
Lidské prionové nemoci v České republice – 10 let zkušeností s diagnostikou
Human Prion Diseases in the Czech Republic – 10 Years of Experience with the Diagnosis
Human prion diseases are rare neurodegenerative diseases. Since 2001, their diagnosis in the Czech Republic is performed at the National Reference Laboratory for human TSE/ CJD, a part of the Department of Pathology and Molecular Medicine, Thomayer Hospital in Prague. The most common is the sporadic form of Creutzfeldt‑ Jakob disease with 111 diagnosed definitive cases during 2002–2012. In addition, an increasing number of genetic forms is being diagnosed, with 24 confirmed cases during 2002–2012. Three cases of Gerstmann‑Sträussler‑ Scheinker syndrome were confirmed in 2009 a 2012. No case of new variant CJD has been diagnosed in the Czech Republic yet. Since 2007, the laboratory also performs obligatory testing of cornea transplant donors. This article recapitulates the ten years of existence of the National Reference Laboratory and summarizes human prion disease incidence and diagnosis in the Czech Republic.
Key words:
prion diseases – Creutzfeldt-Jakob disease – neuropathology – diagnostics
Autoři:
Z. Rohan 1; E. Parobková 1; S. Johanidesová 2; F. Koukolík 1; R. Matěj 1; R. Rusina 2
Působiště autorů:
Thomayerova nemocnice, Praha
Národní referenční laboratoř lidských TSE/CJN při oddělení patologie a molekulární medicíny
1; Thomayerova nemocnice, Praha
Neurologické oddělení
2
Vyšlo v časopise:
Cesk Slov Neurol N 2013; 76/109(3): 300-306
Kategorie:
Přehledný referát
Souhrn
Lidská prionová onemocnění tvoří skupinu vzácných neurodegenerací. Od roku 2001 probíhá jejich diagnostika v České republice v Národní referenční laboratoři (NRL) lidských TSE/ CJN při Oddělení patologie a molekulární medicíny Thomayerovy nemocnice v Praze. Nejčastěji se v České republice vyskytuje sporadická forma Creutzfeldtovy‑ Jakobovy nemoci, jíž bylo v letech 2002– 2012 definitivně potvrzeno 111 případů. Vedle těchto případů je zjišťován i rostoucí podíl genetických forem lidských prionových nemocí, kterých bylo během let 2002– 2012 potvrzeno 24 případů. V roce 2009 a 2012 byly také potvrzeny tři případy Gerstmannova‑ Sträusslerova‑ Scheinkerova syndromu. Výskyt nové varianty Creutzfeldtovy‑ Jakobovy nemoci nebyl v České republice dosud zaznamenán. Od roku 2007 probíhá v rámci NRL i povinné testování vzorků CNS dárců rohovek. Předmětem sdělení je souhrn zkušeností z deseti let práce NRL a přehled situace výskytu a diagnostiky prionových nemocí v České republice.
Klíčová slova:
prionová onemocnění – Creutzfeldtova-Jakobova nemoc – neuropatologie – diagnostika
Úvod
Prionová onemocnění neboli transmisivní spongiformní encefalopatie (TSE) jsou vzácná neurodegenerativní onemocnění způsobená ukládáním patologicky změněného („infekčního“) prionového proteinu (PrPSc) do mozkové tkáně s postupujícím zánikem neuronů a nezvratným poškozením mozku [1,2]. Genetické formy TSE jsou způsobeny patogenní variací (mutací) v PRNP genu uloženém na krátkém rameni 20. chromozomu [3].
Diagnostika lidských prionových onemocnění je založena na kombinaci klinického obrazu a nálezu pomocných vyšetření (EEG, likvor a MR) [4,5]. Definitivní potvrzení vyžaduje komplexní neuropatologické vyšetření s průkazem patogenního prionu v mozkové tkáni bioptického vzorku či vzorku mozkové tkáně získané post mortem.
V roce 2001 byla založena Národní referenční laboratoř pro lidská prionová onemocnění v Thomayerově nemocnici v Praze. Klinická část při Neurologickém oddělení poskytuje možnost klinického potvrzení diagnózy při podezření na prionové onemocnění, podporu rodinným příslušníkům a možnosti genetického skríningu asymptomatických rizikových jedinců. Neuropatologická část při Oddělení patologie a molekulární medicíny disponuje neurohistologickými, molekulárně-genetickými, imunologickými a imunohistochemickými metodami nejen pro diagnostiku prionových chorob, ale i pro široké diferenciálně-diagnostické pole neurodegenerativních onemocnění na současné evropské úrovni. Od roku 2007 jsou rutinně vyšetřováni dárci rohovek k vyloučení TSE. Kontinuálně probíhá získávání epidemiologických dat o výskytu různých forem prionových onemocnění v České republice.
Sporadická Creutzfeldtova- Jakobova nemoc
V roce 2010 byla aktualizována diagnostická kritéria sCJN [4]. Klíčovou změnou oproti předchozím kritériím je doplnění MR nálezu mezi laboratorní markery onemocnění. Kritéria rozlišují možnou, pravděpodobnou a jistou (potvrzenou) diagnózu.
Klinická diagnóza („možná sCJN“) se opírá o rychle se vyvíjející demenci (s trváním nepřesahujícím dva roky), k níž se připojují nejméně dva ze čtyř z následujících klinických projevů: myoklonus; mozečkové a zrakově prostorové postižení; pyramidové a extrapyramidové projevy; akinetický mutizmus. „Pravděpodobná sCJN“ splňuje definici klinického případu a má pozitivní nález alespoň v jednom pomocném vyšetření (EEG, protein 14‑3‑3 v mozkomíšním moku, MR nález). Diagnostickou jistotu („potvrzená sCJN“) poskytne neuropatologické vyšetření mozkové tkáně [4,6].
Ačkoliv je demence základním projevem CJN, v literatuře nejsou (s výjimkou jedné studie porovnávající vývoj u 10 případů vCJN s průřezovými daty od pacientů s histologicky prokázanou sCJN a gCJN s mutací v PRNP genu [7]) dostupné výsledky prospektivního sledování neuropsychologických aspektů u pacientů s CJN. Poznatky o kognitivních projevech lidských prionových onemocnění vycházejí pouze z publikovaných kazuistik a retrospektivních sdělení. Neuropsychologický profil u CJN je často málo specifický a má smíšené kortiko‑ subkortikální projevy [8,9] (kromě Heidenhainovy varianty, pro kterou je charakteristické postižení zrakově‑prostorových funkcí, výskyt vizuálních halucinací a korové slepoty). V oblasti mnestických funkcí se často objevuje porucha výbavnosti i vštípivosti, typický je také frontální syndrom. Vyšetření často komplikuje rychlá progrese onemocnění a afázie.
V rámci Národní referenční laboratoře probíhá od roku 2010 prospektivní sledování souboru pacientů s podezřením na prionové onemocnění. U případů histologicky potvrzené diagnózy (při autopsii) jsme následně zhodnotili neuropsychologický profil. Zatím jsou k dispozici kompletní data 14 pacientů s prokázanou CJN. U všech pacientů byl přítomen globální kognitivní deficit s převažujícími frontálními rysy, alterace exekutivních a fatických funkcí (dysartrie, globální afázie). Narušeny byly také ostatní kognitivní domény (intelektové, mnestické i vizuospaciální funkce, konstrukční apraxie). Typický byl bradypsychizmus, perseverace, grasping, utilizační chování, echopraxie. U většiny pacientů byly zjištěny neuropsychiatrické projevy (depresivita, apatie, iritabilita, anxieta, agresivita, vizuální halucinace, insomnie). Kognitivní profil u našich pacientů se tedy vyznačuje heterogenitou ve své manifestaci, s převažujícím postižením exekutivních a řečových funkcí s významným podílem behaviorálních projevů a výskytem vizuálních halucinací.
U rozvinuté sCJN zobrazí EEG generalizované trifázické nebo polyfázické vlny o délce 100– 300 ms, které se periodicky opakují v intervalech 0,5– 2 s, se senzitivitou 67 % a specificitou 86 %. Vyšetření se v ČR běžně provádí a s výhodou je lze opakovat v případě negativního nálezu a přetrvávání podezření na sCJN.
V NRL pro prionová onemocnění je rutinně stanovována přítomnost proteinu 14‑3‑3 v likvoru technikou western blot. Protein 14‑3‑3 je nespecifický marker neuronálního rozpadu, a tudíž může být detekován u řady parenchymových lézí mozku (např. hemoragie, tumory, rozsáhlejší ischemie), infekčních onemocnění CNS a také u Alzheimerovy nemoci [10].Stanovení 14‑3‑3 má praktický význam pouze v kontextu rychle progredující demence s neurologickými příznaky k potvrzení klinického podezření na sCJN. Naše zkušenosti poukazují na určitou rozporuplnost v přínosu tohoto parametru. Z 62 neuropatologicky vyšetřených pacientů byl 14‑3‑3 protein pozitivní u 36 případů (z toho pět bylo slabě pozitivních) definitivní CJN, u 17 případů definitivní CJN byl 14‑3‑3 negativní. U jiných neurodegenerací pak bylo pozitivních pět případů [11]. Během let 2002– 2012 byl 14‑3‑3 protein analyzován u 62 případů definitivní sCJN. Z toho bylo 40 případů pozitivních (65 %), dva případy slabě pozitivní (3 %) a 20 případů bylo negativních (32 %).
Častým nálezem v likvoru u sCJN je velmi vysoká hladina celkového a fosforylovaného tau proteinu (h‑ tau a p‑ tau) v likvoru. Zejména test hodnoty h‑ tau může být dokonce citlivější než test proteinu 14‑3‑3 a může zvyšovat diagnostickou výtěžnost likvorologické analýzy [12– 14]. V současné době však není součástí platných diagnostických kritérií. V NRL byly ante mortem hodnoty h‑ tau stanoveny u 22 pacientů s neuropatologicky potvrzenou sCJN, z toho 16 (73%) mělo hodnoty h‑ tau velmi zvýšené (nad 1 000 ng/ ml).
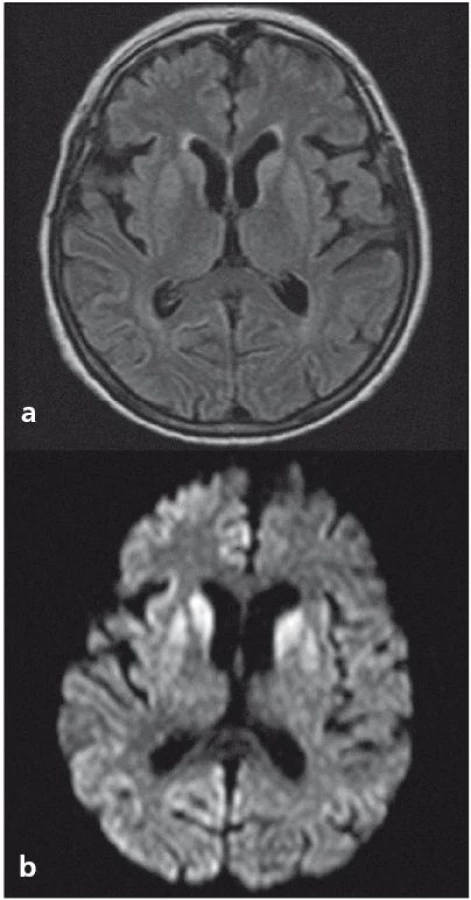
Z analýzy dat získaných v NRL vyplývá, že po aktualizaci diagnostických kritérií pro sCJN se v ČR častěji provádí a hodnotí MR nález u pacientů s podezřením na prionové onemocnění. Typickým nálezem (s 67% senzitivitou a až 93% specificitou [15,16]) bývají hypersignály v difuzním vážení a na FLAIR sekvencích v oblasti bazálních ganglií (typicky v putamen a ncl. caudatus) a korově, s převahou v inzule a frontálně (obr. 1a, b) [17– 19].
V letech 2002– 2012 bylo v NRL vyšetřeno přes 320 autoptických vzorků tkáně CNS zemřelých, u nichž bylo vysloveno podezření na prionové onemocnění na úrovni pravděpodobné či možné CJN, nebo byla CJN uvedena v diferenciálnědiagnostické rozvaze. Ve 138 případech byla neuropatologicky potvrzena definitivní diagnóza prionového onemocnění, nejčastěji sCJN s roční distribucí 1– 2 případy/ milion, což odpovídá přibližně odhadované incidenci sCJN v populaci (graf 1). Diferenciální diagnóza je poměrně široká. V ČR nejčastěji bývá sCJN zaměňována za atypicky rychle probíhající Alzheimerovu nemoc, behaviorální variantu frontotemporálních lobárních degenerací, méně často encefalitidy, tumory mozku, rovněž bylo zachyceno několik případů subkortikální vaskulární demence nebo mnohotné systémové atrofie napodobujících prionové onemocnění [20].

Velmi zajímavé byly případy kombinací několika různých neuropatologických jednotek u téhož pacienta, kdy jsme neuropatologicky diagnostikovali kombinaci Alzheimerovy nemoci s jinou neurodegenerací (např. s nemocí s Lewyho tělísky nebo s frontotemporální lobární degenerací) či atypicky probíhající Wernickeovou encefalopatií. Tyto nálezy potvrzují význam neuropatologické verifikace každého neurodegenerativního onemocnění, zejména při atypickém průběhu.
Nová varianta CJN
Nová varianta CJN (vCJN, též nvCJN, či Willova nemoc) popsaná ve Velké Británii v roce 1996 [21] má úzký vztah k bovinní spongiformní encefalopatii (BSE). Klinické příznaky se liší od klasické sCJN, v popředí je ataxie, poruchy čití a časné psychiatrické projevy. Postupně se rozvíjí demence. EEG u vCJN obvykle nachází jen nespecifický obraz pomalých vln (při velmi pomalé progresi onemocnění byly ojediněle popsány trifázické vlny [22]). Protein 14‑3‑3 v likvoru bývá negativní. U vCJN může MR prokázat až v 90 % případů symetrické hypersignály v zadní části thalamu v sekvencích FLAIR (tzv. pulvinar sign) [5,23]. Patologickou formu prionového proteinu lze u vCJN (na rozdíl od sCJN) prokázat v biopsii orgánů retikuloendotelového systému, rutinně biopsií patrové mandle [24– 26]. V České republice nebyl výskyt vCJN dosud nikdy zaznamenán.
Iatrogenní Creutzfeldtova‑Jakobova nemoc
Náhodně přenesená (iatrogenní) Creutzfeldtova‑ Jakobova nemoc může být způsobena transplantací tvrdé pleny, užitím růstového hormonu a gonadotropinu získaných z lidských hypofýz, transplantací rohovky a užitím nedostatečně sterilizovaných intracerebrálních elektrod. Ve světě bylo popsáno přes 400 případů iatrogenní CJN, s inkubací 12– 24 měsíců přenosu při invazivních neurochirurgických výkonech a až 30 let při přenosu kontaminovaným durálním štěpem. Inkubační doba se po podání lidského růstového hormonu pohybuje mezi 4 a 25 lety [27]. O riziku přenosu krevní transfuzí se uvažuje pouze u vCJN. Ve Velké Británii byly neuropatologicky potvrzeny tři případy úmrtí v souvislosti s vCJN u osob, které obdržely transfuzi od v době darování asymptomatických dárců, u nichž se poté vyvinula vCJN. Inkubační doba byla 6,5– 7,8 let a průběh onemocnění se nelišil od jiných případů vCJN [28].
Hrubý statistický odhad potenciálního rizika přenosu CJN při transplantacích je v České republice 3 promile (2 případy prionových chorob/ 100 000 lidí/ rok při 1 500 transplantacích) [29]. Naše laboratoř zastihla dva případy potenciálního přenosu CJN po biopsii mozku na neurochirurgickém pracovišti při nejasném neurologickém onemocnění, které jsme po úmrtí pacientky diagnostikovali jako sCJN. Retrospektivně jsme prokázali priony i v zapůjčeném bioptickém vzorku. Nástroje byly sterilizovány standardním způsobem, což priony neovlivňuje. Jeden ze dvou následně operovaných pacientů zemřel z jiného důvodu. Druhý je, pokud je nám známo, živ a v odstupu dvou let bez příznaků prionového onemocnění.
Dodržováním preventivních opatření při chirurgických zákrocích, označením biologicky nebezpečných vzorků, správným opatřením při čištění a dekontaminaci nástrojů by měl být iatrogenní přenos účinně omezen na nejnižší možnou míru.
Problematika mozkové biopsie u CJN
U pacientů s atypickou demencí a rychlým horšením bývá zvažována možnost mozkové biopsie k určení příčiny. V nedávné retrospektivní studii na 135 pacientech s progredujícím neurologickým postižením bez nálezu expanzivního procesu [30]byla senzitivita otevřené mozkové biopsie pouze 35 %, nejčastější zvažovaná předoperační diagnóza byla vaskulitida a nejčastějším bioptickým nálezem byla CJN a amyloidová angiopatie. Další terapeutický postup byl ovlivněn biopsií pouze v 8 % případů; a pouze u čtyř pacientů ze 100 měl bioptický nález vliv na další vývoj onemocnění. Lze tedy shrnout, že vzhledem k podobným zkušenostem a při riziku iatrogenního přenosu v případě prionového onemocnění zůstává biopsie mozku u rychle progredujících demencí s nálezem pouze atrofie na MR eticky velmi problematickým a medicínsky jen málo přínosným zákrokem. Měla by být proto indikována velmi uvážlivě, nikoliv k potvrzení předpokládané CJN, ale pouze pokud existuje jasná diagnostická hypotéza léčitelného onemocnění.
Genetická prionová onemocnění
Asi 10– 15 % lidských prionových onemocnění má genetický podklad a jsou způsobena mutacemi genu PRNP na 20. chromozomu [3]. Ty buď vedou k záměně jedné aminokyseliny za jinou (bodové mutace), nebo vedou ke změně v počtu oktapeptidových repetic na N‑ konci prionového proteinu (inzerce či delece) [3]. V genetice prionových onemocnění dále hrají významnou roli polymorfizmy kodonů 129 a 219 [3]. Na kodonu 129 může být kódována aminokyselina metionin (M) nebo valin (V). Největší vnímavost vůči prionovým onemocněním mají homozygoti (MM nebo VV). Předpokládá se, že v patogenezi prionových onemocnění je důležitým faktorem dimerizace až oligomerizace proteinu, ke které dochází mnohem snáze u homozygotů než u heterozygotů [31].
Přibližně 71 % pacientů se sporadickou formou CJN mají na 129. kodonu MM [32],nová varianta CJN má toto uspořádání ve 100 % [33,34]. V případech diagnostikovaných v ČR je průměrné rozložení polymorfizmů na kodonu 129 73,5 % MM, 16,9 % MV a 9,6 % VV (graf 2). Molekulárně-genetické vyšetření polymorfizmů PRNP na kodonu 129 provedené ante mortem není jednoznačně diagnostické, přesto může pomoci odhadnout míru rizika a další průběh CJN [32].

V některých geografických oblastech je výskyt genetických forem CJN (gCJN) nápadně vyšší, například na Slovensku na Oravě [35], v Izraeli u etnické skupiny Židů libyjského původu [36] nebo v Kalábrii a Kampánii v jižní Itálii [37,38].
Genetické formy Creutzfeldtovy‑Jakobovy nemoci
Celkem je známo přibližně 20 mutací PRNP genu, z toho v ČR byly zaznamenány mutace E200K, D178N a R208H. Penetrance těchto mutací je vysoká a zvyšuje se s věkem, v některých populacích je po 85. roce života úplná [39], v jiných, například na Slovensku či v Itálii, dosahuje 60 % [35,37]. Analýzu hereditárních mutací PRNP genu je možné v ČR provést v NRL TSE/ CJN u osob s prokázanou rodinnou zátěží gCJN.
Mutace E200K je s 19 zaznamenanými případy v České republice nejrozšířenější. Klinicky i neuropatologicky se onemocnění podobá sCJN, většina případů má vazby na rodiny v socioekonomických clusterech na Slovensku.
Mutace D178N byla nalezena ve čtyřech případech. Na klinický fenotyp má v tomto případě vliv polymorfizmus na 129. kodonu. Mutace D178N a přítomnost valinu na kodonu 129 způsobí gCJN. Je‑li na kodonu 129 metionin, rozvine se fatální familiární insomnie (FFI) [3,40].
Mutace R208H je extrémně vzácná [41,42], klinická prezentace bývá podobná sCJN. V České republice byla nedávno zjištěna tato mutace u pacientky s atypickým průběhem prionového onemocnění, které v počáteční fázi nápadně připomínalo progresivní supranukleární obrnu a až v pozdním stadiu se objevily klinické a laboratorní markery evokující Creutzfeldtovu‑ Jakobovu nemoc (EEG, 14‑3‑3 i MR nález) [43]. Tento atypický průběh i negativní rodinná anamnéza ilustrují diferenciálně diagnostickou obtížnost v kontextu prionových onemocnění.
Gerstmannova-Sträusslerova-Scheinkerova nemoc (GSS)
GSS je velmi vzácné autozomálně dominantní onemocnění podmíněné několika druhy mutací PRNP, popsané prvně v roce 1936. Vyskytuje se spíše v mladším věku, od 4. decenia, průběh je pomalejší než u sCJN a trvá v průměru 49– 57 měsíců.
Klasický klinický obraz probíhá pod obrazem ataxie s poruchami chůze, časté bývají bolestivé dysestezie a parestezie, demence je zpravidla pozdním projevem [44].Mohou se vyskytovat parkinsonské projevy a spasticita na dolních končetinách. Má poměrně charakteristický neurohistologický obraz zejména s přítomností tvarově i tinkčně typických amyloidových plak, které se objevují i v bílé hmotě. Nejčastější (80 % případů GSS) a zároveň první rozpoznanou mutací je P102L, která byla rovněž zjištěna u původní rodiny popsané Gerstmannem. Další mutace jsou mnohem vzácnější, s rozmanitým klinickým průběhem připomínajícím progresivní supranukleární obrnu (mutace A133V) [45] nebo sporadickou formu CJN s časnou demencí [46].
V České republice byla koncem 80. let minulého století zachycena rodina s tímto onemocněním, molekulárně-genetické vyšetření tehdy nebylo provedeno. Nedávno byl v ČR diagnostikován případ 44leté ženy s mutací P102L a velmi atypickým průběhem. Již v počátečních stadiích onemocnění se objevily poruchy chování a změny osobnosti, pak se přidaly parestezie s ataxií a prohloubila se demence, v pozdní fázi onemocnění se rozvinula spasticita s rigiditou a myoklonie. Nálezy pomocných vyšetření byly podobné jako u sCJN (přítomnost periodických trifázických vln na EEG a typické subkortikální signálové změny na MR) [44]. Do současnosti byly autopticky verifikovány tři případy GSS s průkazem patogenní variace P102L v PRNP, jeden pacient s molekulárněgeneticky potvrzenou GSS je stále živ.
Fatální familiární a sporadická insomnie
Příčinou fatální familiární insomnie je D178N mutace asociovaná s metioninem na pozici 129, existuje i velmi vzácná sporadická forma tohoto onemocnění. V klinickém obrazu převažuje nespavost a známky dysautonomie, později se objevuje ataxie, dysartrie a myoklonie; časté bývají pyramidové příznaky. V závěru onemocnění je insomnie úplná, doprovází ji demence, rigidita, dystonie a mutizmus [47,48]. V České republice nebyla žádná forma tohoto onemocnění dosud prokázána.
Neuropatologie prionových onemocnění
Definitivní diagnóza lidských prionových chorob je založena na neurohistologickém vyšetření mozkové tkáně doplněném metodami imunohistochemie a western blot [49]. Součástí vyšetřování je molekulárně-genetická analýza, kdy se sekvenuje gen PRNP a hledá se kauzální patogenní variace.
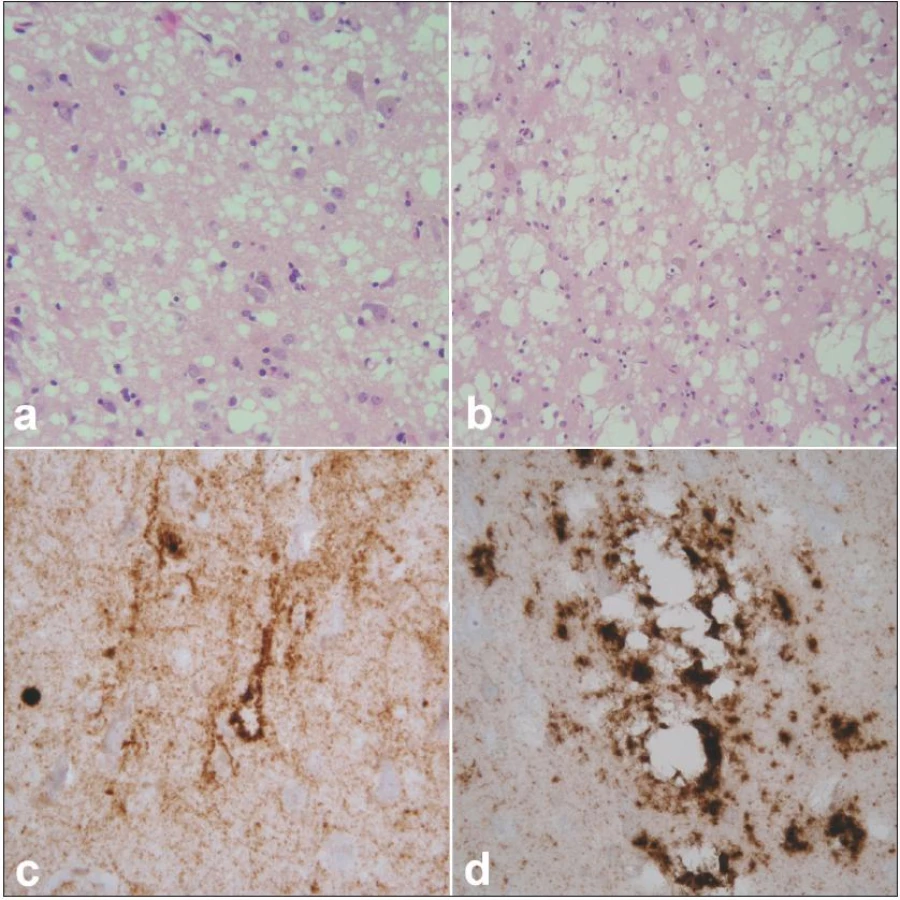
Klasickou neurohistologickou trojicí změn při prionových nemocích je spongiformní dystrofie, numerická atrofie neuronů a sekundární isomorfní astroglióza (obr. 2a, b). Spongiformní dystrofii charakterizují vakuoly v neuropilu, jejichž průměr je 2– 20 µm. Imunohistochemické vyšetření užívá různé typy protilátek ozřejmujících výskyt patogenních prionů ve tkáni (obr. 2c, d). Uvedené změny postihují zejména kortex, subkortikální šedou hmotu (téměř vždy je lze nalézt v caput nuclei caudati) a stratum moleculare mozečku, postižení je však v rámci jednotlivých podtypů variabilní. Podrobnější výčet změn charakteristických pro jednotlivé podtypy sCJN je však nad rámec tohoto textu. Posléze je rovněž biochemicky verifikována přítomnost a určen subtyp patologické formy prionového proteinu metodou western blot.
Novou variantu CJN charakterizuje velký počet „floridních“ plak v kůře mozku i mozečku a pokročilá spongiformní dystrofie, úbytek neuronů a glióza zejména v zadních jádrech thalamu a v kůře mozku a mozečku. V imunohistochemické reakci proti PrPSc jsou typická perineuronální a axonální depozita ve striatu a shluky depozit PrPSc, která lze dále prokázat v mezencefalu a v šedé hmotě kmene a prodloužené míchy. Specifický je rovněž biochemický profil patologického prionového proteinu [50].
Neuropatologické vyšetření a následná analýza PRNP genu za účelem vyhledání možné kauzální mutace jsou nezbytné u všech podezření na sporadickou Creutzfeldtovu‑ Jakobovu nemoc a u atypicky probíhajících neurodegenerací za účelem průkazu (nebo vyloučení) genetické příčiny onemocnění. Stanovení polymorfizmu PRNP genu není součástí doporučených rutinních vyšetřovacích postupů.
V České republice je autoptická verifikace všech podezření na prionové onemocnění povinná a podléhá hygienicko‑epidemiologické surveillance.
Správné rozpoznání prionového onemocnění má zásadní význam pro pacienta samotného i jeho rodinu. Při neexistenci kauzální terapie lze ušetřit pacienta zbytečných dalších pomocných vyšetření, mnohdy invazivních postupů a je možné začít se včasnou a cílenou paliativní péčí. V případě prokázaného dědičného onemocnění je pak genetické poradenství a podpora rodinných příslušníků věcí zásadního významu.
Laboratoř spolupracuje s referenčními laboratořemi prionových nemocí WHO v Edinburgu a ve Vídni. Všechna oddělení patologie ČR byla písemně informována o způsobu spolupráce v případě podezření na lidské prionové onemocnění.
Od 1. 1. 2007 začala NRL na základě doplňku transplantačního zákona povinně testovat mozkovou tkáň všech dárců rohovek na přítomnost patologické formy prionového proteinu metodou western blot. NRL spolupracuje se všemi očními bankami v České republice a od doby spuštění testování bylo vyšetřeno již přes 3 000 vzorků s negativním výsledkem s nálezem jednoho suspektního vzorku, který však byl po konzultaci s National CJD Research and Surveillance Unit v Edinburgu definitivně dovyšetřen jako negativní [29].
Autoři tímto děkují všem, kteří se na činnosti NRL v jejím průběhu podíleli či podílejí. V abecedním pořadí pak zejména MUDr. Pavlu Bočanovi, CSc., prim. MUDr. Vladimíru Gregorovi, CSc., Mgr. Ivetě Hnathové, doc. MUDr. Otakaru Kellerovi, CSc., RNDr. Miladě Matějčkové, Ing. Janě Novákové, CSc., MUDr. Jakubu Sikorovi, Ing. Magdaléně Smětákové a Ing. Aleně Srbové. Za zapůjčení snímků MR děkujeme prim. MUDr. Petru Lhotákovi z Radiologického oddělení Nemocnice České Budějovice, a.s.
Práce byla částečně podpořena granty IGA MZ ČR NT 12094-5/2011, IGA NT13543-4/2012 a IGA NT/14145/2013.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Radoslav Matěj, Ph.D.
Národní referenční laboratoř lidských TSE/CJN
Oddělení patologie a molekulární medicíny
Thomayerova nemocnice
Vídeňská 800
140 59 Praha 4
e-mail: radoslav.matej@ftn.cz
Přijato k recenzi: 27. 2. 2013
Přijato do tisku: 9. 4. 2013
Zdroje
1. Aguzzi A, Falsig J. Prion propagation, toxicity and degradation. Nat Neurosci2012; 15(7): 936– 939.
2. Pastore A, Zagari A. A structural overview of the vertebrate prion proteins. Prion2007; 1(3): 185– 197.
3. Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P. Genetic Creutzfeldt‑ Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 2011; 121(1): 21– 37.
4. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U et al.Updated clinical diagnostic criteria for sporadic Creutzfeldt‑ Jakob disease. Brain 2009; 132(Pt 10): 2659– 2668.
5. Heath CA, Cooper SA, Murray K, Lowman A, Henry C,MacLeod MA et al.Validation of diagnostic criteria for variant Creutzfeldt‑ Jakob disease. Ann Neurol 2010; 67(6): 761– 770.
6. WHO. WHO manual for surveillance of human transmissible spongiform encephalopathies including variant Cretzfeldt‑ Jakob disease. 2003. Available from: http:/ / whqlibdoc.who.int/ publications/ 2003/ 9241545887.pdf.
7. Cordery RJ, Alner K, Cipolotti L, Ron M, Kennedy A,Collinge J et al.The neuropsychology of variant CJD: a comparative study with inherited and sporadic forms of prion disease. J Neurol Neurosurg Psychiatry 2005; 76(3): 330– 336.
8. Snowden JS, Mann DM, Neary D. Distinct neuropsychological characteristics in Creutzfeldt‑ Jakob disease. J Neurol Neurosurg Psychiatry 2002; 73(6): 686– 694.
9. Alner K, Hyare H, Mead S, Rudge P, Wroe S, Rohrer JD et al. Distinct neuropsychological profiles correspond to distribution of cortical thinning in inherited prion disease caused by insertional mutation. J Neurol Neurosurg Psychiatry 2012; 83(1): 109– 114.
10. Steinacker P, Aitken A, Otto M. 14‑3‑3 Proteins in neurodegeneration. Semin Cell Dev Biol 2011; 22(7): 696– 704.
11. Matej R, Novakova J, Fiala J, Koukolik F, Rusina R. Vyšetřování proteinu 14‑3‑3 v mozkomíšním moku – klinicko‑patologická korelace. Cesk Slov Neurol N 2008; 71/ 104(6): 695– 699.
12. Hamlin C, Puoti G, Berri S, Sting E, Harris C, Cohen M et al.A comparison of tau and 14‑3‑3 protein in the diagnosis of Creutzfeldt‑ Jakob disease. Neurology 2012; 79(6): 547– 552.
13. Muayqil T, Gronseth G, Camicioli R. Evidence‑based guideline: diagnostic accuracy of CSF 14‑3‑3 protein in sporadic Creutzfeldt‑ Jakob disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology 2012; 79(14): 1499– 1506.
14. Stoeck K, Sanchez‑ Juan P, Gawinecka J, Green A,Ladogana A, Pocchiari M et al. Cerebrospinal fluid biomarker supported diagnosis of Creutzfeldt‑ Jakob disease and rapid dementias: a longitudinal multicentre study over 10 years. Brain 2012; 135(Pt 10): 3051– 3061.
15. Shiga Y, Miyazawa K, Sato S, Fukushima R, Shibuya S, Sato Y et al. Diffusion‑ weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt‑ Jakob disease. Neurology 2004; 63(3): 443– 449.
16. Young GS, Geschwind MD, Fischbein NJ, Martindale JL, Henry RG, Liu S et al. Diffusion‑ weighted and fluid‑ attenuated inversion recovery imaging in Creutzfeldt‑ Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol 2005; 26(6): 1551– 1562.
17. Wang LH, Bucelli RC, Patrick E, Rajderkar D, Alvarez Iii E, Lim MM et al. Role of magnetic resonance imaging, cerebrospinal fluid, and electroencephalogram in diagnosis of sporadic Creutzfeldt‑ Jakob disease. J Neurol 2013; 260(2): 498– 506.
18. Tschampa HJ, Zerr I, Urbach H. Radiological assessment of Creutzfeldt‑ Jakob disease. Eur Radiol 2007; 17(5): 1200– 1211.
19. Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres‑ Chae C et al. Diffusion‑ weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology 2011; 76(20): 1711– 1719.
20. Matej R, Rusina R, Koukolik F. 5 let činnosti Národní referenční laboratoře lidských prionových onemocnění při Oddělení patologie a molekulární medicíny FTNsP: naše zkušenosti a přehled literatury. Cesk Slov Neurol N 2007; 70/ 103(6): 637– 642.
21. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A et al. A new variant of Creutzfeldt‑ Jakob disease in the UK. Lancet1996; 347(9006): 921– 925.
22. Shinde A, Kunieda T, Kinoshita Y, Wate R, Nakano S, Ito H et al.The first Japanese patient with variant Creutzfeldt‑ Jakob disease (vCJD). Neuropathology 2009; 29(6): 713– 719.
23. Zeidler M, Sellar RJ, Collie DA, Knight R, Stewart G,Macleod MA et al.The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt‑ Jakob disease. Lancet 2000; 355(9213): 1412– 1418.
24. Hill AF, Butterworth RJ, Joiner S, Jackson G, Rossor MN, Thomas DJ et al. Investigation of variant Creutzfeldt‑ Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 1999; 353(9148): 183– 189.
25. Hill AF, Zeidler M, Ironside J, Collinge J. Diagnosis of new variant Creutzfeldt‑ Jakob disease by tonsil biopsy. Lancet 1997; 349(9045): 99– 100.
26. Bruce ME, McConnell I, Will RG, Ironside JW. Detection of variant Creutzfeldt‑ Jakob disease infectivity in extraneural tissues. Lancet 2001; 358(9277): 208– 209.
27. Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG et al. Iatrogenic Creutzfeldt‑ Jakob disease, final assessment. Emerg Infect Dis 2012; 18(6): 901– 907.
28. Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Three reported cases of variant Creutzfeldt‑ Jakob disease transmission following transfusion of labile blood components. Vox Sang 2006; 91(4): 348.
29. Jirsova K, Krabcova I, Novakova J, Hnathova I, Koukolik F, Kubesova B et al. The assessment of pathogenic prions in the brains of eye tissue donors: 2‑years experience in the Czech Republic. Cornea 2010; 29(9): 996– 999.
30. Schuette AJ, Taub JS, Hadjipanayis CG, Olson JJ. Open biopsy in patients with acute progressive neurologic decline and absence of mass lesion. Neurology 2010; 75(5): 419– 424.
31. Tahiri‑ Alaoui A, Gill AC, Disterer P, James W. Methionine 129 variant of human prion protein oligomerizes more rapidly than the valine 129 variant: implications for disease susceptibility to Creutzfeldt‑ Jakob disease. J Biol Chem 2004; 279(30): 31390– 31397.
32. Alperovitch A, Zerr I, Pocchiari M, Mitrova E, de Pedro Cuesta J, Hegyi I et al. Codon 129 prion protein genotype and sporadic Creutzfeldt‑ Jakob disease. Lancet1999; 353(9165): 1673– 1674.
33. Mackay GA, Knight RS, Ironside JW. The molecular epidemiology of variant CJD. Int J Mol Epidemiol Genet 2011; 2(3): 217– 227.
34. Zeidler M, Stewart G, Cousens SN, Estibeiro K, Will RG. Codon 129 genotype and new variant CJD. Lancet 1997; 350(9078): 668.
35. Mitrová E, Belay G. Creutzfeldt‑ Jakob disease with E200K mutation in Slovakia: characterization and development. Acta Virol 2002; 46(1): 31– 39.
36. Hsiao K, Meiner Z, Kahana E, Cass C, Kahana I, Avrahami D et al. Mutation of the prion protein in Libyan Jews with Creutzfeldt‑ Jakob disease. N Engl J Med 1991; 324(16): 1091– 1097.
37. D‘Alessandro M, Petraroli R, Ladogana A, Pocchiari M.High incidence of Creutzfeldt‑ Jakob disease in rural Calabria, Italy. Lancet 1998; 352(9145): 1989– 1990.
38. Ladogana A, Puopolo M, Poleggi A, Almonti S, Mellina V, Equestre M et al. High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology 2005; 64(9): 1592– 1597.
39. Chapman J, Ben‑ Israel J, Goldhammer Y, Korczyn AD. The risk of developing Creutzfeldt‑ Jakob disease in subjects with the PRNP gene codon 200 point mutation. Neurology 1994; 44(9): 1683– 1686.
40. Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P et al. Fatal familial insomnia and familial Creutzfeldt‑ Jakob disease: disease phenotype determined by a DNA polymorphism. Science 1992; 258(5083): 806– 808.
41. Mastrianni JA, Iannicola C, Myers RM, DeArmond S,Prusiner SB. Mutation of the prion protein gene at codon 208 in familial Creutzfeldt‑ Jakob disease. Neurology 1996; 47(5): 1305– 1312.
42. Capellari S, Cardone F, Notari S, Schinina ME, Maras B, Sita D et al.Creutzfeldt‑ Jakob disease associated with the R208H mutation in the prion protein gene. Neurology 2005; 64(5): 905– 907.
43. Matej R, Kovacs GG, Johanidesova S, Keller J, Matejckova M, Novakova J et al. Genetic Creutzfeldt‑ Jakob disease with R208H mutation presenting as progressive supranuclear palsy. Mov Disord 2012; 27(4): 476– 479.
44. Rusina R, Fiala J, Holada K, Matejckova M, Novakova J, Ampapa R et al.Gerstmann‑Sträussler‑ Scheinker syndrome with the P102L pathogenic mutation presenting as familial Creutzfeldt‑ Jakob disease: a case report and review of the literature. Neurocase 2013; 19(1): 41– 53.
45. Rowe DB, Lewis V, Needham M, Rodriguez M, Boyd A, McLean C et al. Novel prion protein gene mutation presenting with subacute PSP‑like syndrome. Neurology 2007; 68(11): 868– 870.
46. Majtenyi C, Brown P, Cervenakova L, Goldfarb LG, Tateishi J. A three‑ sister sibship of Gerstmann‑Sträussler‑ Scheinker disease with a CJD phenotype. Neurology 2000; 54(11): 2133– 2137.
47. Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A et al. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 1986; 315(16): 997– 1003.
48. Montagna P, Gambetti P, Cortelli P, Lugaresi E. Familial and sporadic fatal insomnia. Lancet Neurol 2003; 2(3): 167– 176.
49. Parchi P, de Boni L, Saverioni D, Cohen ML, Ferrer I,Gambetti P et al.Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter‑ rater study among surveillance centres in Europe and USA. Acta Neuropathol 2012; 124(4): 517– 529.
50. Ironside JW, Head MW. Neuropathology and molecular biology of variant Creutzfeldt‑ Jakob disease. Curr Top Microbiol Immunol 2004; 284: 133– 159.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2013 Číslo 3
Nejčtenější v tomto čísle
- Mechanizmy spasticity a její hodnocení
- Lidské prionové nemoci v České republice – 10 let zkušeností s diagnostikou
- Myozitida s inkluzními tělísky se slabostí šíjových svalů a pozitivním efektem imunoglobulinu – kazuistika
- Extrakraniálně metastazující meningeomy