
Late-onset Tay-Sachs Disease Can Mimic Spinal Muscular Atrophy Type III – Two Case Reports
Pozdní forma Tay-Sachsovy choroby napodobuje spinální svalovou atrofii III. typu – dvě kazuistiky
Pozdní forma Tay-Sachsovy choroby patří mezi GM2 gangliosidózy. První příznaky se objevují během dospívání nebo v časném dospělém věku. Uvádíme kazuistiky dvou sester ve věku 19 a 29 let s typickým klinickým obrazem a výsledky pomocných vyšetření. Počátečními příznaky byla porucha řeči a neobratnost, postupně se objevila progredující slabost pletencového svalstva dolních končetin. Elektromyografie prokazovala postižení na úrovni míšního motoneuronu. Molekulárně genetickým vyšetřením nebyla zjištěna delece SMN1 (survival of motor neuron) genu. Na magnetické rezonanci mozku se zobrazila výrazná mozečková atrofie. V séru, plazmě a leukocytech byl prokázán významný pokles aktivity β-hexosaminidázy A. DNA analýza HEXA genu identifikovala heterozygotní mutaci: c.805G>A and c.1123delG. U starší z obou sester trvalo stanovení správné diagnózy 12 let. Při klinických příznacích spinální svalové atrofie nebo spinocerebellární ataxie doporučujeme zvažovat v diferenciální diagnostice pozdní formu Tay-Sachsovy choroby. Správná diagnostika může mít význam z hlediska budoucích možností léčby.
Klíčová slova:
Tay-Sachs disease – hexosaminidase A deficiency – spinal muscular atrophy
Authors:
I. Prihodova 1; T. Kalincik 1; H. Poupětová 2; H. Jahnová 2; S. Nevsimalova 1
Authors place of work:
Department of Neurology and Centre for Clinical Neurosciences
1; Charles University, st Medical Faculty and General Teaching Hospital:
1; Institute of Inherited Metabolic Disorders
2
Published in the journal:
Cesk Slov Neurol N 2013; 76/109(2): 221-224
Category:
Kazuistika
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Summary
Late-onset Tay-Sachs disease is a form of GM2 gangliosidosis with the first manifestation in adolescence to young adulthood. We present clinical and paraclinical findings in two sisters (19 and 29 years). Speech impairment and clumsiness as the initial signs were followed by progressive proximal weakness in the lower extremities. Electromyography showed involvement of the lower motor neuron. Molecular genetic testing brought no evidence of deletion within the SMN1 (survival of motor neuron) gene. Magnetic resonance imaging of the brain revealed marked cerebellar atrophy. Marked decrease in β-hexosaminidase A activity was found in the serum, plasma and leukocytes. DNA analysis of the HEXA gene identified heterozygous mutations: c.805G>A and c.1123delG. It took 12 years to establish the diagnosis in the older sister. We recommend considering late-onset Tay-Sachs disease for the differential diagnosis of patients with clinical signs suggesting spinal muscular atrophy or spinocerebellar ataxia. Accurate diagnosis may be of prospective significance in terms of therapeutic management.
Key words:
Tay-Sachsova choroba – deficit hexosaminidázy A – spinální svalová atrofie
Introduction
GM2 gangliosidoses (Tay-Sachs disease, Sandhoff disease and GM2 activator deficiency) are autosomal recessive lysosomal storage disorders characterized by GM2 ganglioside accumulation within neurons [1]. Tay-Sachs disease is caused by mutations in the HEXA gene (chromosomal locus 15q23–q24) that codes the α-subunit of lysosomal enzyme β-hexosaminidase A. The most common infantile form is associated with absence of β-hexosaminidase A activity. It is characterized by rapid infaust course and manifests with early hypotonia, deteriorating psychomotor development, seizures and blindness. The late-onset chronic form first occurs in adolescence and adulthood, and takes a slower course with progressive proximal limb weakness and lower motor neuron signs in combination with cerebellar and occasionally also extrapyramidal signs [2]. The activity of β-hexosaminidase A is reduced to below 10% of the normal values. This form may mimic spinal muscular atrophy (SMA) or diseases arising from spinocerebellar degeneration [3]. In this report, we present the cases of two sisters, one of whom was followed for SMA (a non-deletion form) for 12 years.
Case reports
Patient 1
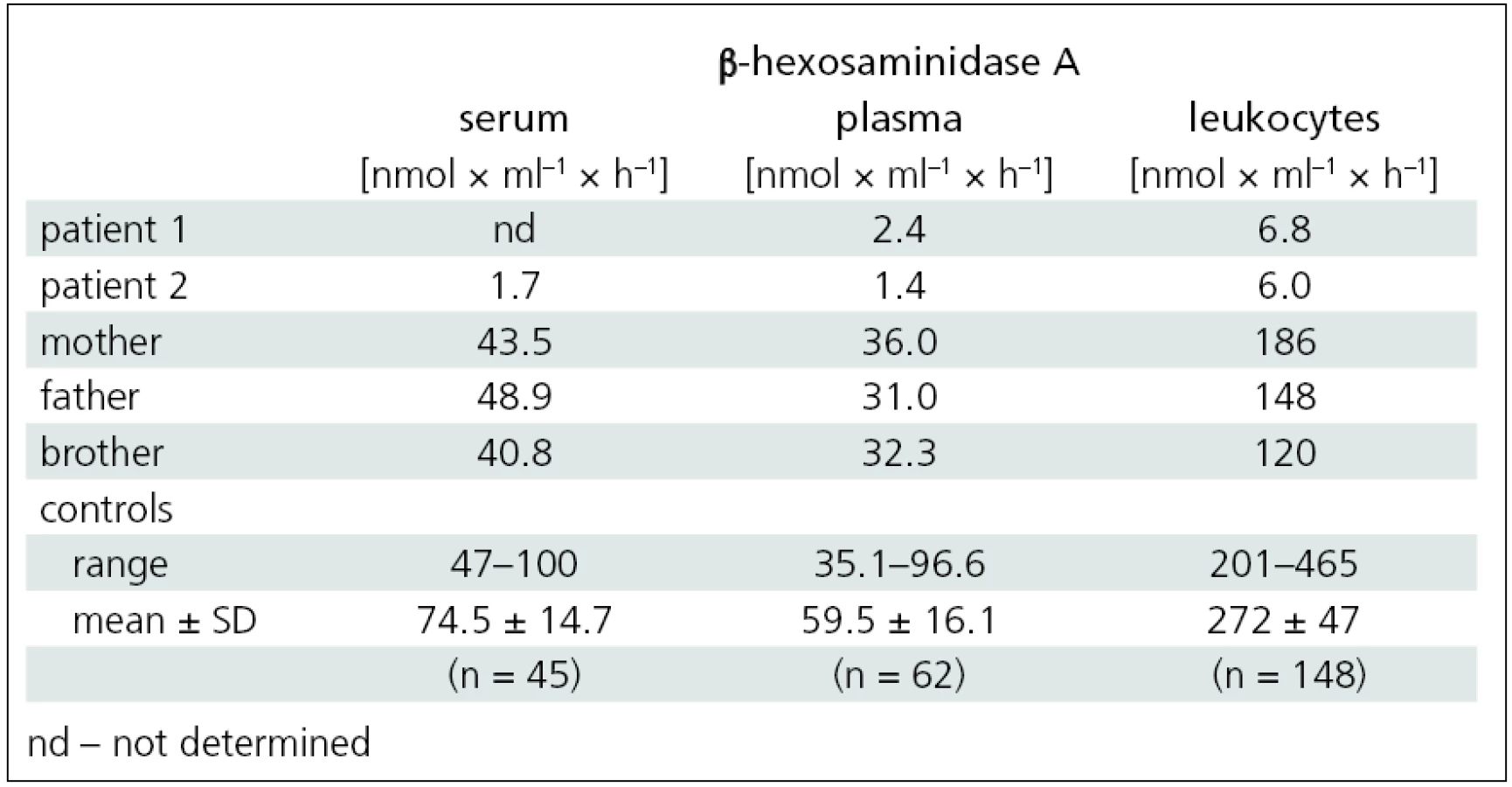
A 17-year-old girl was referred to the Department of Neurology of the General University Hospital for progressive weakness of her lower extremities. This was also observed in her older sister (patient 2). During her school years, the girl was followed up for Attention Deficit Hyperactivity Disorder (ADHD). From 10 years of age, she started to experience weakening of the lower limbs during exercise. At the age of 14, she developed spluttered speech with rapid speech rate, classified as tumultus sermonis, with no response to subsequent speech therapy. At the age of 16, she developed gait problems, especially when climbing stairs; on an even surface, she was able to walk a little over a kilometer. In addition, impaired fine motor skills impeded her hairdresser training. Neurological examination at our department revealed dysarthria, rapid speech rate, neo-cerebellar syndrome and proximal weakness of the lower limbs with normal reflexes. Full blood count, urea and creatinine, liver function tests, levels of muscle enzymes and myoglobin, electrophoresis of lipoproteins, serum lactate, vitamin E level and thyroid hormones were all normal. Electroencephalography (EEG) showed no abnormalities. Electromyography (EMG) revealed chronic diffuse regenerative lesion of the lower motor neuron, mainly in the lower extremities. Nerve conduction studies were normal. Needle EMG examination revealed signs of denervation, fasciculations, polyphasic motor unit potentials (MUP) with higher amplitude and longer duration. Cardiological and ophthalmological examinations were normal. Brain magnetic resonance imaging (MRI) showed symmetrical lamellar atrophy of the cerebellum and discrete atrophy of the mesencephalon (fig. 1). Psychological examination corroborated the diagnosis of ADHD, no discernible signs of depression or psychosis were present. Routine screening for inborn errors of metabolism was negative. Genetic testing for Friedreich’s ataxia, spinocerebellar ataxia – types 1 and 2, and for SMA yielded negative findings. The activity of β-hexosaminidase A in the plasma and leukocytes was significantly decreased and confirmed the diagnosis of Tay-Sachs disease (tab. 1).

Patient 2
In the older sister of patient 1, 29 years, the disease first manifested with clumsiness at the age of 10, followed by speech disturbance (also described as tumultus sermonis) at the age of 12. From 17 years of age, the girl was examined repeatedly for progressive weakness of the lower limbs. Her laboratory examinations were as follows: urea and electrolytes, muscle enzymes, myoglobin and thyroid hormone levels were within the normal ranges. EMG indicated disperse lesion in the anterior horns of the spinal cord. The clinical and EMG findings raised suspicion of a lower motor neuron disease. However, molecular genetic testing for SMA showed no deletion in the SMN1 (Survival of Motor Neuron) gene.
Disease progression then accelerated and, at the age of 26, she was able to walk a little over one kilometer unaided. At 28, she began using forearm crutches for support. Upon presentation at our department, she was able to walk about 200 meters unaided only, with frequent stumbles and falls. Neurological examination showed dysarthria, mild neocerebellar syndrome and peripheral quadruparesis, with a maximum in the proximal lower limbs (fig. 2). EMG indicated diffuse lesion of the lower motor neuron. Examination findings were the same as for her younger sister: nerve conduction studies were normal and needle EMG examination revealed signs of denervation, fasciculations, polyphasic MUPs with higher amplitude and longer duration. Brain MRI revealed marked atrophy of the cerebellum and brainstem (fig. 1). Ophthalmological examination was normal. Psychological testing revealed no cognitive impairment, intellectual skills were within the range of lower average, and there were no signs of depression or psychosis. The activity of β-hexosaminidase A in the serum, plasma and leukocytes was significantly decreased and confirmed the diagnosis of Tay-Sachs disease (tab. 1).

In both sisters, DNA analysis of the HEXA gene identified heterozygous mutations: c.805G>A and c.1123delG. The activity of β-hexosaminidase A in the serum, plasma and leukocytes of the family members (mother, father and brother) indicated the carrier status for Tay-Sachs disease (tab. 1).
Discussion
Tay-Sachs disease, a form of GM2 gangliosidosis, is a rare autosomal recessive lysosomal storage disease seen mainly in the Jewish population (especially Askhenazi). Its prevalence at birth in the Czech population is 1 per 289,345 live births [4]. In the Czech Republic, there are a total of 14 patients diagnosed with Tay-Sachs disease, thereof six with the late onset form. Information about the incidence of the late onset form is scarce worldwide. A study of the prevalence of lysosomal diseases in Portugal provided data on the prevalence of the juvenile form (30 cases, age 2–14 years) and of the chronic adult forms (9 cases, age 10–44 years) [5]. The prevalence in Portugal is higher than, for example, in the Netherlands where there are 31 patients [6], or in Australia with 21 diagnosed patients (proportions of particular forms have not been reported) [7]. Publications by Shapiro et al refer to cohorts of 30–44 patients [8,9].
The rapidly progressing, infaust infantile form first described independently by ophthalmologist Warren Tay and neurologist Bernard Sachs, is the most frequent [1]. In 1980ies, its late-onset variants, with subacute (juvenile) and chronic (adult) form, became known. The subacute form manifests in childhood with blindness, growing spasticity, rigidity, epilepsy and dementia, with progression to apallic state within 5 to 15 years [2,10]. The age at onset, course and prognosis of the chronic (adult) form is more variable. This variability may be apparent even among members of the same family [3]. The patients with the chronic form may present with combined impairment of the spinal motor neuron, cerebellum and, occasionally, the extrapyramidal system. After the age of 20, cerebellar signs (ataxia, dysmetria, tremor) develop. The combination of ataxia and muscle weakness results in impaired gait, though ambulation can remain preserved until the 5th or 6th decade.
Dysphagia, hyperreflexia and pyramidal signs may occur [2].
Ophthalmological examination in the late onset form of the disease shows no cherry-red spot in the fundus oculi, a sign typical for the infantile form. The spot results from retinal cell apoptosis, an event never seen in the late form of the disease because of the high residual enzyme activity.
The clinical course observed in our two patients can be considered as typical: inconspicuous signs (clumsiness, atypical gait, more frequent falls) were noticed throughout their childhood. Both developed speech disorders misclassified as spluttered speech that failed to respond to speech therapy. Speech impairment (childhood stutter) had been described as one of the initial presenting symptoms [11]. Gradual development of proximal weakness in the lower limbs during adolescence was observed in both sisters. They also developed mild neocerebellar syndrome. The cerebellar signs may often be rather subtle and easily overlooked, as they were in our two cases. Pronounced cerebellar atrophy is a characteristic MRI feature that was also found in both sisters. However, the extent of cerebellar atrophy had not been found to correlate with the severity of neocerebellar syndrome [12].
Neither of our two patients showed any signs of a psychiatric disorder, even though this may develop in up to a half of those affected, and usually take the form of episodic psychosis, depression or anxiety [13]. Their cognitive functions were within the broader normal range; these seldom deteriorate as a result of Tay--Sachs disease (though sporadic cases of dementia have been reported).
Tay-Sachs disease is caused by mutations of the HEXA gene coding the α-subunit of β-hexosaminidase A, an enzyme that takes part in the degradation of GM2 ganglioside. At present, over 80 mutations are known to cause the disease [14]. The majority of these mutations lead to major changes in the enzyme structure and severe impairment of its function, thus causing the severe infantile form. Several prevalent mutations, frequently in homozygeous arrangement, occur within geographically and ethnically isolated populations, such as Askhenazi Jews. The mutations responsible for the infantile form of the disease may be panethnic as well as restricted to a single family. Common variants that do not affect active sites of the enzyme tend to cause the late-onset forms of the disease. The majority of patients with the late-onset chronic form are compound heterozygotes carrying one severe and one mild mutation (a mild mutations is predominantly c.805G>A). The clinical course of the disease is then determined by the less affected allele [2].
Our two patients’ ancestors came from different regions, and their parents denied any consanguinity. This was confirmed by the presence of compound parental heterozygosity (each parent carried a different HEXA gene mutation).
So far, no effective treatment for Tay--Sachs disease has been found. Enzyme replacement therapy and bone marrow transplantation showed no benefit [2]. Current therapies focus mainly on substrate deprivation therapy (miglustat) and stabilization of the impaired enzyme with chemical chaperones (pyrimethamine) [8,15].
Antenatal diagnosis has markedly helped to limit the incidence of the infantile form [10]. Yet, a number of the late-onset Tay-Sachs disease cases probably remain undiagnosed [16]. In our case reports, the time from disease onset to its correct diagnosis was 12 years in the older sister. Adolescent patients may be misdiagnosed as having SMA or spinocerebellar ataxia [17]. In adulthood, the disease may mimic an atypical motor neuron disorder, amyotrophic lateral sclerosis or a psychiatric disorder. In case of Tay-Sachs disease mimicking SMA, the EMG findings may misleadingly support the wrong diagnosis [3]. Therefore, in the relevant cases with no evidence of deletion in the SMN1 gene, appropriate enzymatic and genetic tests for Tay-Sachs disease should be performed.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Iva Prihodova, MD, PhD
Department of Neurology
Charles University 1st Medical Faculty and General Teaching Hospital
Katerinska St. 30
128 00 Prague
e-mail: iprih@lf1.cuni.cz
Accepted for review: 9. 1. 2012
Accepted for print: 15. 8. 2012
This work was supported by the Czech Ministry of Education (research program MSM 0021620849 and MSM 0021620806) and PRVOUK-P26/LF1/4.
Zdroje
1. Gravel R, Kaback M, Proia R et al. The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS et al (eds). The metabolic and molecular bases of inherited disease. New York: Mc Graw-Hill 2001: 3827–3876.
2. Neudorfer O, Kolodny EH. Late-onset Tay-Sachs disease. Isr Med Assoc J 2004; 6(2): 107–111.
3. Navon R, Argov Z, Frisch A. Hexosaminidase A deficiency in adults. Am J Med Genet 1986; 24(1): 179–196.
4. Poupetova H, Ledvinova J, Berna L, Dvorakova L, Kozich V, Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010; 33(4): 387–396.
5. Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H et al. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet 2004; 12(2): 87–92.
6. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet 1999; 105(1–2): 151–156.
7. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999; 281(3): 249–254.
8. Shapiro BE, Pastores GM, Gianutsos J, Luzy C, Kolodny EH. Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet Med 2009; 11(6): 425–433.
9. Shapiro BE, Hatters-Friedman S, Fernandes-Filho JA, Anthony K, Natowicz MR. Late-onset Tay-Sachs disease: adverse effects of medications and implications for treatment. Neurology 2006; 67(5): 875–877.
10. Kaback MM, Desnick RJ. Tay-Sachs disease: from clinical description to molecular defect. Adv Genet 2001; 44: 1–9.
11. Shapiro BE, Natowicz MR. Late-onset Tay-Sachs disease presenting as a childhood stutter. J Neurol Neurosurg Psychiatry 2009; 80(1): 94–95.
12. Streifler JY, Gornish M, Hadar H, Gadoth N. Brain imaging in late-onset GM2 gangliosidosis. Neurology 1993; 43(10): 2055–2058.
13. Streifler J, Golomb M, Gadoth N. Psychiatric features of adult GM2 gangliosidosis. Br J Psychiatry 1989; 155: 410–413.
14. Myerowitz R. Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene. Hum Mutat 1997; 9(3): 195–208.
15. Clarke JT, Mahuran DJ, Sathe S, Kolodny EH, Rigat BA, Raiman JA et al. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay--Sachs or Sandhoff variants). Mol Genet Metab 2011; 102(1): 6–12.
16. Neudorfer O, Pastores GM, Zeng BJ, Gianutsos J, Zaroff CM, Kolodny EH. Late-onset Tay-Sachs disease: phenotypic characterization and genotypic correlation in 21 affected patients. Genet Med 2005; 7(2): 119–123.
17. Praline J, Guennoc AM, Vourc‘h P, Sedel F, Andres CR, Corcia P. Late onset Tay-Sachs disease may mimic adult SMA. Rev Neurol (Paris) 2011; 167(6–7): 549–550.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2013 Číslo 2
Nejčtenější v tomto čísle
- Creutzfeldt-Jacob disease
- Spinocerebellar Ataxia 7 – a Case Report
- Lyme Borreliosis as a Cause of Bilateral Neuroretinitis with Pronounced Unilateral Stellate Maculopathy in a 8-Year Old Girl
- Electrophysiological Examination of the Pelvic Floor