
Význam zadněprovazcové symptomatiky v diferenciální diagnostice hereditárních ataxií
The Importance of Posterior Column Signs for Differential Diagnosis of Hereditary Ataxias
Hereditary spinocerebellar diseases have high inter‑ and intra‑ familiar variability in their onset, course and clinical manifestations. Therefore, recent scientific developments enabled and led to the current trend of verifying hereditary spinocerebellar ataxias at the molecular genetic level; detailed clinical and neurological analysis, on the basis of which these diseases were originally named and described, loses its importance. The goal of this research was to asctertain with posturographic testing whether posterior column involvement is so typical for Friedreich’s ataxia that it may lead to targeted DNA diagnosis of spinocerebellar degeneration. Autosomal dominant spinocerebellar ataxia type 2 and Friedreich’s ataxia are among the most common hereditary ataxias in the Czech Republic. Therefore, 17 patients with spinocerebellar ataxia type 2, 12 patients with Friedreich’s ataxia and 10 healthy controls were subjected to standard posturographic examination. There was no difference between patients with spinocerebellar ataxia type 2 and Friedreich’s ataxia in standing position with visual control but the examination clearly demonstrated a difference between patients and healthy controls (mediolateral deviation, anteroposterior deviation p < 0.01). Upright stance without visual control distinguished patients from healthy controls as well as patients with spinocerebellar ataxia type 2 and Friedreich’s ataxia (mediolateral deviation p < 0.01). Our results indicate that, after evaluation of family history and other symptoms, clinical examination focused on cerebellar afferents and, even more appropriately, posturographic examination can be used to direct the first phase of DNA diagnosis as an objective correlate of neurological findings.
Key words:
ataxia – cerebellar ataxia – Friedreich ataxia – sensory ataxia – spinocerebellar ataxia type 2
Autoři:
J. Schwabová 1,2; T. Malý 3; F. Zahálka 3; Z. Mušová 2,4; L. Apltová 2,4; V. Komárek 5; A. Zumrová 2,5
Působiště autorů:
Neurologická klinika 2. LF UK a FN v Motole, Praha
1; Centrum hereditárních ataxií, FN v Motole, Praha
2; Laboratoř sportovní motoriky FTVS UK v Praze
3; Ústav biologie a lékařské genetiky 2. LF UK a FN v Motole, Praha
4; Klinika dětské neurologie 2. LF UK a FN v Motole, Praha
5
Vyšlo v časopise:
Cesk Slov Neurol N 2013; 76/109(3): 336-342
Kategorie:
Původní práce
Souhrn
Vysoká variabilita nástupu obtíží, průběhu onemocnění i klinického obrazu je u dědičných spinocerebelárních chorob intra‑ i interfamiliárně běžná. Proto je v důsledku vědeckého pokroku současným trendem verifikace typu dědičných spinocerebelárních ataxií na molekulárněgenetické úrovni. Význam podrobného klinického neurologického vyšetření, na jehož základu byly původně choroby pod nejrůznějšími jmény popsány, ustupuje do pozadí. Cílem práce bylo na základě výsledků posturografického vyšetření verifikovat, zda postižení zadních provazců je pro Friedreichovu ataxii natolik typické, že může vést k zacílení DNA diagnostiky u spinocerebelárních degenerací. Mezi nejčastější hereditární ataxie v České republice patří autozomálně dominantní spinocerebelární ataxie typu 2 a ataxie Friedreichova. Klasickým posturografickým vyšetřením jsme proto vyšetřili 17 pacientů se spinocerebelární ataxií typu 2, 12 pacientů s Friedreichovou ataxií a 10 zdravých kontrol. Pomocí stoje se zrakovou kontrolou byli jasně odlišeni pacienti od zdravých kontrol (mediolaterální výchylka, předozadní výchylka p < 0,01), pacienty se spinocerebelární ataxií typu 2 a ataxií Friedreichovou toto vyšetření neodlišilo. Stoj bez zrakové kontroly odlišil jak pacienty od zdravých kontrol, tak jednoznačně i obě sledované skupiny mezi sebou (mediolaterální výchylka p < 0,01). Výsledky ukazují, že po zhodnocení rodinné anamnézy a dalších příznaků onemocnění lze u ataktických pacientů použít v nasměrování první fáze DNA diagnostiky nejen cílené klinické vyšetření zaměřené na aferentaci cerebela, ale vhodněji i vyšetření posturografické jako objektivní korelát neurologického nálezu.
Klíčová slova:
ataxie – cerebelární ataxie – Friedreichova ataxie – senzorická ataxie – spinocerebelární ataxie typu 2
Podporováno: GAUK 96909; IGA MZCR 100005- 4; MSM 002160864, VZ FNM MZO 0064203- 6505 a GACR P407/ 11/ P784-PRVOUK P38.
Úvod
Výraz „ataxie“ pochází z řečtiny, kde „αταξία“ znamená nepravidelnost, neuspořádanost. Ataxie vzniká v důsledku poruchy mozečku či jeho centripetálních a/ nebo centrifugálních drah. Z tohoto pohledu lze teoreticky dělit ataxii na cerebelární a na senzorickou (např. historicky typické postižení při tabes dorsalis) včetně vestibulární.
Klinický obraz pacientů s hereditární ataxií je však obvykle smíšený – má v různé variabilní míře složku jak cerebelární, tak senzorickou, což ztěžuje diferenciálnědiagnostický postup.
Příčiny cerebelární ataxie jsou buď dědičné, nebo získané. Získané příčiny lze většinou vyloučit běžně používanými vyšetřovacími metodami (MR, vyšetření likvoru, vyšetření protilátek proti neurotropním virům atd.), avšak oblast dědičných poruch je stále rozsáhlá a nepřehledná. Přitom pokroky v molekulárněgenetické diagnostice jsou pro pacienta a jeho rodinu stále větší šancí najít určitý typ kauzální mutace, a tím možností nejen prenatální diagnostiky, ale i určité predikce vývoje pacientova onemocnění.
Friedreichova ataxie (FRDA) je autozomálně recesivně dědičné onemocnění manifestující se v důsledku expanze repeatů (repeat, z angl. repaete = opakování, je část DNA, která se skládá z opakujících se sekvencí nukleotidů). U FRDA a SCA2 se jedná o trojici neukleotidů, tzv. triplet repeatů, kódujících aminokyselinu glutamin) v FXN (X25) genu na chromozomu 9 [1].V naprosté většině jde o expanzi GAA repeatů v oblasti intronu 1, pouze zhruba ve 2 % případů se jedná o tzv. compound heterozygoty, kdy na jedné alele dochází k expanzi GAA repeatů a na druhé k jinému typu mutace [2– 4]. Tato oblast DNA kóduje bílkovinu frataxin, která je zapojena do mitochondrálního metabolizmu železa [1,3]. U Friedreichovy ataxie dochází kromě redukce počtu Purkyňových buněk zvláště v oblasti horního vermis mozečku a počtu buněk v nucleus dentatus k progresivnímu úbytku buněk v gangliích zadních kořenů míšních a zadních rozích míšních, méně v jádrech hlavových nervů. Dále dochází k úbytku velkých myelinizovaných axonů periferních nervů se sekundární gliózou, atrofizaci zadních provazců míšních, ventrálního i laterálního spinocerebelárního traktu i traktu kortikospinálního [2,3,5]. Klinický obraz zahrnuje obvykle progresivní ataxii, dysartrii, spasticitu dolních končetin, v různé míře skoliózu a autonomní obtíže. Areflexie na dolních končetinách spolu s výbavným jevem Babinského bývá typická, postupně dochází k poruše vibračního čití a polohocitu. Přibližně dvě třetiny pacientů s FRDA mají kardiomyopatii, třetina diabetes mellitus [6]. V minulosti bylo toto onemocnění spojováno s dětským věkem, v současnosti patří i do diferenciální diagnostiky hereditárních ataxií dospělého věku [6]. Zhruba u 25 % na úrovni DNA verifikovaných pacientů je popisován atypický průběh s pozdějším nástupem prvních příznaků a zachovanou výbavností šlachosvalových reflexů [7]. V České republice bylo dosud diagnostikováno 47 pacientů s FRDA.
Autozomálně dominantně dědičná spinocerebelární atrofie typu 2 (SCA2) je způsobena expanzí CAG repeatů v ATXN2 genu [8,9]. Důsledkem je alterace genového produktu, ataxinu 2, jehož změněná funkce vede k zániku neuronů. Na změněný produkt jsou zejména citlivé Purkyňovy a granulární buňky mozečku, zatímco Golgiho a košíčkové buňky jsou relativně ušetřeny stejně jako neurony v nucleus dentatus a v ostatních mozečkových jádrech. Dále dochází k degeneraci dolních oliv, pontinních jader a pontocerebelárních vláken. Degenerace zadních provazců, méně spinocerebelárních drah, stejně jako úbytek buněk v substantia nigra byla popsána pouze u části pacientů [10– 12]. Klinický průběh je variabilní, zahrnuje progresivní cerebelární ataxii, nystagmus, včetně pomalých sakadovaných očních pohybů, u některých pacientů byla pozorována paréza pohledu nebo projevy parkinsonské. Šlachosvalové reflexy jsou zpočátku živé, později vyhasínají, pyramidové jevy bývají přítomny [13– 15]. V České republice bylo dosud diagnostikováno 42 pacientů s SCA2.
Klinický obraz obou chorob se tedy částečně překrývá, i když u ataxie Friedreichovy je statisticky podíl senzorické ataxie nepochybně výraznější.
Posturografie je elektrofyziologická metoda, která kvantifikuje a kvalitativně hodnotí posturální stabilitu na podkladě snímání středu tlakového působení objektu v ploše kontaktu (Centre Of Pressure, COP). Na rozdíl od klinického vyšetření včetně objektivizačních škál se jedná o metodu objektivní, tedy nezatíženou subjektivní (dez)interpretací, jejíž výsledky je možno dokumentovat graficky a především numericky, což umožňuje přesnější hodnocení poruch rovnováhy, porovnávání a archivaci výsledků [16– 18].
Literární údaje potvrzují u pacientů s ataxií větší výchylky COP ve stoji přímém [19,20], protože pacienti nemají schopnost na výchylky ve stoji přiměřeně reagovat a mají tendenci přestřelovat [21,22]. Potvrzen byl také význam testu Rombergova [18, 23,24].
Cílem našeho projektu bylo pomocí posturografického vyšetření zjistit, zda je při klinickém vyšetření pozitivní Rombergův test dostatečným kritériem, odlišujícím pacienty dvou nejčastějších typů spinocerebelárních ataxií v České republice (FRDA a SCA2) a zjistit senzitivitu klinického vyšetření oproti objektivní posturografické metodě.
Soubor a metodika
Celkem bylo vyšetřeno 17 pacientů s SCA2, 12 pacientů s FRDA a 10 zdravých kontrol. Diagnóza pacientů byla potvrzena analýzou DNA. Předpokladem studie byla schopnost pacientů vydržet ve stoji s/ bez zrakové kontroly bez nutnosti opory, další výběr byl náhodný tak, aby co nejvíce simuloval klinickou praxi.
Průměrný věk 11 mužů a 6 žen s SCA2 byl 43,8 let (18– 58), průměrný počátek klinických příznaků 10,8 let (1– 29). Sedm mužů a pět žen pacientů s FRDA mělo průměrný věk 31,9 let (19– 59), průměrný počátek jejich klinických příznaků byl 10,9 let (5– 27). Testovaní zdraví jedinci byli vybíráni tak, aby věkově, pohlavím a váhou odpovídali pacientům a zároveň neměli žádnou ortopedickou ani neurologickou anamnézu, jejich neurologický nález byl v normě. Klinický nález pacientů i zdravých kontrol byl hodnocen pomocí škály Cooperative Ataxia Rating Scale (ICARS) [25]. Průměrná hodnota ICARS u pacientů s FRDA byla 32,25, u pacientů s SCA2 24,61. Výsledek ICARS u zdravých jedinců byl 0.
Posturografické vyšetření probíhalo na multisenzorické plošině FootScan (RScan, Belgie) o velikosti 0,5 m × 0,4 m s 4 100 sen-zory s citlivostí 0,1 N/cm2 a snímkovací frekvencí 500 Hz. Snímací deska vyhodnocuje tlakové zatížení jednotlivých senzorů a následně vypočítává středy tlakového působení objektu v ploše kontaktu, tedy COP (Centre Of Pressure) pomocí softwaru FootScan Balance 7.7, což je specializovaný software pro měření a hodnocení posturální stability (obr. 1).

Jako objektivizační test byl vybrán standardizovaný stoj o široké bázi se zrakovou kontrolou, poté bez zrakové kontroly. Šíři stoje byla vymezena šířkou boků probanda, která byla po odměření antropometrem přenesena na podložku, a udala tak vzdálenost vnější strany chodidel. Mezi chodidla a tlakovou desku FOOTSCAN byla vložena průhledná fólie pro zakreslení polohy chodidel při stoji tak, aby byla zajištěna individuální shoda při opakovaném vyšetření. Stoj se měřil při otevřených očích podle standardizačních postupů v délce 30 s [26]. Hodnotícími parametry byly: celková dráha centra tlakového působení (TTW), rozsah centra tlakového působení v ose mediolaterální (DeltaX), rozsah centra tlakového působení v ose předozadní (DeltaY), směrodatná odchylka rychlosti centra tlakového působení po dobu měření (RychlostSD) [27]. Tyto parametry byly vybrány na základě naší předešlé práce, která ověřovala různé indikátory stability a jejich afinitu ke klinickým škálám [16]. Výsledky byly statisticky zpracovány. Signifikantní rozdíly mezi sledovanými proměnnými byly hodnoceny pomocí mnohonásobné analýzy rozptylu s fixovanými efekty s designem „double multivariate model“. Fixované efekty (typ skupiny – SCA2, FRDA a KONTROLA) a VIZUÁLNÍ KONTROLA (otevřené a zavřené oči) byly nezávislé proměnné. Indikátory posturální stability jako TTW, DeltaX, DeltaY a RychlostSD byly závislé proměnné.
Hodnotili jsme účinek jak hlavních faktorů (efektů) DIAGNÓZA a VIZUÁLNÍ KONTROLA, tak účinek jejich vzájemného působení – interakce mezi nimi (DIAGNÓZA versus VIZUÁLNÍ KONTROLA). V případě statistického významu hlavního faktoru jsme posuzovali interakce mezi parametry pomocí Bonferonniho post hoc testů. Zamítnutí nulové hypotézy jsme posuzovali s rizikem p < 0,05. Pro hodnocení věcné významnosti jsme použili koeficient η2(Eta square), který vysvětluje procento rozptylu hlavního faktoru. Statistická významnost byla vypočtena pomocí software IBM SPSS 20.0.
Autoři prohlašují, že studie na lidských subjektech popsaná v manuskriptu byla provedena v souladu s etickými standardy příslušné komise (institucionální a národní) odpovědné za provádění klinických studií a Helsinskou deklarací z roku 1975, revidovanou v r. 2000.
Výsledky
Mnohonásobná analýza rozptylu prokázala signifikantní účinek hlavního efektu (DIAGNÓZA) při sledování vybraných indikátorů posturální stability (F8,66 = 3,38, p < 0,01, η2 = 0,290). Pacienti s FRDA dosahovali v testu nižší hodnoty (44,1 mm, 5,1 %) TTW ve srovnání s pacienty s SCA2. I tak nebyl zjištěn signifikantní rozdíl ani mezi oběma skupinami, ani ve srovnání s kontrolní skupinou (tab. 1). Při porovnání výchylek DeltaX a DeltaY byly prokázány rozdíly mezi skupinami pacientů (FRDA, SCA2) a kontrolní skupinou (DeltaX: p < 0,05, DeltaY: p < 0,01) (obr. 2).
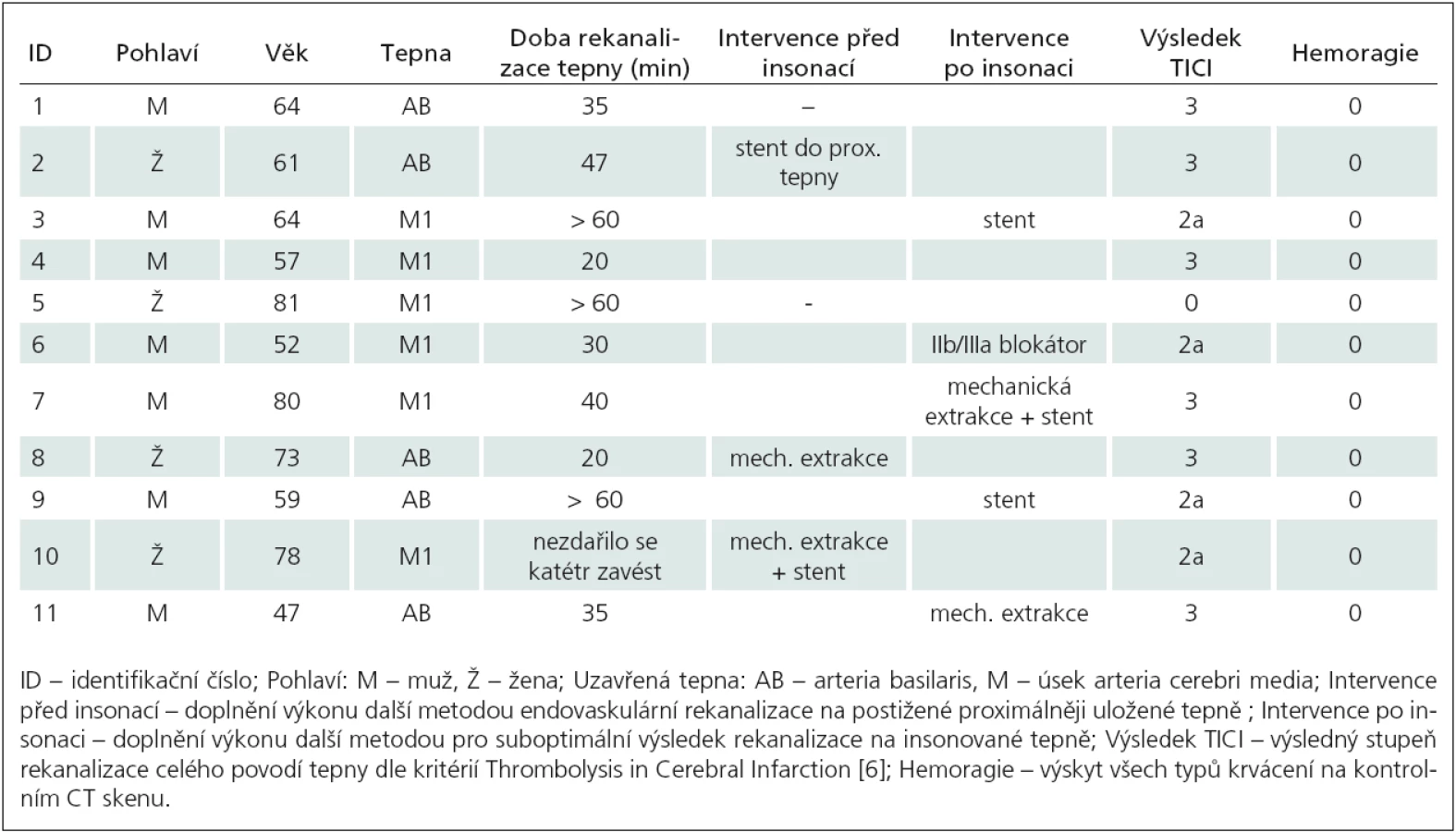
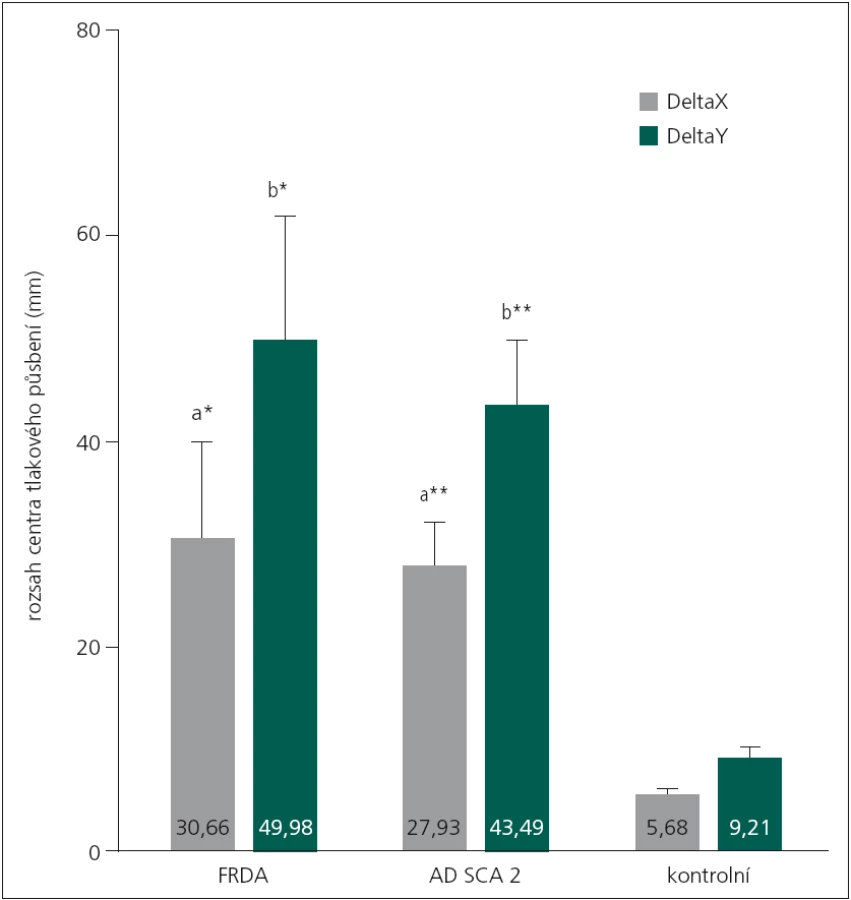
Avšak ani tyto parametry nedokázaly vzájemně odlišit pacienty FRDA od pacientů s SCA2. Nevýznamné rozdíly mezi skupinou FRDA vs SCA2 byly nalezeny také v parametru směrodatné odchylky rychlosti COP po dobu měření (p > 0,05).
Při zjišťování vnitroskupinovéhp efektu hlavního faktoru „VIZUÁLNÍ KONTROLA“ byl zjištěn signifikantní účinek na sledované parametry posturální stability (F4,32 = 4,55, p < 0,01). Při zjišťování vnitroskupinového efektu hlavního faktoru „VIZUÁLNÍ KONTROLA“ byl zjištěn signifikantní účinek na sledované parametry posturální stability (F4,32 = 4,55, p < 0,01, η2 = 0,363). Ve všech sledovaných indikátorech posturální stability byl efekt vizuální kontroly signifikantní (p < 0,01) (tab. 2). V rámci sledovaných indikátorů měl tento faktor minimální podíl na společné varianci při výchylce DeltaX (20,1 %) a maximální podíl na výchylce DeltaY (34,2 %).

Analýza interakčních efektů obou hlavních faktorů (DIAGNÓZA, VIZUÁLNÍ KONTROLA) prokázala signifikantní efekt (F8,66 = 2,94, p < 0,01, η2 = 0,262). Jednoduchá analýza rozptylu prokázala signifikantní rozdíl interakce u všech sledovaných parametrů.
Bonferroniho post hoc analýza testů prokázala u pacientů signifikantní rozdíly u sledovaných parametrů, mimo parametr DeltaX ve skupině pacientů SCA2 (tab. 3). Na základě tohoto rozdílu v motorickém projevu (rozsah centra tlakového působení v mediolaterální ose) mezi skupinou FRDA a SCA2 jsme identifikovali signifikantní rozdíl, který může vypovídat o typologickém motorickém projevu neurologického pacienta. V kontrolní skupině jsme nezjistili signifikantní účinek efektu interakce ani v jednom ze sledovaných parametrů.

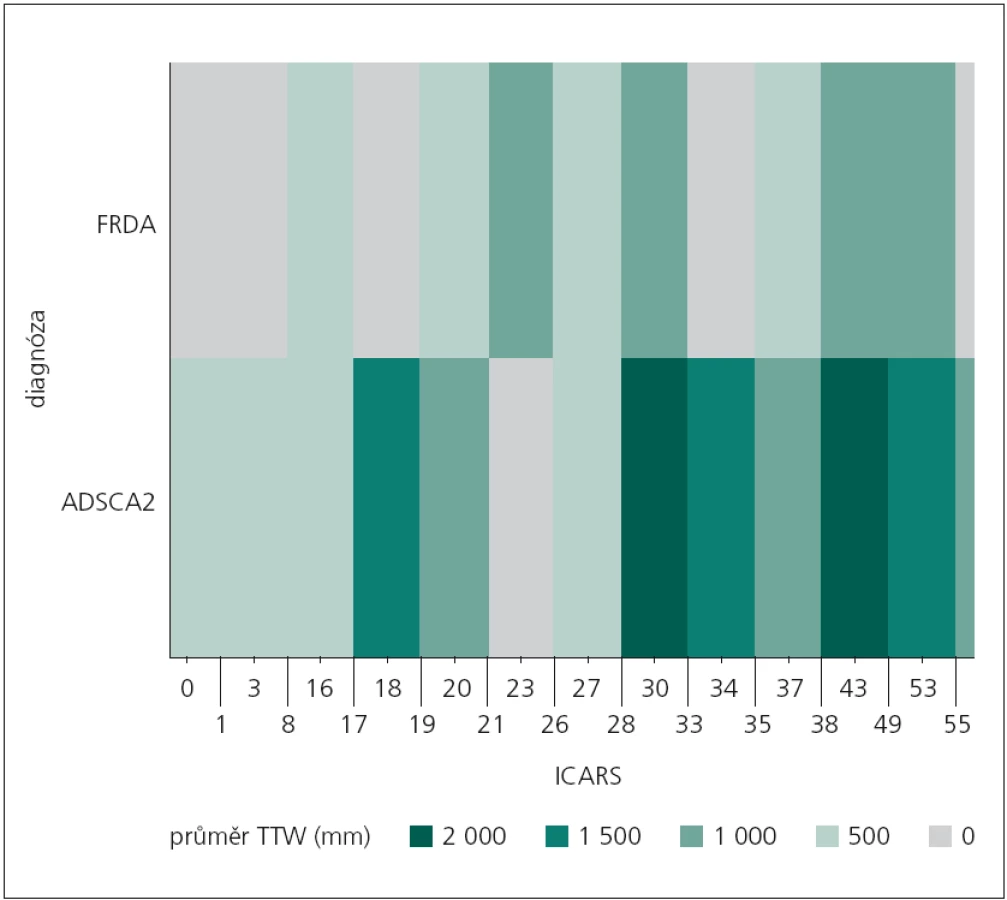
V případě stoje s vizuální kontrolou byl pomocí „teplotní mapy“ zobrazen výskyt úrovně průměrné hodnoty celkové dráhy (TTW) vzhledem k dosaženému výsledku v klinické škále ICARS u obou neurologických skupin (obr. 3). Z předešlé analýzy nebyly zjištěny signifikantní rozdíly mezi sledovanými skupinami. V uvedeném případě se ukazuje, že možný inflexní bod získaného skóre na škále ICARS vzhledem k diferenciální diagnostice z pohledu hodnocení celkové dráhy bude 28. Až do dosažení tohoto bodu (stupeň postižení) se obě skupiny z pohledu celkové dráhy projevují srovnatelně (obr. 3).
Diskuze
Stoj přímý se zrakovou kontrolou nebyl po kvantitativním zhodnocení sledovaných posturografických parametrů prokázán jako test dostatečně senzitivní k odlišení pacientů s FRDA od SCA2, ale jasně odlišil pacienty od zdravých kontrol (mediolaterální výchylka, předozadní výchylka p < 0,01; směrodatná odchylka rychlosti p < 0,05), což je v souladu s literaturou [19,20]. Je známo, že pro pacienty s postižením předního laloku mozečku jsou typické větší výchylky v předozadním směru, kdežto u pacientů s postižením vestibulocerebela, včetně pacientů s FRDA, jsou typické vysoké výchylky bez predominujícího směru, tedy i ve směru mediolaterálním [28].
Naše výsledky vyšetření ve stoji přímém s otevřenýma očima prokázaly větší výchylky jak v předozadní, tak mediolaterální ose u obou skupin pacientů (p < 0,01). Nález může tedy anatomicky odpovídat skupině pacientů s difuzním mozečkovým postižením nebo s postižením předního laloku mozečku, obojí v kombinaci s poruchou aferentních spinálních drah. Tato skutečnost je v souladu se znalostmi o patofyziologii vyšetřovaných chorob, jak bylo v úvodu popsáno. Dle výše uvedeného a také dle pozorování Diener et al [28] by se dalo dále předpokládat, že pacienti s SCA2 budou dosahovat větších výchylek v předozadním směru, kdežto pacienti s FRDA budou dosahovat větších výchylek jak v předozadním směru, tak ve směru mediolaterálním.
Na základě námi pozorovaného však nelze na analýzu výchylek, zejména na poměr mezi předozadními a mediolaterálními výchylkami, zcela spoléhat, i když Diener et al [28] prezentují tyto parametry jako užitečné pro diferenciaci ataxie.
Jiným zajímavým aspektem, který vyplynul z výsledků, je, že ve stoji přímém s otevřenýma očima nemají pacienti s FRDA horší výsledky než pacienti s SCA2, i když dosahují horší hodnoty v klinických hodnotících škálách (ICARS: FRDA 32,5; SCA2 24,61; kontrola 0). Stoj samotný je totiž ovlivněn poruchou propriocepce daleko více než postižením mozečkovým. Diskrepanci mezi laboratorním vyšetřením a vyšetřením klinickou škálou si vysvětlujeme tím, že škála ICARS obsahuje některé položky vycházející ze situace bez zrakové kontroly. Pacienti s FRDA mohou proto ve srovnání se skupinou SCA2 dosahovat v těchto testech horších výsledků, i když subjektivně i objektivně vnímané motorické postižení v běžném životě (pod zrakovou kontrolou) není tak výrazné. Domníváme se proto, že klinické škály lze využít ke komparaci mezi pacienty s jedním onemocněním nebo s jedním typem ataxie, avšak srovnávat klinický stav pacientů s různými typy ataxie dle výše uvedených škál vhodné není.
Stoj přímý se zrakovou kontrolou nebyl dostatečně senzitivní k odlišení pacientů s FRDA od SCA2, avšak po vztažení průměrné hodnoty celkové dráhy k výsledku ve škále ICARS, lze od určitého bodu, tedy od určité míry neurologického postižení, pacienty FRDA od pacientů SCA2 odlišit. To znamená, že posturografie nemusí pouze nahrazovat klinické škály nebo působit jako jejich objektivnější korelát, ale naopak v kombinaci s informací, které dávají klinické testy, může přinášet informaci pro klinika novou a i takto napomáhat v diferenciální diagnostice například hereditárních ataxií. Při zavření očí ve stoji přímém došlo k odebrání důležitého informačního zdroje pro udržení rovnováhy, což jasně odlišilo nejen pacienty od zdravých kontrol, ale také pacienty s FRDA od pacientů s SCA2 (mediolaterální výchylka p < 0,01). Tím byl potvrzen předpoklad, že u pacientů s FRDA hraje na výsledném klinickém obrazu důležitou roli porucha senzorické aferentace, i když postižení této části nervové soustavy může být součástí neurologického obrazu i u SCA2, avšak patrně v daleko menší míře. Proto lze předpokládat, že u pacienta s progredující ataxií a s významnou instabilitou v Rombergově testu je nutné v diferenciální rozvaze hereditárních ataxií zvažovat na prvním místě ataxii Friedreichovu. V opačném případě je potřebné soustředit se spíše na diferenciální diagnostiku spinocerebelárních ataxií autozomálně dominantně dědičných, samozřejmě i s ohledem na rodinnou a osobní anamnézu. Avšak řada studií i naše osobní zkušenosti ukazují, že údaje o rodinné anamnéze bývají často němé i při dominantním přenosu onemocnění (anticipace u těchto repeatových chorob, absence údajů o rodině). Nelze zapomínat ani na široký věk manifestace nejenom AD SCA, ale i ataxie Friedreichovy – pacientce s nejpozdějším nástupem obtíží v České republice bylo 57 let [29].
Závěry
V diferenciální diagnostice a zacílení DNA analýzy pacientů se suspekcí na hereditární ataxii (po zhodnocení anamnézy, dalších symptomů pacienta a výsledků pomocných laboratorních vyšetření) lze pro rozlišení ataxie Friedreichovy od nejčastější autozomálně dominantní ataxie typu 2 použít cílené klinické vyšetření zaměřené na aferentaci cerebela.
Na základě výsledků studie doporučujeme objektivizovat klinický nález vyšetřením posturografickým, které vyšetřením stoje bez zrakové kontroly, tedy korelátem Rombergova testu, odliší jak pacienty od zdravých kontrol, tak jednoznačně i obě sledované skupiny mezi sebou (mediolaterální výchylka p < 0,01).
Kromě toho umožňuje objektivita a přesnost vyšetření velmi dobrou kontrolu průběhu onemocnění či efektivity fyzioterapie jako jediné zatím dostupné léčby.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 5. 3. 2012
Přijato do tisku: 5. 4. 2013
MUDr. Jaroslava Schwabová
Neurologická klinika
2. LF UK a FN v Motole
V úvalu 84
150 00 Praha
e-mail: schwabova@gmail.com
Zdroje
1. Campuzano V, Montermini L, Moltò MD, Pianese L,Cossée M, Cavalcanti F et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996; 271(5254): 1423– 1427.
2. Koeppen AH. Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci 2011; 303(1– 2): 1– 12.
3. Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J Neurol 2009; 256 (Suppl 1): 9– 17.
4. De Castro M, García‑ Planells J, Monrós E, Cañizares J,Vázquez‑ Manrique R, Vílchez JJ et al. Genotype and phenotype analysis of Friedreich’s ataxia compound heterozygous patients. Hum Genet 2000; 106(1): 86– 92.
5. Della Nave R, Ginestroni A, Tessa C, Salvatore E, Bartolomei I, Salvi F et al. Brain white matter tracts degeneration in Friedreich ataxia. An in vivo MRI study using tract‑based spatial statistics and voxel‑based morphometry. Neuroimage 2008; 40(1): 19– 25.
6. Pandolfo M. Friedreich ataxia: the clinical picture. J Neurol 2009; 256 (Suppl 1): 3– 8.
7. Diehl B, Lee MS, Reid JR, Nielsen CD, Natowicz MR. Atypical, perhaps under‑ recognized? An unusual phenotype of Friedreich ataxia. Neurogenetics 2010; 11(2): 261– 265.
8. Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes‑ Cendes I et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996; 14(3): 269– 276.
9. Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 1996; 14(3): 277– 284.
10. Rüb U, Del Turco D, Del Tredici K, de Vos RA, Brunt ER, Reifenberger G et al. Thalamic involvement in a spinocerebellar ataxia type 2 (SCA2) and a spinocerebellar ataxia type 3 (SCA3) patient, and its clinical relevance. Brain 2003; 126(Pt 10): 2257– 2272.
11. Rüb U, Del Turco D, Bürk K, Diaz GO, Auburger G, Mittelbronn M et al. Extended pathoanatomical studies point to a consistent affection of the thalamus in spinocerebellar ataxia type 2. Neuropathol Appl Neurobiol 2005; 31(2): 127– 140.
12. Rüb U, Bürk K, Schöls L, Brunt ER, de Vos RA, Diaz GO et al. Damage to the reticulotegmental nucleus of the pons in spinocerebellar ataxia type 1, 2, and 3. Neurology 2004; 63(7): 1258– 1263.
13. Orozco Diaz G, Nodarse Fleites A, Cordovés Sagaz R,Auburger G. Autosomal dominant cerebellar ataxia: clinical analysis of 263 patients from a homogeneous population in Holguin, Cuba. Neurology 1990; 40(9): 1369– 1375.
14. Engel KC, Anderson JH, Gomez CM, Soechting JF. Deficits in ocular and manual tracking due to episodic ataxia type 2. Mov Disord 2004; 19(7): 778– 787.
15. Cancel G, Dürr A, Didierjean O, Imbert G, Bürk K, Lezin A et al. Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families. Hum Mol Genet 1997; 6(5): 709– 715.
16. Schwabová J, Zahálka F, Komárek V, Malý T, Hráský P, Gryc T et al. Validita mezinárodní škály pro pacienty s ataxií – scale for the assessment and rating of ataxia. Cesk Slov Neurol N 2010; 73/ 106(1): 689– 693.
17. Schwabová J, Zahálka F, Komárek V et al. Activities of daily living scale – the tool for clinical state monitoring of spinocerebellar ataxia and Friedreich ataxia patients [online]. Archives: The International Journal of Medicine 2009. Available from: http:/ / www.thefreelibrary.com/ Activities+of+daily+living+scale‑ – the+tool+for+clinical+state...– a0216632324.
18. Schwabova J, Zahalka F, Komarek V, Maly T,Hrasky P, Gryc T et al. Uses of the postural stability test for differential diagnosis of hereditary ataxias. J Neurol Sci 2012; 316(1– 2): 79– 85.
19. Mauritz KH, Dichgans J, Hufschmidt A. Quantitative analysis of stance in late cortical cerebellar atrophy of the anterior lobe and other forms of cerebellar ataxia. Brain 1979; 102(3): 461– 482.
20. Diener HC, Dichgans J. Pathophysiology of cerebellar ataxia. Mov Disord 1992; 7(2): 95– 109.
21. Diener HC, Dichgans J, Bacher M, Guschlbauer B. Characteristic alterations of long‑loop „reflexes“ in patients with Friedreich’s disease and late atrophy of the cerebellar anterior lobe. J Neurol Neurosurg Psychiatry 1984; 47(7): 679– 685.
22. Horak FB, Diener HC. Cerebellar control of postural scaling and central set in stance. J Neurophysiol 1994; 72(2): 479– 493.
23. Jansen EC, Larsen RE, Olesen MB. Quantitative Romberg’s test. Measurement and computer calculation of postural stability. Acta Neurol Scand 1982; 66(1): 93– 99.
24. Bastian AJ. Mechanisms of ataxia. Phys Ther 1997; 77(6): 672– 675.
25. Trouillas P, Takayanagi T, Hallett M, Currier RD, Subramony SH, Wessel K et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neuropharmacology Committee of the World Federation of Neurology. J Neurol Sci 1997; 145(2): 205– 211.
26. Kapteyn TS, Bles W, Njiokiktjien CJ, Kodde L, Massen CH, Mol JM. Standardization in platform stabilometry being a part of posturography. Agressologie 1983; 24(7): 321– 326.
27. Winter DA. Human balance and posture control during standing and walking. Gait Posture 1995; 3(4): 193– 214.
28. Diener HC, Dichgans J, Bacher M, Gompf B. Quantification of postural sway in normals and patients with cerebellar diseases. Electroencephalogr Clin Neurophysiol 1984; 57(2): 134– 142.
29. Zumrova A, Mazanec R, Vyhnalek M, Krepelova A, Musova Z, Krilova S et al. Concomitancy of mutation in FRDA gene and FMR1 premutation in 58 year‑ old woman. Neuro Endocrinol Lett 2005; 26(1): 71– 74.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2013 Číslo 3
Nejčtenější v tomto čísle
- Mechanizmy spasticity a její hodnocení
- Lidské prionové nemoci v České republice – 10 let zkušeností s diagnostikou
- Myozitida s inkluzními tělísky se slabostí šíjových svalů a pozitivním efektem imunoglobulinu – kazuistika
- Extrakraniálně metastazující meningeomy