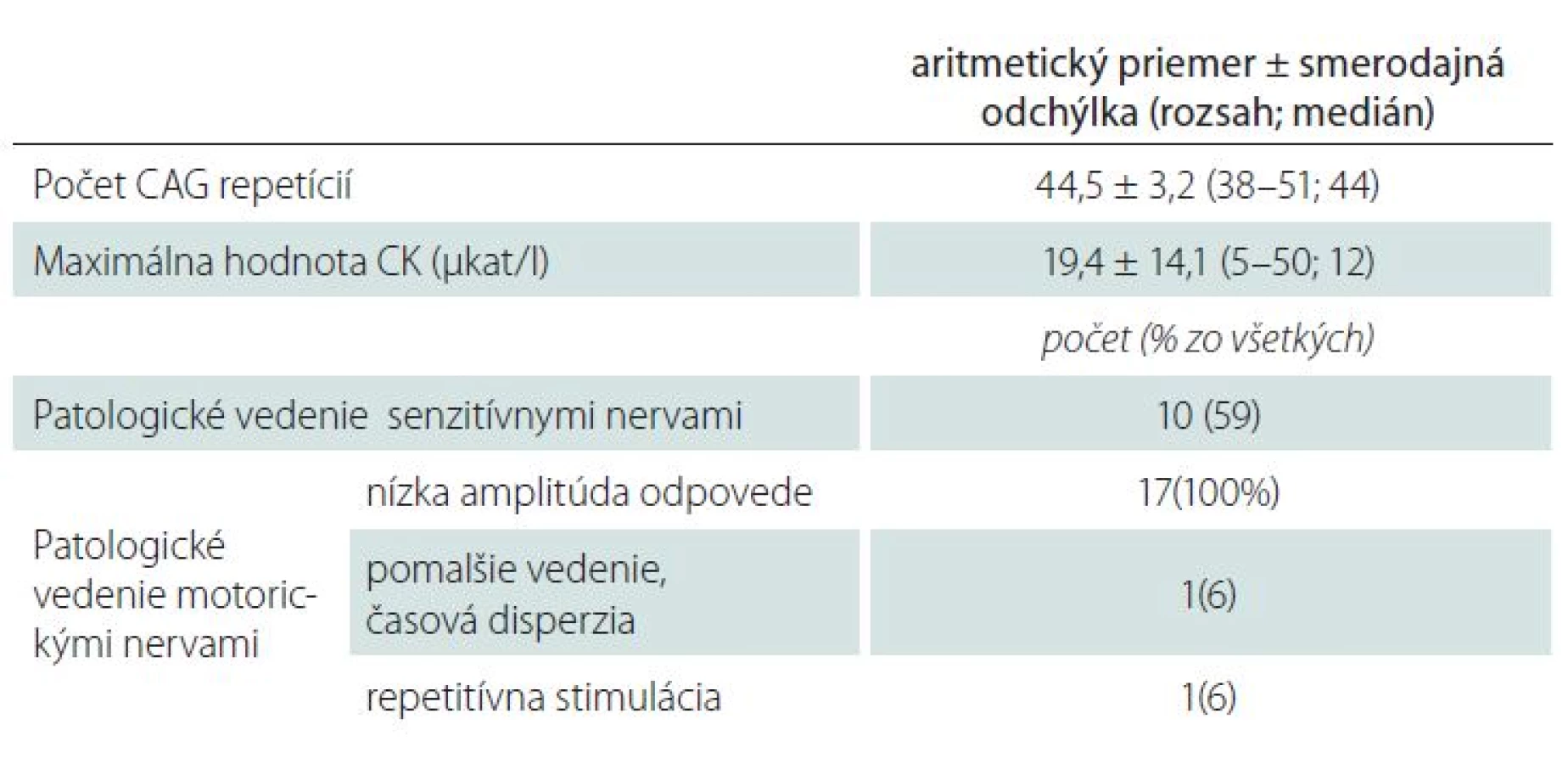
Pri vyhodnotení laboratórnych parametrov (tab. 2) sme zistili priemernú hodnotu počtu repetícií CAG 44,4 ± 3,2, priemerná hodnota maximálnej nameranej hladiny kreatínfosfokinázy (CK) bola 19,4 ± 14,1 µkat/ l. U 10 pacientov (59 %) bol zistený patologický nález (nízka amplitúda, chýbanie odpovede alebo spomalenie vedenia) pri vyšetrení vedenia aspoň v jednom z vyšetrených senzitívnych nervov, klinické prejavy poruchy citlivosti z toho mali piati pacienti (u dvoch z nich boli spôsobené úžinovým syndrómom v karpálnom kanáli). Pri vyšetrení vedenia motorickými vláknami bolo u všetkých pacientov zistené zníženie amplitúdy odpovede v aspoň jednom motorickom nerve, u jedného pacienta sa vyskytlo zistené i spomalenie vedenia a fenomén časovej disperzie, u jedného pacienta signifikantný pokles odpovedí pri repetitívnej stimulácii. V ihlovej elektromyografii bol u pacientov zistený obraz subakútne priebiehajúcej periférne neurogénnej lézie s nukleárnymi črtami, ktorého rozsah závisel od trvania ochorenia.
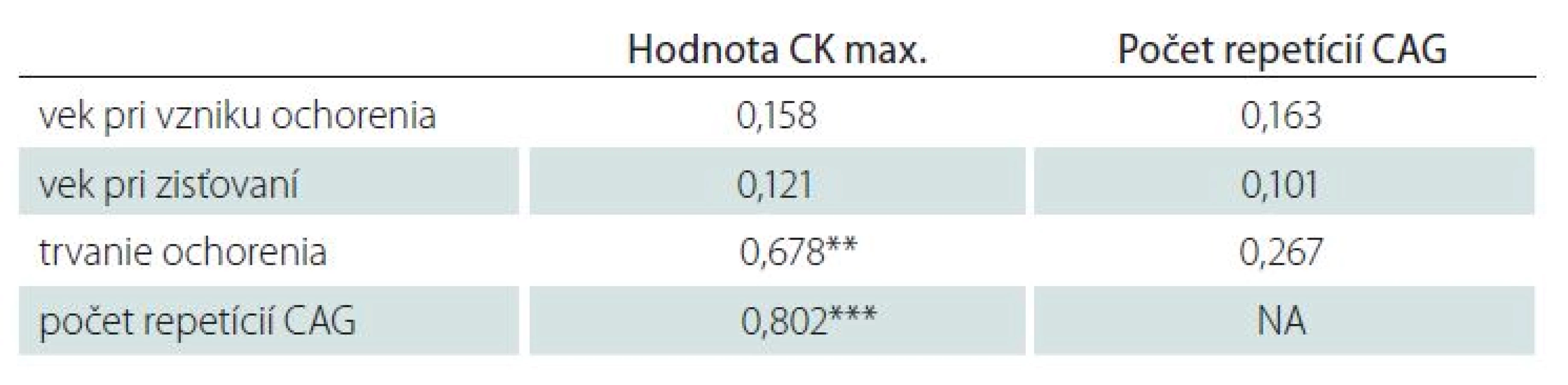
Vybrané dáta sme podrobili korelačnej analýze. Veľmi silnú koreláciu (rho 0,802) sme zistili medzi počtom repetícií CAG a najvyššou nameranou hodnotou CK, silnú koreláciu (rho 0,678) medzi trvaním ochorenia a najvyššou nameranou hodnotou CK. Iné významnejšie korelácie medzi sledovanými parametrami neboli zistené (tab. 3).
Diskusia
Kennedyho spinálna a bulbárna atrofia je považovaná za najčastejšiu formu spinálnej muskulárnej atrofie so vznikom v dospelom veku s predpokladanou prevalenciou 1,6 na 100 000 obyvateľov [7]. Pri uvedenej prevalencii by v Slovenskej republike malo byť približne 85 chorých, naše centrum s celoslovenskou pôsobnosťou ich zachytilo 17. Tento záchyt je pri porovnaní počtu obyvateľov mierne vyšší ako v súbore prospektívne aktívne vyhľadaných pacientov neurologických pracovísk vo Veľkej Británii. Porovnanie vybraných parametrov z nášho súboru a troch zahraničných súborov prináša tab. 4.
![Porovnanie údajov súborov pacientov – spracované podľa [4–6].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/738d4596efb2692cad94360ab1e09694.png)
Pri porovnaní demografických charakteristík sme zistili u väčšiny priemerných hodnôt výraznú podobnosť v súboroch z rôznych geografických lokalít. Priemerný vek pacientov v dobe spracovávania údajov bol 53– 57,6 roka, priemerný vek pacienta v dobe zistenia prvých klinických príznakov kolísal medzi 41 až 43,4 roka. V súboroch z Veľkej Británie a USA boli zaznamenané záchyty prvých príznakov ochorenia aj v mladom veku (14, respektíve 18 rokov), čo sa prejavilo i v priemernom trvaní ochorenia v súboroch – zatiaľ čo slovenský a japonský súbor vykázal podobné priemerné trvanie ochorenia 9,2 a 9,8 roka, v súbore z USA to bolo 11,8 a v súbore z Veľkej Británie dokonca 16,6 roka. Reprezentatívnejší z hľadiska skutočného zastúpenia chorých by bol údaj o mediáne, ktorý však z iných prác nemáme k dispozícii. V našej práci bol medián trvania osem rokov – rozdiel v porovnaní s aritmetickým priemerom je spôsobený trvaním ochorenia u jediného pacienta, ktoré je výrazne dlhšie ako u ostatných pacientov. Doba od vzniku ťažkostí po presné určenie diagnózy je v našom súbore podobná ako v súbore z USA – 5,2 oproti 5,5 roka, medián v našom súbore bol štyri roky. Podobne ako v iných súboroch aj v našom súbore dominovali pri hodnotení prvých príznakov ťažkosti na dolných končatinách sledované hornými končatinami a bulbárnou symptomatológiou. Väčšina ťažkostí na HK začala v proximálnej – pletencovej lokalizácii, u dvoch pacientov sa však jednalo o predkolenia. Považujeme za dôležité poukázať aj na tento menej typický priebeh ochorenia, ktorý môže imitovať obraz polyneuropatie. U jediného z našich pacientov sme mohli sledovať klinické príznaky v dlhšom časovom období – viac ako 20 rokov. Priebeh zneschopnenia u pacienta zodpovedá popisovaným priebehom v japonskej a britskej štúdii – pacient má aktuálne 58 rokov a je schopný chôdze za pomoci palíc alebo chodidla. Medián veku pacientov, kedy začínajú byť pripútaní na invalidný vozík, je v japonskej a britskej štúdii udávaný okolo 60 rokov. Na rozdiel od pomerne uniformne najmä japonskými autormi popisovaného priebehu ochorenia v skorších štádiách bola heterogenita príznakov u našich pacientov podstatne väčšia, čo sa týka ich výskytu v čase aj intenzite.
Výrazný rozdiel bol v prítomnosti pozitívnej rodinnej anamnézy – v našom súbore sme ju zistili v 35 % pacientov, v súbore z USA bola udávaná hodnota 68 % a z Veľkej Británie až 71,8 %. Príčinou tohto rozdielu je nejasná, najskôr je spôsobený interpretáciou ťažkostí a nedostatočnými údajmi o príbuzenstve u nami vyšetrovaných jedincov. Na druhej strane v súbore pacientov z Kórei [8] s veľkosťou podobnou nášmu bol popísaný familiárny výskyt u 50 % pacientov, čo je číslo bližšie naším zisteniam.
Priemerný počet CAG repetícií sa v zahraničných prácach pohyboval v rozpätí od 45,5 do 46,7, najmenší zistený počet bol 40. V našej práci sme zistili podobnú priemernú hodnotu počtu repetícií – priemerne 44,4, zachytili sme i pacienta s počtom repetícií 38. Nezistili sme koreláciu medzi vekom vzniku ťažkostí a počtom CAG repetícií, ktorá bola popísaná v zahraničných súboroch [4,6].
Objavili sme však silnú koreláciu medzi počtom repetícií, respektíve dĺžkou trvania ochorenia a najvyššou nameranou hodnotou CK. Vysoká hladina sérovej kreatínfosfokinázy je jav uniformne popisovaný u pacientov s Kennedyho chorobou a je u iných neurogénnych ochorení vzácny. Definitívne objasnenie tohoto javu nie je známe. Niektorí autori ho vysvetľujú „myopatickými zmenami“ popisovanými pri dlhšie trvajúcej neuropatii s porušením nervových vlákien a bunkovej odpovede vo svale. Pre tento mechanizmus by mohla nepriamo svedčiť i nami zistená silná korelácia medzi trvaním ochorenia v čase odberu a nameranou maximálnou hodnotou CK. Na druhej strane autori prác venujúcich sa morfologickým zmenám vo svaloch pacientov s Kennedyho chorobou upozorňujú, že rozsah „myopatických“ zmien nekoreluje s trvaním ochorenia, ale skôr s poruchou funkcie [9]. Podľa ich názoru sú „myopatické“ zmeny u pacientov s Kennedyho chorobou spôsobené poruchou aktivácie satelitných buniek, ktoré majú kľúčový význam v procesoch opravy a prevencie svalového poškodenia.
U jedného pacienta z nášho súboru sme zistili známky poruchy neuromuskulárneho prevodu pri elektrofyziologickom vyšetrení (typický pokles odpovedí po repetitívnej stimulácii) a liečebnom teste – v úvode úprava ťažkostí pri farmakologickej liečbe a dlhodobejší ústup ťažkostí po tymektómii. Koincidenciu geneticky verifikovanej Kennedyho choroby s myastenickými príznakmi (únavnosť, pokles odpovede pri repetitívnej stimulácii a zlepšenie únavnosti po venózne podanom edrofóniu) prvý krát popísali Yamada et al v roku 1997 [10]. Vzhľadom ku chýbajúcej terapeutickej odpovedi na tymektómiu a podávanie pyridostigmínu per os, ako aj chýbaniu protilátok proti acetylcholínovému receptoru, hľadali príčinu elektrofyziologického nálezu pri repetitívnej stimulácii v procese chronickej denervácie a reinervácie pri Kennedyho chorobe. Podobnú kazuistiku uvádza aj Stevic s kolektívom v roku 2014 [11]. Napriek pozitívnej odozve na liečbu inhibítormi acetylcholínesterázy sa autori v súlade s nálezmi Inoueho et al [12] domnievajú, že podkladom pre patologické nálezy pri single fiber elektromyografii je pri Kennedyho chorobe znížená schopnosť motoneurónov k reinervácii, prípadne ich odumretie pred vytvorením terminálneho vetvenia. Boz et al [13] v roku 2007 referovali kazuistiku pacienta s kolísavými prejavmi diplopie, ptózy a porúch prehĺtania a dlhšie trvajúcimi svalovými krampami. Elektrofyziologickým vyšetrením bola potvrdená porucha neuromuskulárneho prevodu, okulárne a bulbárne príznaky ustúpili po podaní inhibítorov acetylcholínesterázy. Genetickým vyšetrením bola potvrdená porucha typická pre Kennedyho chorobu. Autori stav uzatvorili ako prvý popis koincidencie okulárnej formy myasténie gravis a Kennedyho choroby. Koincidenciu séropozitívnej myasténie gravis a Kennedyho choroby u jedného pacienta popísali neskôr Jokela et al [14]. V prípade nášho pacienta sa domnievame, že vzhľadom ku terapeutickej odozve a dlhodobému obdobiu remisie klinických príznakov po imunosupresívnej liečbe sa jedná o tretí popísaný prípad koincidencie myasténie gravis a Kennedyho choroby.
Kennedyho spinálna a bulbárna atrofia je najčastejšou formou spinálnej muskulárnej atrofie so vznikom v dospelom veku. Typický vek začiatku jej príznakov je 40 rokov, vyskytuje sa u mužov a začína najčastejšie slabosťou, fascikuláciami a krampami na DK, predilekčne v pletencoch. Klinické symptómy však môžu byť variabilné, na čo sme poukázali i v našej práci. Správna diagnóza pomocou genetickej diagnostiky je v dnešnej dobe dôležitá najmä pri odlíšení iných amyotrofií s výrazne horšou prognózou, nakoľko účinnú liečbu Kennedyho choroby dnes nemáme k dispozícii. Klinické štúdie s leuprorelínom (release hormónom luteinizujúceho hormónu) a dutasteridom (inhibítorom 5‑alfa‑reduktázy) nepriniesli očakávaný efekt. Ako perspektívna sa ukazuje cesta farmakologickej aktivácie obranných mechanizmov bunky – molekulárnych chaperónov, ubikvitín-proteazomóvého systému a autofágie [15].
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Prijato k recenzii: 9. 2. 2015
Prijato do tlače: 28. 4. 2015
MUDr. František Cibulčík, CSc.
Neurologická klinika
LF SZU UN Bratislava Ružinov
Ružinovská 6
826 06 Bratislava
e-mail: cibulcik@hotmail.com
Zdroje
1. Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset: a sex‑linked recessive trait. Neurology 1968; 18(7): 671– 680.
2. LaSpada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X‑linked spinal and bulbar mucular atrophy. Nature 1991; 352(6330): 77– 79.
3. Mayer M, Kojecký Z, Urbánek K, Bartoušek J, Vlachová I. Kennedyho choroba – méně obvyklá forma spinální svalové atrofie. Cesk Slov Neurol N 1995; 58/ 91(4): 180– 183.
4. Atsuta N, Watanabe H, Ito M, Banno H, Suzuki K, Katsuno M et al. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain 2006; 129(6): 1446– 1455.
5. Rhodes LE, Freeman BK, Auh S, Kokkinis AD, La Pean A,Chen Ch et al. Clinical features of spinal and bulbar muscular atrophy. Brain 2009; 132(12): 3242– 3251. doi: 10.1093/ brain/ awp258.
6. Fratta P, Nirmalananthan N, Masset L, Skorupinska I, Collins T, Cortese A et al. Correlation of clinical and molecular features in spinal bulbar muscular atrophy. Neurology 2014; 82(23): 2077– 2084. doi: 10.1212/ WNL.0000000000000507.
7. Guidetti D, Sabadini R, Ferlini A, Torrente I. Epidemiological survey of X‑linked bulbar and spinal muscular atrophy, or Kennedy disease, in the province of Reggio Emilia, Italy. Eur J Epidemiol 2001; 17(6): 587– 591.
8. Lee JH, Shin JH, Park KP, Kim IJ, Kim JG, Lim JG et al. Phenotypic variability in Kennedyś disease: implications of the early diagnostic features. Acta Neurol Scand 2005; 112(1): 57– 63.
9. Sorarú G, D’Ascenzo C, Polo A, Palmieri A, Baggio L, Vergnani L et al. Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci 2008; 264(1– 2): 100– 105.
10. Yamada M, Inaba A, Shiojiri T. X‑linked spinal and bulbar muscular atrophy with myasthenic symptoms. J Neurol Sci 1997; 146(2): 183– 185.
11. Stevic Z, Peric S, Pavlovic S, Basta I, Lavrnic D. Myasthenic symptoms in a patient with Kennedy’s disease. Acta Neurol Belg 2014; 114(1): 71– 73.
12. Inoue K, Hemmi S, Miyashi M, Kutoku Y, Murakami T,Kuokawa K et al. Muscular fatigue and decremental response to repetitive nerve stimulation in X‑linked spinobulbar muscular atrophy. Eur J Neurol 2009; 16(1): 76– 80. doi: 10.1111/ j.1468‑ 1331.2008.02349.x.
13. Boz C, Kalay E, Sahin N, Velioglu S, Ozmenoglu M, Karaguzel A. Ocular myasthenia gravis associated with X-linked recessive spinal and bulbar muscular atrophy. J Clin Neuromuscul Dis 2004; 5(3): 115– 118.
14. Jokela M, Udd B, Päivärinta M. Double trouble: spinal muscular atrophy type II and seropositive myasthenia gravis in the same patient. Neuromusc Disord 2012; 22(2): 129– 130. doi: 10.1016/ j.nmd.2011.07.011.
15. Katsuno M, Tanaka F, Adachi H, Banno H, Suzuki K, Watanabe H et al. Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA). Prog Neurobiol 2012; 99(3): 246– 256. doi: 10.1016/ j.pneurobio.2012.05.007.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2015 Číslo 3
Nejčtenější v tomto čísle
- Addenbrookský kognitivní test – orientační normy pro českou populaci
- Míšní šok – od patofyziologie ke klinickým projevům
- Diagnostika epileptických záchvatů
- Vzduchová embolie mozku – kazuistika