
Kongenitální myastenie jako příčina respiračního selhání u dvou kojenců a batolete – kazuistiky
Congenital Myasthenia as a Cause of Respiratory Failure in two Infants and a Toddler – Case Reports
Congenital myasthenic syndromes (CMS) are rare autosomal recessive disorders of neuromuscular transmission. They manifest early, but although symptoms can be improved with treatment, they often remain unrecognized. In our region, CMS with mutations of CHNE gene is well known and is particularly common in the gypsy ethnic group. However, there also are other, much more severe types, such as COLQ CMS that may be complicated by respiratory failure. In COLQ CMS, decomposition of acetylcholine at a synapse is disrupted. The synapse is overloaded and, therefore, syntostigmine as well as acetylcholinesterase inhibitors worsen the symptoms. Ephedrine is the treatment of choice. Apnoea monitor and resuscitation training in parents can be life-saving. In cases where both pathogenic mutations are known, prenatal or preimplantation diagnostics are available. We present three CMS patients who suffered from respiratory crises at an early age. The outcome was variable – a boy died at 6 months of age, a 7-year old girl needs nocturnal ventilation support and walks 10 meters, an adult patient has minimal sequelae. In two patients, COLQ mutations have been confirmed as a cause of CMS and COLQ CMS is suspected in the remaining patient (patient P2).
Key words:
congenital myasthenic syndrome – respiratory failure – neuromuscular transmission disorders – muscle hypotonia – muscle weakness
Autoři:
M. Adamovičova 1; K. Fabríciová 2; M. Jakubíková 3; I. Příhodová 3; P. Klement 1; P. Sýkora 4; M. Dusl 5; A. Abicht 5; J. Zeman 1; P. Seeman 6,7; T. Honzík 1
Působiště autorů:
Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
1; I. detská klinika LF UK a DFNsP Bratislava
2; Neurologická klinika a Centrum klinických neurověd 1. LF UK a VFN v Praze
3; Klinika detskej neurológie LF UK a DFNsP Bratislava
4; Univerzita Ludvika Maximiliana, Laboratoř molekulární myologie Mnichov
5; Klinika dětské neurologie 2. LF UK a FN v Motole, Praha
6; Centrum lékařské genetiky Gennet, Praha
7
Vyšlo v časopise:
Cesk Slov Neurol N 2012; 75/108(6): 757-762
Kategorie:
Kazuistika
Poděkování Prof. dr. med. Hannsi Lochmullerovi a dr. med. Violetě Mihaylové, Univerzita Ludvika Maximiliana, Laboratoř molekulární myologie a Neurologická klinika, Mnichov za provedená DNA vyšetření COLQ genu, prof. dr. med. Andrew G. Engelovi, Mayo Clinic, USA a dr. med Ulrike Schara z Neuropediatrické kliniky v Essenu za cenné rady při nastavování léčby efedrinem. Práce vznikla s podporou těchto projektů: PRVOUK P24/LF/3, RVOVFN64165, VZ MSM 0021620849, PRVOUK-P26/LF1/4, GA UK v rámci projektu Klinická, laboratorní a socioekonomická analýza u pacientů s myasthenia gravis v ČR, č. 351011/2011.
Souhrn
Kongenitální myastenické syndromy (CMS) jsou vzácné, autozomálně recesivně dědičné choroby nervosvalového přenosu. Manifestace je časná, a ač jsou léčbou ovlivnitelné, nezřídka uniknou diagnóze. U nás je znám CMS podmíněný mutacemi v CHRNE genu, častý u romského etnika. Existují však i jiné, těžší typy, komplikované respiračním selháním, například CMS s mutací COLQ. U tohoto typu CMS je porušeno rozkládání acetylcholinu na synapsi. Synapse je přetížena, a proto syntostigminový test i inhibitory acetylcholinesterázy potíže zhoršují. Lékem volby je efedrin. Apnea monitor a zácvik rodičů do resuscitace mohou být život zachraňující. Při znalosti obou patogenních mutací je dostupná prenatální či preimplantační diagnostika. Prezentujeme tři pacienty s CMS, kteří prodělali v časném věku respirační krize. Průběh onemocnění byl různý – chlapec zemřel v šesti měsících, sedmiletá dívka vyžaduje noční ventilační podporu a ujde 10 metrů, dospělá pacientka má minimální potíže. U dvou pacientů byly prokázány jako příčina CMS mutace COLQ genu, u jedné pacientky (pacientka P2) je tato diagnóza suspektní.
Klíčová slova:
kongenitální myastenický syndrom – respirační selhání – nervosvalový přenos – hypotonie svalů – svalová slabost
Úvod
Kongenitální myastenické syndromy (CMS) představují heterogenní skupinu onemocnění nervosvalového přenosu a s výjimkou slow channel syndromů jsou AR dědičné [1,2]. U pacientů s CMS je svalová únavnost a slabost způsobena mutacemi genů pro některou z bílkovin, které se podílejí na nervosvalovém přenosu. U jednotlivých typů se liší místo postižení synapse, funkční důsledky poruchy, klinické projevy i léčba. Dosud je známo 14 genů spojených s CMS, zhruba 40 % pacientů s CMS není geneticky objasněno [1,2]. Manifestace je většinou kongenitální nebo do 2 let věku [1]. V našem regionu je nejčastější CMS s homozygotní mutací CHRNE genu c. 1267delG. Pacienti bývají romského etnika, dominuje ptóza, oftalmoplegie a mírná až středně těžká pletencová slabost. K respiračnímu selhání dochází výjimečně [3]. Tento CMS je u nás znám již 10 let a jeho diagnóza by neměla činit potíže [4,5]. Existují však CMS s těžším průběhem, často se projevují i respiračním selháním novorozence nebo kojence (s mutacemi v genech CHAT, RAPSN, COLQ, DOK-7 či fast channel CMS) [1]. U českých a slovenských pacientů byly kromě mutací v CHRNE [4,5] nalezeny pouze mutace v RAPSN u kojence a v COLQ u školáka [6].
U COLQ myastenie, poprvé popsané roku 1977 [7], je onemocnění způsobeno nedostatkem acetylcholinesterázy (AChE) na synaptické štěrbině. Gen COLQ, objevený v roce 1998 [8,9], kóduje podjednotku AChE, která je nezbytná pro ukotvení enzymu v prostoru synapse. Nerozložený acetylcholin opakovaně stimuluje receptory a dochází k přetížení synapse [10]. COLQ myastenie způsobuje obvykle těžké postižení od novorozeneckého či kojeneckého věku s generalizovanou slabostí a často s respiračními krizemi [10–12].
Představujeme tři pacienty s CMS, u kterých byly prokázány mutace v COLQ – u pacientů P1 a P3 byly nalezeny obě patogenní mutace, u pacientky P2 byla nalezena pouze jedna patogenní mutace. Dívka P2 má klinicky nepochybně CMS a projevy jsou typické pro COLQ myastenii (zejména tíže postižení, odpověď na syntostigmin a na efedrin a pro COLQ specifická obleněná fotoreakce). Z genetického hlediska však není diagnóza na DNA úrovni zcela objasněna, protože druhou patogenní mutaci se u dívky nepodařilo sekvenováním všech kódujících exonů prokázat. Diagnózu by bylo možné upřesnit vyšetřením RNA ve svalu, nicméně nutné by bylo zopakovat svalovou biopsii, což rodiče odmítli.
Metoda a pacienti
DNA diagnostika
Bylo sekvenováno všech 17 kódujících exonů COLQ genu, včetně přilehlých intronových sekvencí. Jako reference a pro označení nalezených mutací byla použita referenční sekvence: NM_005677.3.
Pacienti
Referujeme o třech nepříbuzných pacientech kavkazského etnika (obr. 1). Pacient P1 zemřel v 6 měsících, pacientka P2 je nyní 7letá a pacientka P3 30letá. V rodinách se nevyskytuje nervosvalové onemocnění ani konsanguinita. Pre- a perinatální údaje pacientů jsou bez nápadností, s výjimkou akutního císařského řezu pro alteraci srdečních ozev u pacienta P1. Pacientka P2 byla drobnější (PH 2 620, délka 48 cm).
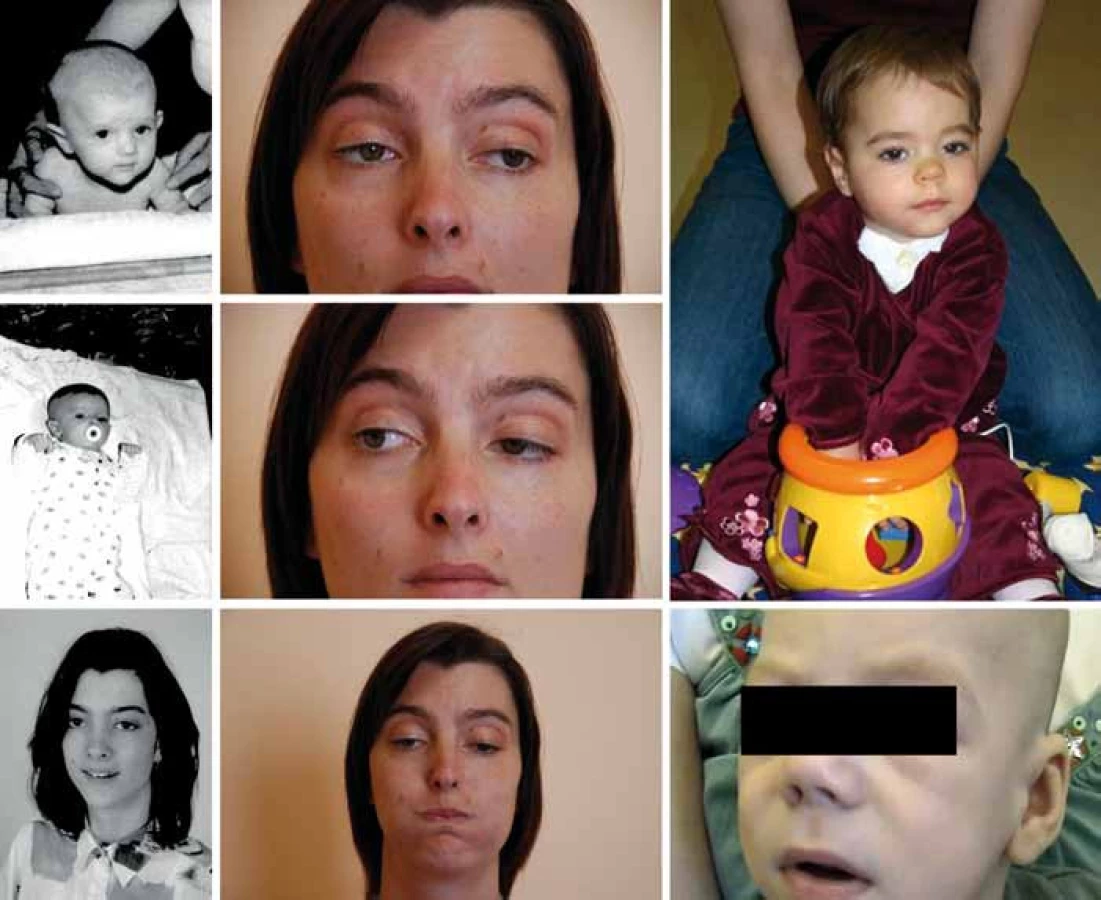
Klinické projevy
Vybrané klinické údaje jsou pro přehlednost uvedeny v tab. 1.
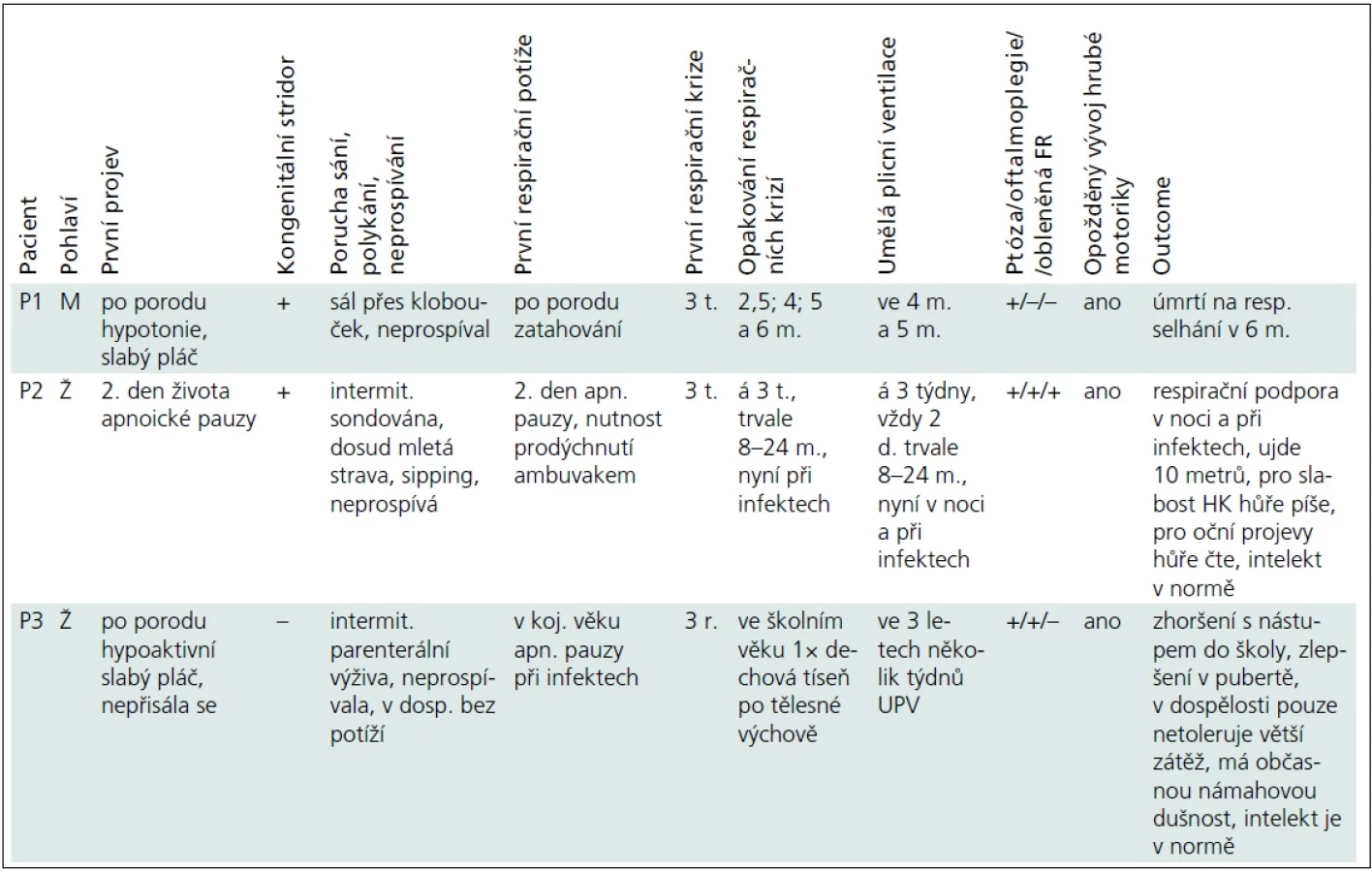
Pacient P1 byl od narození hypotonický, slabě plakal, sál pouze přes klobouček a neprospíval. Od 3 týdnů měl opakované respirační krize charakteru hypoventilace a lapavého dýchání s cirkumorální cyanózou. První dvě odezněly spontánně po 20 min, při dalších docházelo po hodinách potíží k prudkému zhoršení, které si dvakrát vynutilo umělou plicní ventilaci, v 6 měsících na respirační selhání zemřel.
Pacientka P2 měla dobrou poporodní adaptaci. Od 2. dne života se objevily apnoické pauzy a od 3 týdnů opakovaně respirační krize, které začínaly neklidem, dráždivostí a odmítáním stravy, pokračovaly apatií, hypoventilací, stáčením bulbů, rytmickými automatizmy tváře, nekoordinovanými pohyby končetin, hypersalivací a vždy progredovaly do respiračního selhání s nutností 1–2denní UPV. Dívce byla zavedena tracheostomie. V etiologii příhod byla zvažována epilepsie, nicméně EEG bylo bez specifických změn a přechodné podávání fenobarbitalu potíže neovlivnilo. Od osmi měsíců, kdy došlo při zavádění centrálního žilního katétru k ruptuře a. subclavia s nutností chirurgického řešení, byla dívka ventilována trvale až do 24 měsíců. Zlom přinesla léčba efedrinem, nadále však vyžaduje respirační podporu v noci či při infektu.
Pacientka P3 byla po porodu hypoaktivní, slabě plakala a nepřisála se. V kojeneckém věku neprospívala, měla časté respirační infekty a při nich apnoické pauzy. Respirační krizi s nutností několikatýdenní UPV prodělala ve třech letech. Potíže byly dávány do souvislosti s vakcinací proti spalničkám a hodnoceny jako „nejasná epizoda bezvědomí s přechodným edémem mozku při febrilních křečích“, dívka pak užívala pět let antiepileptika.
Oční příznaky (tab. 1) se u pacientů rozvíjely postupně. U P1 a P3 od novorozeneckého věku. U P2 od šesti měsíců a kromě ptózy a obleněné fotoreakce byla nápadná i nedostatečná farmakologická mydriáza. Aktuálně má tato dívka oboustrannou miózu, ptózu a oftalmoplegii.
Všichni naši pacienti měli opožděný vývoj hrubé motoriky. Pacient P1, který zemřel v šesti měsících, neudržel v pěti měsících v trakci hlavičku, ale hrál si s hračkou. Pacientka P2 chodila s oporou od 2,5 roku, samostatné krůčky zvládla od čtyř let, kdy pro skoliózu našlapovala na celou levou nohu a rovnováhu udržovala špičkou pravé nohy. Po operaci skoliózy zatěžuje obě nohy stejně, přesto v sedmi letech ujde 10 metrů. Pacientka P3 chodila samostatně od 19 měsíců, v dospělosti má pouze intoleranci fyzické námahy. Potíže pacientek P2 a P3 kolísají, zhoršují se během dne, při fyzické námaze či při infektu. Všichni pacienti v našem souboru měli skoliózu, u pacientky P2 byla řešena operačně s dobrým efektem.
Vyšetření
Výsledky vybraných vyšetření jsou pro přehlednost uvedeny v tab. 2.

U všech pacientů byla hodnota CK v normě, protilátky proti acetylcholinovým receptorům nebyly prokázány, všichni měli normální rychlost vedení vzruchu periferním nervem, pouze u pacientky P2 byla ve třech měsících nižší amplituda motorické odpovědi. U všech byla provedena svalová biopsie. U pacienta P1 bylo histologické, imunohistochemické a elektronoptické vyšetření s normálním nálezem, vyšetření parametrů oxidativní fosforylace nesvědčilo pro primární mitochondriální poruchu. U pacientky P2 bylo hodnoceno jen množství a uspořádání mitochondrií, které bylo v normě. Biopsie u pacientky P3 byla provedena před 22 lety a pro „vadnou diferenciaci typů svalových vláken“ bylo vysloveno podezření na neprogredující kongenitální myopatii.
Největší přínos pro diagnózu mělo vyšetření repetitivní stimulace a syntostigminový test, diagnostický byl nález mutací v COLQ genu (tab. 2). Naopak zavádějící byl nález ve svalové biopsii pacientky P3 a výsledek lokálního kokainového testu provedený u pacientky P2 pro podezření na poruchu centrálního sympatiku, kdy pravé oko reagovalo lépe než levé, se závěrem: pravděpodobná kmenová léze – Hornerův syndrom vlevo.
Léčba
U pacientky P2 neovlivnil 3,4-diaminopyridin klinické ani elektrofyziologické projevy. Efedrin užívala pouze pacientka P2: 2,5 mg/kg/den s postupným navýšením na 5 mg/kg/den. Po třech měsících léčby bylo možno dívku odpojit od ventilátoru přes den. Další přehled léčby je uveden v tab. 2.
Diskuze
Klinické příznaky
Kongenitální myastenické syndromy se dají léčbou ovlivnit. Vzhledem k jejich vzácnosti však chybí zkušenost a navíc mají velkou variabilitu příznaků, a tak je jejich rozpoznání často obtížné. Manifestují se převážně v novorozeneckém či kojeneckém věku, diagnostická prodleva je však spíše pravidlem než výjimkou [13]. I v našem souboru byla latence k diagnóze od 21 měsíců (P2) do 30 let (P3).
Od u nás dobře známé CMS podmíněné mutacemi CHRNE genu se COLQ myastenie liší těžším průběhem, výskytem respiračních krizí, atypickou reakcí na podání syntostigminu či inhibitorů acetylcholinesterázy a zhruba u poloviny pacientů i chyběním oftalmoplegie.
Klasická infantilní forma COLQ myastenie má časné a těžké projevy s respiračními krizemi, mnohdy je fatální (P1) či těžce hendikepující (P2) [1]. Mírnější forma začíná obvykle v dětském věku a má pomalou progresi (v ČR byla již zachycena [6]). Variabilita je však větší – řada těžce postižených kojenců má později jen minimální obtíže (P3) a naopak pozdní začátek nevylučuje rychlou progresi onemocnění včetně atak respiračních krizí [11,13]. Do klinického obrazu COLQ myastenie patří hypotonie, slabý pláč, porucha sání a polykání, neprospívání, život ohrožující respirační krize (vše u P1–P3), kongenitální stridor (P1, P2), u většiny pacientů ptóza (P1–P3), asi u poloviny oftalmoplegie (P2, P3) [11,13], pro COLQ specifická obleněná fotoreakce [1,11] je zhruba u čtvrtiny pacientů (P2). Dále bývá svalová slabost a hypotrofie, skolióza (P1–P3), šlachosvalové reflexy mohou být snížené. Asi polovina pacientů má opožděný motorický vývoj (P1–P3), většina dosáhne samostatné chůze (P2, P3) [1,11,13]. Potíže kolísají během dne, ale i v průběhu týdnů až měsíců, například v závislosti na fyzické námaze, akutních infektech (P2, P3), léčbě inhibitory AChE (P3) či v pubertě (P3) a v těhotenství [12].
Vyšetření
U COLQ CMS vybaví repetitivní stimulace 2–3Hz dekrement (P2, P3) a SF-EMG zvýšený jitter [1,11]. U COLQ myastenie mohou být v EMG i myopatické změny (P1, P3) – jsou způsobeny přetížením postsynaptické oblasti kationty. CK bývá v normě (P1–P3) [11]. Pouze u dvou typů CMS – u COLQ a u slow channel syndromů – vyvolá jediný podnět opakované sumační svalové akční potenciály (CMAPS), nález však mnohdy chybí [11,13] a u žádného z našich pacientů zachycen nebyl. Ve svalové biopsii je obvykle pouze nespecifický nález, nejčastěji atrofie vláken 2. typu [11,13], změny na synapsi lze zobrazit jen speciálními technikami. U pacientů (P1, P2) byla biopsie svalu popsána jako normální, u pacientky (P3) bylo vysloveno podezření na neprogredující kongenitální myopatii. Pokud chybí zkušenosti, je výsledek syntostigminového testu u COLQ myastenie spíše zavádějící: odpověď chybí (P3) nebo dojde ke zhoršení slabosti (P1, P2). Léčba inhibitory acetylcholinesterázy je rovněž neúspěšná a vede ke zhoršení stavu (P3) [1]. Většina pacientů se zlepší při podávání efedrinu, obvykle ti s mírnější formou. Efekt nastupuje během týdnů až měsíců (P2) a mechanizmus není znám. Pravděpodobně jako beta2-agonista působí anabolicky na sval – mohl by tak zmírňovat sekundární myopatii [14]. Úspěšně se používá i další beta2-agonista – albuterol [1].
Dle HGMD Professional Database [15] bylo v COLQ genu dosud popsáno nejméně 39 mutací. Korelace genotyp-fenotyp je minimální, i sourozenci mohou mít různě těžké klinické projevy [16].
Diferenciální diagnostika
V novorozeneckém či kojeneckém věku ještě nemusí být rozvinuty oční příznaky, klinickému obrazu dominuje hypotonie a porucha sání, případně s respirační insuficiencí či bulbárními příznaky [13]. V takovém případě je nutné vyloučit např. tranzitorní novorozeneckou myasthenia gravis, spinální svalovou atrofii, kongenitální svalové dystrofie, zejména kongenitální myotonickou dystrofii 1. typu, kongenitální myopatie, mitochondriální myopatie, abnormity mozkového kmene, Moebiův syndrom, infantilní botulizmus či morbus Pompe [17].
Náhlý rozvoj respiračních krizí a případně sekundární anoxické křeče mohou vést k mylné diagnóze epilepsie (P2) či febrilních křečí (P3) a k užívání antiepileptik (P2, P3) [13].
Obecně je u kongenitálních myastenických syndromů diagnostika obtížná i pro nekonstantní výbavnost dekrementu při EMG vyšetření či pro zavádějící výsledky syntostigminového testu (tab. 3). Citlivým vyšetřením je single fibre EMG, ale jeho dostupnost, zejména pro malé děti, je značně omezená [13].
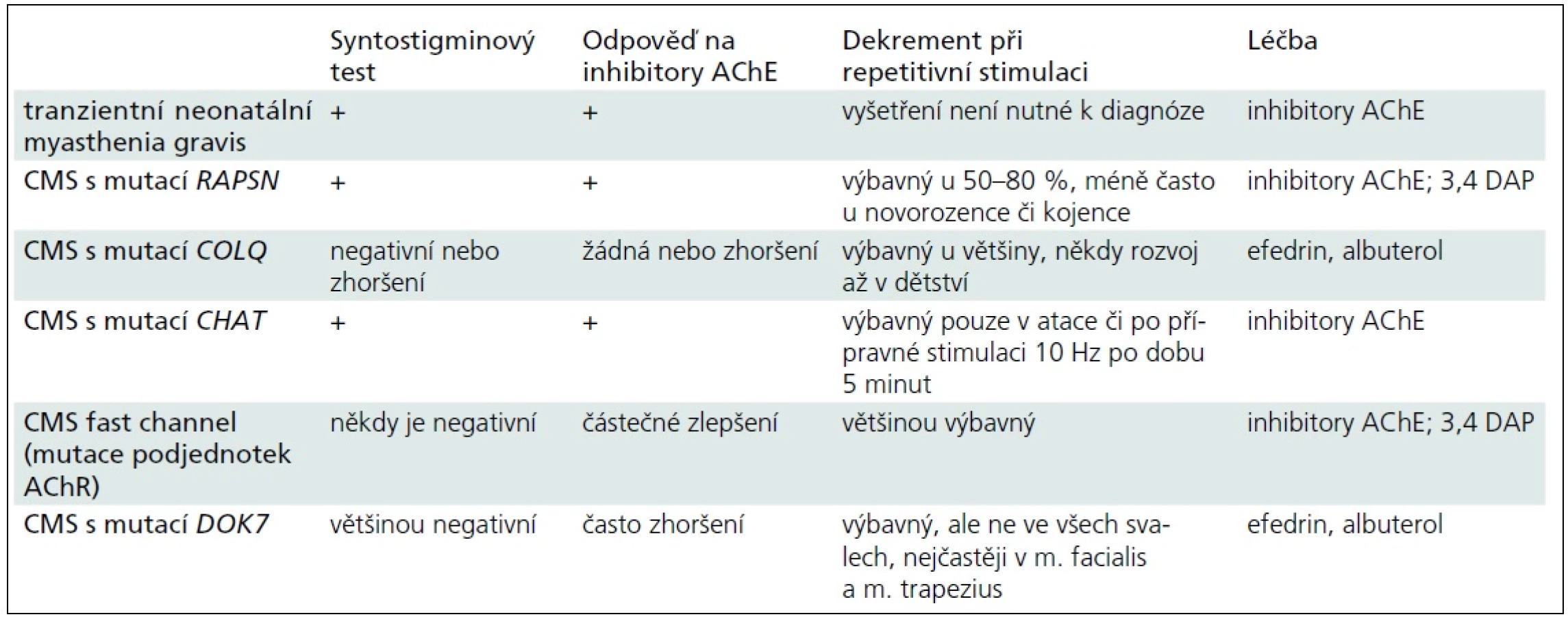
Závěr
CMS zůstávají často nerozpoznány, pacienti jsou vedeni pod jinými diagnózami, například jako kongenitální myopatie, svalová dystrofie či metabolická myopatie [13]. Zatímco u naprosté většiny výše uvedených diagnóz nelze průběh onemocnění výrazněji ovlivnit, u CMS může být efekt léčby významný. Znalost a předcházení možných komplikací (respirační krize, riziko aspirací) jsou mnohdy klíčové pro přežití.
Zvážení CMS by tedy mělo být prioritou nejen u novorozenců nebo kojenců se svalovou slabostí a případně s respiračním selháním, ale i u starších dětí či dospělých s neobjasněným myastenickým či myopatickým syndromem. Chybění ptózy, oftalmoplegie, dekrementu či negativní syntostigminový test diagnózu nevylučují. Základem úspěšné diagnózy je klinické podezření, spolupráce se zkušeným elektrofyziologem a následné cílené DNA vyšetření. Při znalosti obou kauzálních mutací je možno nabídnout rodině cílenou genetickou prevenci ve formě prenatální či preimplantační diagnostiky.
Použité zkratky
CMS kongenitální myastenický syndrom/syndromy
AR autozomálně recesivní
ChAT cholinacetyltransferáza
AChR acetylcholinový receptor
AChE acetylcholinesteráza
COLQ gen kódující COLQ podjednotku AChE, která zajišťuje ukotvení enzymu v prostoru synapse
CHRNE gen kódující epsilon podjednotku AChR
RAPSN gen kódující rapsyn
DOK7 Downstream Of Kinase gen
PH porodní hmotnost
UPV umělá plicní ventilace
CMAP Compound Muscle Action Potential (sumační svalový akční potenciál)
CMAPS Compound Muscle Action Potentials (sumační svalové akční potenciály)
MUDr. Miriam Adamovičová
Klinika dětského a dorostového lékařství
1. LF UK a VFN
Ke Karlovu 2
121 09 Praha 2
e-mail: adamovicova.m@seznam.cz
Přijato k recenzi: 29. 5. 2012
Přijato do tisku: 1. 8. 2012
Zdroje
1. Engel AG. Current status of the congenital myasthenic syndromes. Neuromuscul Disord 2012; 22(2): 99–111.
2. Jakubíková M. Kongenitální myastenické syndromy a myasthenie dětského věku. Neurol Prax 2010; 11(2): 100–103.
3. Kolníková M, Sýkora P, Seeman P. Kongenitálny myastenický syndróm. Pediatr Prax 2009; 10(5): 268–269
4. Seeman P, Sisková D. Autosomal recessive ethnic diseases of Czech Gypsies. Čas Lék Česk 2006; 145(7): 557–560.
5. Hálek J, Neklanová M, Seeman P. Kongenitální myastenický syndrom (CMS). Čes-slov Pediat 2007; 62(6): 393–397.
6. Adamovičová M, Šišková D. Kongenitální myastenické syndromy – kazuistiky. Cesk Slov Neurol N 2010; 73/106(1): 62–67.
7. Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann Neurol 1977; 1(4): 315–330.
8. Donger C, Krejci E, Serradell AP, Eymard B, Bon S, Nicole S et al. Mutation in the human acetylcholinesterase-associated collagen gene, COLQ, is responsible for congenital myasthenic syndrome with end-plate acetylcholinesterase deficiency (Type Ic). Am J Hum Genet 1998; 63(4): 967–975.
9. Ohno K, Brengman J, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagenlike tail subunit (ColQ) of the asymmetric enzyme. Proc Natl Acad Sci USA 1998; 95(16): 9654–9659.
10. Hutchinson DO, Walls TJ, Nakano S. Congenital endplate acetylcholinesterase deficienty. Brain 1993; 116(3): 633–653.
11. Mihaylova V, Müller JS, Vilchez JJ, Salih MA, Kabiraj MM, D‘Amico A et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain 2008; 131(3): 747–759.
12. Wargon I , Richard P, Kuntzer T, Sternberg D, Nafissi S, Gaudon K et al. Long-term follow-up of patients with congenital myasthenic syndrome caused by COLQ mutations. Neuromuscul Disord 2012; 22(4): 318–324.
13. Kinali M, Beeson D, Pitt MC, Jungbluth H, Simonds AK, Aloysius A et al. Congenital myasthenic syndromes in childhood: diagnostic and management challenges. J Neuroimmunol 2008; 15(201–202): 6–12.
14. Lashley D, Palace J, Jayawant S, Robb S, Beeson D. Ephedrine treatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology 2010; 74(19): 1517–1523.
15. HGMD Professional Database. On-line. Available from URL: https://portal.biobase-international.com/hgmd.
16. Yeung WL, Lam CW, Ng PC. Intra-familial variation in clinical manifestations and response to ephedrine in siblings with congenital myasthenic syndrome caused by novel COLQ mutations. Dev Med Child Neurol 2010; 52(10): e243–e244.
17. Jakubíková M, Seeman P. Kongenitální myastenické syndromy. In: Piťha J et al (eds). Myasthenia gravis a ostatní poruchy nervosvalového přenosu. Praha: Maxdorf Jessenius 2010: 319–341.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2012 Číslo 6
Nejčtenější v tomto čísle
- Epidemie roztroušené sklerózy ve světě?
- Spinální kongres
- Kortikální patologie u roztroušené sklerózy – morfologické, imunopatologické a klinické souvislosti
- Fázový model neurorehabilitace