
Morbus Lhermitte-Duclos – kazuistika
Lhermitte-Duclos Disease – a Case Report
Dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) is a rare benign lesion. According to the the most recent WHO classification (2007), the disease is considered a hamartoma or tumour (WHO grade 1). A number of authors incline to the opinion that it is of hamartogenous origin, basing their view on the results of recent genetic studies. Clinical symptoms of the disease include cerebellar symptoms and signs of compression of the brain stem and cranial nerves, while the disease may also be associated with intracranial hypertension syndrome. Magnetic resonance examination is the best diagnostic imaging technique, since as it reveals a unique pattern for the disorder. Dysplastic gangliocytoma of the cerebellum may be associated with Cowden disease. This is an autosomal dominant condition that results mainly from a mutation of the PTEN gene. Multiple hamartomas are specific to Cowden disease, which is associated with high risk of systemic malignancies. Radical surgery is the optimum treatment approach but may not be feasible in all cases. Genetic examination is always essential in order to exclude Cowden disease. The authors present the case report of a 32-year-old man with a history of cerebellar signs and obstructive hydrocephalus. On the basis of the results of magnetic resonance imaging, radical resection of a tumour was performed and the patient has been without signs of residuum of the tumour or recurrence since. The results of the genetic examination were negative.
Key words:
Lhermitte-Duclos disease – dysplastic gangliocytoma of the cerebellum – Cowden syndrome
Autoři:
V. Vybíhal 1
![]() ; P. Fadrus 1; M. Duba 1; M. Vidlák 1; A. Šprláková-Puková 2; L. Křen 3
; P. Fadrus 1; M. Duba 1; M. Vidlák 1; A. Šprláková-Puková 2; L. Křen 3
Působiště autorů:
LF MU a FN Brno
Neurochirurgická klinika
1; LF MU a FN Brno
Radiologická klinika
2; LF MU a FN Brno
Ústav patologie
3
Vyšlo v časopise:
Cesk Slov Neurol N 2010; 73/106(5): 563-567
Kategorie:
Kazuistika
Souhrn
Dysplastický gangliocytom mozečku (morbus Lhermitte-Duclos) patří mezi vzácné benigní léze. Podle poslední klasifikace WHO z roku 2007 je onemocnění považováno za hamartom nebo nádor (WHO gradus I). Na základě genetických studií se řada autorů v poslední době přiklání k hamartogennímu původu. Klinicky se onemocnění projevuje cerebelárními příznaky, event. příznaky z komprese mozkového kmene a hlavových nervů. Může se také manifestovat syndromem nitrolební hypertenze. Základní zobrazovací metoda je magnetická rezonance, na které lze spatřit typický obraz onemocnění. Dysplastický gangliocytom mozečku může být součástí syndromu Cowdenové. Jedná se o autozomálně dominantní onemocnění, jehož hlavní příčinou je mutace genu PTEN. Jeho charakteristickým projevem je výskyt mnohočetných hamartomů a vysoké riziko vzniku systémových malignit. Nejefektivnější léčba je radikální chirurgická resekce, která ale nemusí být vždy možná. Nezbytné je provedení genetického vyšetření k vyloučení syndromu Cowdenové. V práci autoři prezentují kazuistiku 32letého muže s anamnézou cerebelární symptomatiky a obstrukčním hydrocefalem. Po provedení magnetické rezonance pacient podstoupil radikální resekci tumoru a dosud je bez známek rezidua či recidivy. Genetické vyšetření bylo negativní.
Klíčová slova:
morbus Lhermitte-Duclos – dysplastický gangliocytom mozečku – syndrom Cowdenové
Úvod
Morbus Lhermitte-Duclos (LDD) neboli dysplastický gangliocytom mozečku patří mezi vzácné benigní léze. Podle poslední WHO klasifikace z roku 2007 se jedná o hamartom nebo o nádor (WHO gradus I), přesná histogeneze onemocnění není zcela jasná. Může být součástí autozomálně dominantního onemocnění – syndromu Cowdenové (CS), které patří mezi fakomatózy charakterizované patognomickými dermatologickými změnami, mnohočetnými hamartomy a vysokým rizikem vzniku systémových malignit, především karcinomu prsu, karcinomu štítné žlázy a karcinomu endometria.
Kazuistika
Pacient, 32letý muž, pravák, si stěžuje na dva týdny trvající bolesti hlavy s maximem parietálně oboustranně. Dále udává zvýšenou únavnost a poslední den opakovaně zvrací. Při vstupním neurologickém vyšetření na sektorové neurologii je přítomna diskrétní kvantitativní porucha vědomí, nejsou přítomny meningeální příznaky, je normální nález na hlavových nervech, reflexy na horních i dolních končetinách jsou symetrické, stoj stabilní, ale je lehce nejistá chůze a lehká pravostranná dysmetrie. Kožní morfy typické pro syndrom Cowdenové nebyly nalezeny. Na akutně indikovaném vyšetření výpočetní tomografií (CT) mozku se znázorňuje rozsáhlá hypodenzní tumorózní expanze infratentoriálně v pravé mozečkové hemisféře způsobující obstrukční hydrocefalus. Na očním pozadí je fyziologický nález, papily bez známek městnání.
Po přijetí na naše pracoviště byla pacientovi zavedena zevní komorová drenáž kvůli obstrukčnímu hydrocefalu. Provedená magnetická rezonance (MR) zobrazila rozsáhlou tumorózní lézi (rozšiřující se folia cerebelli – hypointenzní na T1 vážených snímcích, hyperintenzní na T2 vážených snímcích a nesytící se po aplikaci gadolinia) zaujímající celou pravou mozečkovou hemisféru a jdoucí částečně i přes střední čáru na druhou stranu s komprimovanou IV. komorou. Na difuzně vážených snímcích bylo ložisko lehce hyperintezní a na ADC (apparent diffusion coeficient) mapách nebyla prokázána restrikce difuze (obr. 1).
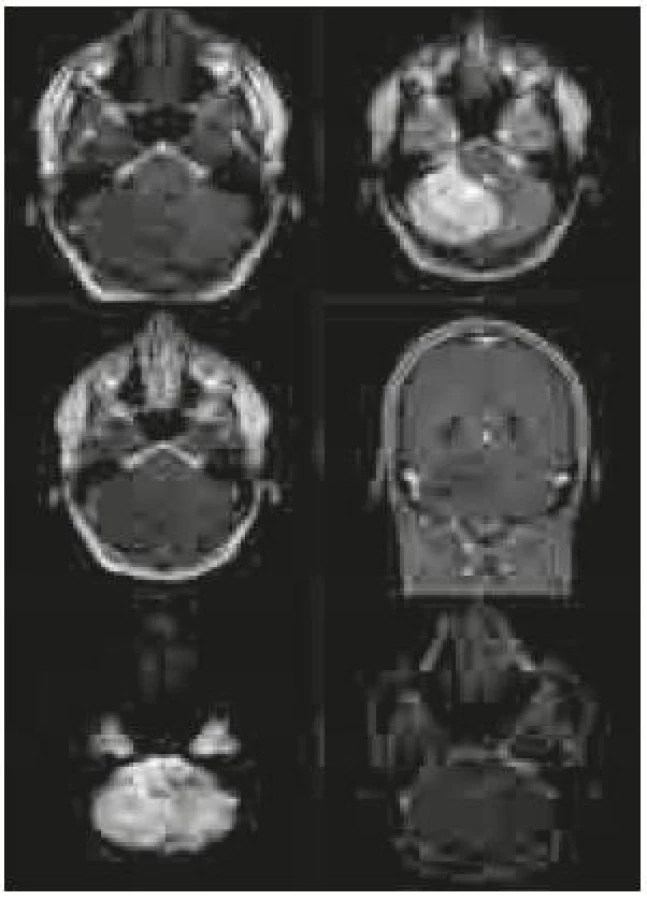
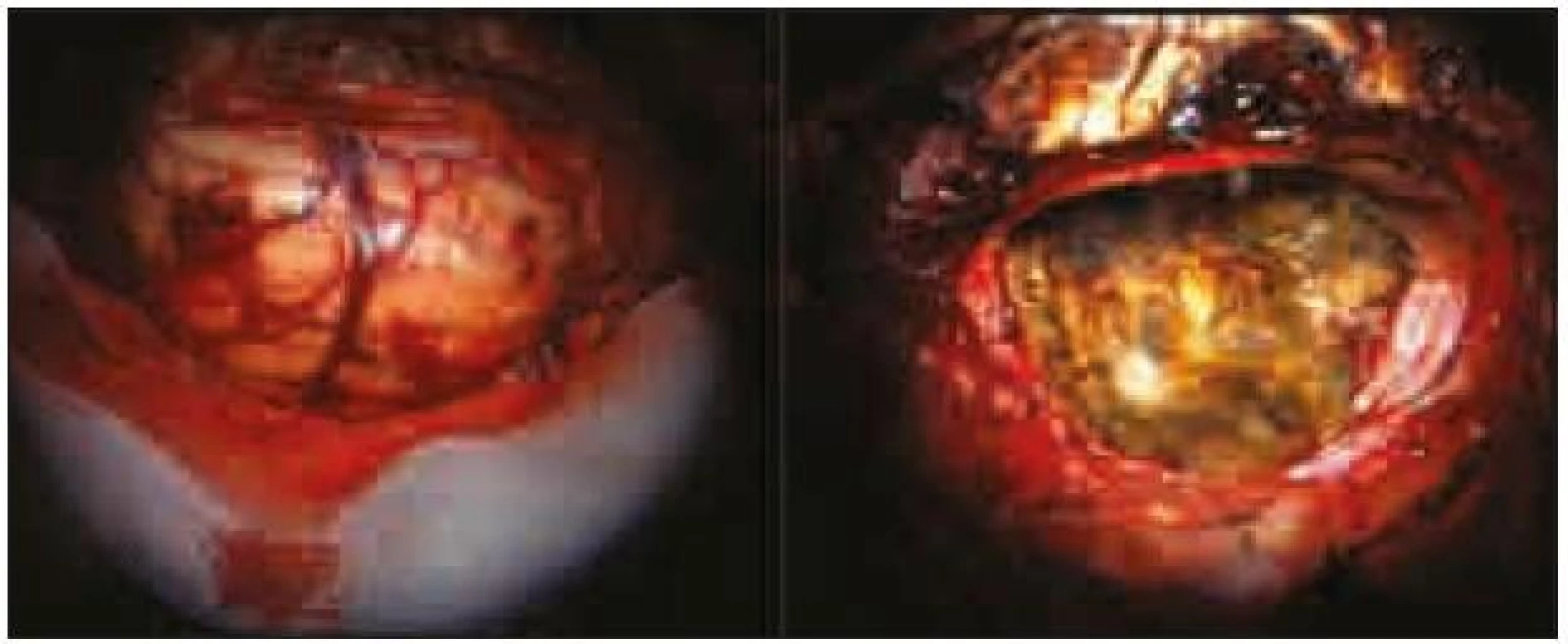
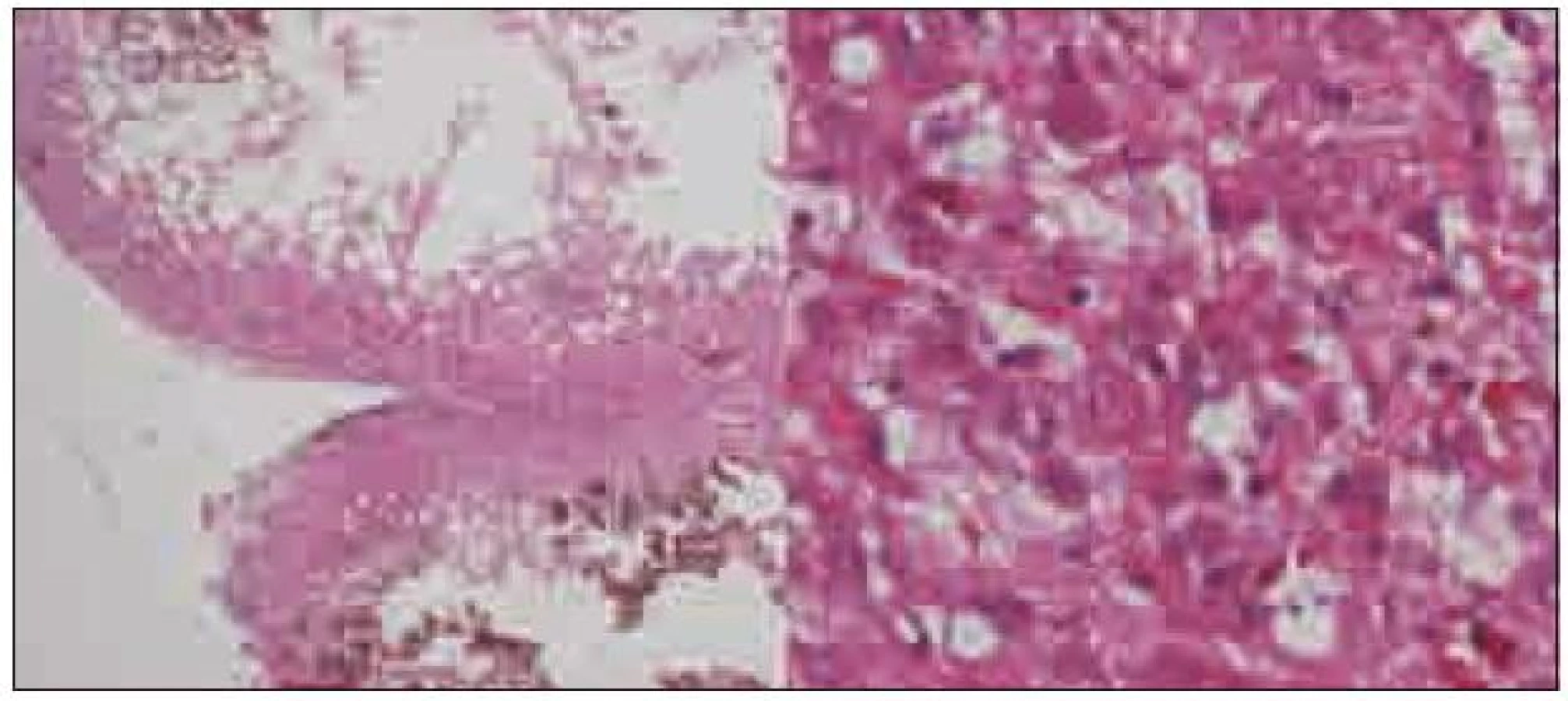
Expanze byla totálně resekována z pravostranné paramediální subokcipitální kraniotomie (obr. 2). Od okolní zdravé tkáně nebyla zřetelně ohraničena, měla křehčí konzistenci, diskrétně bělavější zbarvení a atypickou cévní kresbu. Výkon proběhl bez komplikací. Na časné pooperační magnetické rezonanci nebylo prokázáno reziduum tumoru. Rána se zhojila per primam a původní mozečková symptomatika regredovala. Histologicky byl potvrzen dysplastický gangliocytom mozečku WHO gradus I (obr. 3).
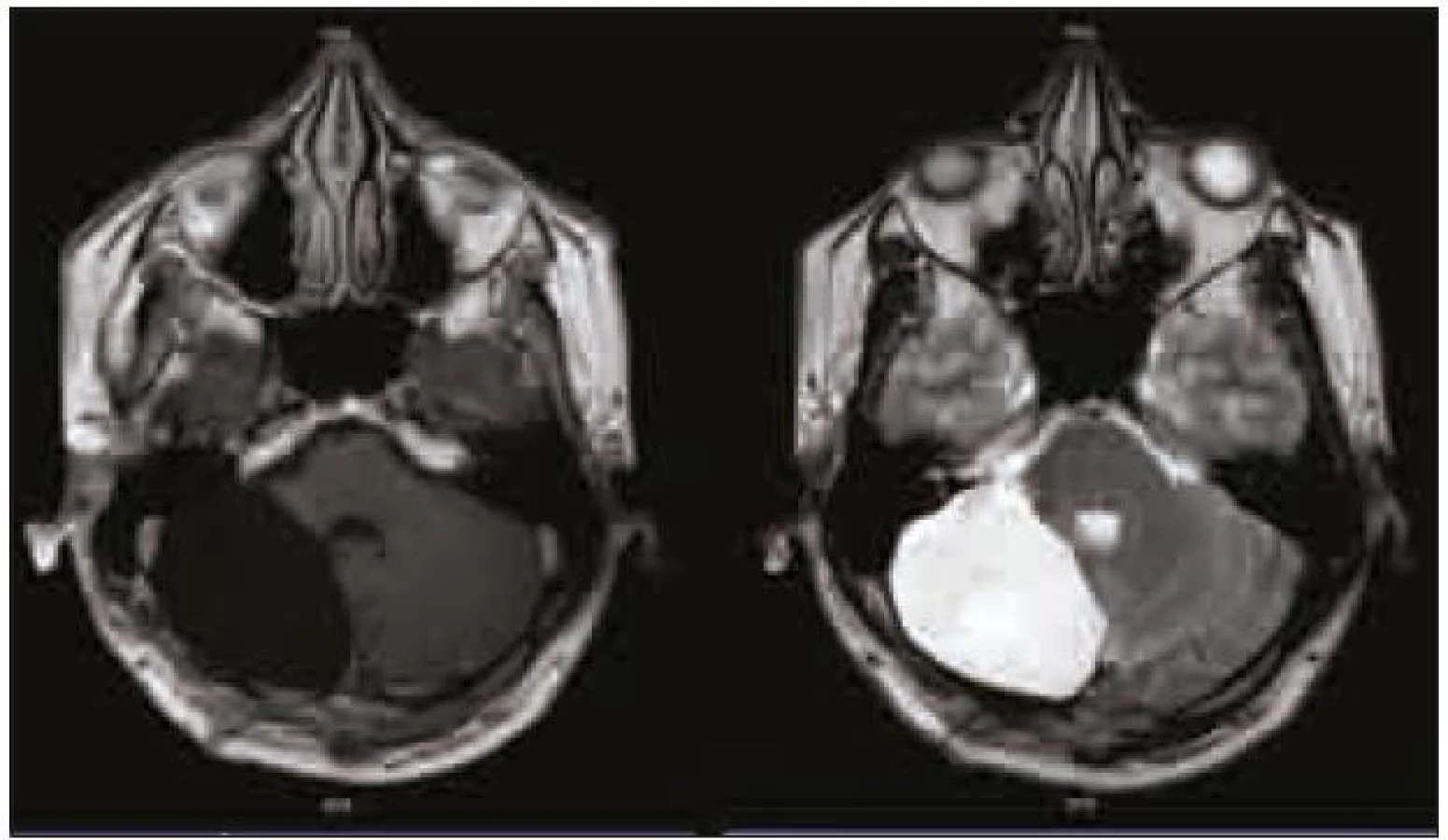
Na dalších kontrolních MR mozku za tři a devět měsíců (obr. 4) po operaci nebylo prokázáno reziduum tumoru. Při komplexním vyšetření pacienta nebyla zjištěna žádná další patologie. Genetické vyšetření na chorobu Cowdenové bylo negativní.
Diskuze
Dysplastický gangliocytom mozečku poprvé popsali v roce 1920 Lhermitte a Duclos, podle nichž se nemoc nazývá [1].
První operaci pacienta s LDD provedli v roce 1930 Bielschowsky a Simons [2]. První úspěšná resekce se uskutečnila v roce 1937. Do roku 1955 přežili po operaci pouze tři pacienti a do roku 1994 umírala jedna třetina pacientů na komplikace jejich choroby [2]. V současnosti je prognóza pacientů s tímto onemocněním díky moderním zobrazovacím metodám a mikrochirurgickým technikám výrazně lepší.
Dysplastický gangliocytom mozečku bývá většinou lokalizován v jedné mozečkové hemisféře. Klinicky se projevuje vznikem mozečkových příznaků, tlak na mozkový kmen a hlavové nervy způsobuje poruchu funkce příslušných hlavových nervů a kmenové příznaky. Onemocnění se může také projevit akutně vzniklým syndromem nitrolební hypertenze. Není predilekce pohlaví. LDD se může vyskytovat v kterémkoli věku s maximem výskytu mezi 30.–40. rokem života.
Významnou roli v diagnostice a péči o pacienty sehrávají moderní zobrazovací metody, a to především MR. V T1 váženém obraze je léze hypointenzní s žádným nebo minimálním sycením po aplikaci kontrastní látky, které nevylučuje tuto diagnózu, pokud je jinak obraz typický pro LDD [3]. Na T2 vážených snímcích je patrná hyperintenzní, dobře ohraničená léze s typickým proužkováním (izointenzní pruhy v hyperintenzní oblasti). Na difuzně vážených snímcích je léze hyperintenzní a izointenzní na ADC mapách [4].
Při vyšetření MR spektroskopií (MRS) nacházíme redukovaný poměr N-acetylasparátu a cholinu spolu s redukovaným poměrem N-acetylasparátu a kreatininu a zvýšenou hodnotu laktátu ve srovnání s normální mozečkovou tkání [5].
Unikátní obraz na MR může být diagnostický a u asymptomatických pacientů nemusí být nezbytné provedení biopsie [2]. Nicméně je nutno upozornit, že velmi zřídka může mít meduloblastom dosti podobný obraz jako LDD. V těchto případech může k upřesnění diagnózy pomoci MRS, kdy meduloblastom vykazuje na rozdíl od dyplastického gangliocytomu mozečku zvýšený poměr cholinu a kreatininu a také cholinu a N-acetylasparátu. Autoři v tomto případě trvají na histologické verifikaci [6].
Vyšetření pomocí pozitronové emisní tomografie (PET) s použitím [18F]2-fluoro-2-deoxy-D-glukózy a 11C značeným metioninem zjistila zvýšený metabolizmus jako u zhoubných nádorů [7]. Naopak studie s 15O značenou vodou zaznamenaly metabolický obrat kyslíku podobný normální mozečkové tkáni [8].
Při vyšetření jednofotonovou emisní tomografií (SPECT) pomocí technecia99 byly léze hyperaktivní bez výraznějšího poškození hematoencefalické bariéry [9].
Při histopatologickém vyšetření nalézáme difuzní rozšíření molekulární a vnitřní granulární vrstvy mozečku při relativně zachovalé cerebelární architektuře. Folia jsou rozšířená, zkroucená a objemnější, ale zachovaná. Makroskopicky bývají bledší než normální kortex. V zevní molekulární vrstvě jsou často pozorovány svazky paralelně probíhajících abnormálně myelinizovaných axonů hypertrofických granulárních buněk. Purkyňovy buňky jsou početně redukovány nebo mohou dokonce i chybět. Často bývají přítomny kalcifikace. Předpokládá se, že dysplastické buňky mají původ v mozečkových granulárních neuronech a kombinace jejich aberantní migrace a jejich hypertrofie je zodpovědná za formaci léze [10]. Při operaci jsou hranice mezi lézí a normální mozečkovou tkání často nejasné vzhledem k pozvolnému přechodu patologické tkáně do normální zdravé tkáně [10]. Onemocnění nevykazuje žádnou proliferační aktivitu a dosud nebyla popsána maligní transformace.
CS je autozomálně dominantní onemocnění s věkově závislou a variabilní penetrací. Byla pojmenována Lloydem a Dennisem v roce 1963 podle Rachel Cowdenové – první pacientce s tímto syndromem, která umřela ve věku 30 let na infiltrující duktální karcinom prsu [11]. Za hlavní příčinu vzniku CS je považována mutace genu PTEN (synonyma MMAC1, TEP1) na lokusu 10q23.3 [12]. Vyskytuje se u 80 % pacientů a v dalších 10 % je přítomna v oblasti promotorové oblasti genu. Mutace tohoto genu byly ještě popsány u syndromu Bannayan-Riley-Ruvalcaba, Proteus syndromu a Proteus-like syndromu. Na základě současných genetických studií a imunohistochemických analýz, kdy pacienti s LDD vykazují sníženou expresi PTEN u hypetrofických neuronů doprovázenou zvýšenou expresí P-Akt1 a aktivací mTOR (schéma 1), se onemocnění považuje za hamartom [13].
![Schéma 1. Schematické znázornění aktivace enzymatické kaskády cestou serin/treoninové kinázy při ztrátě funkce PTEN (phosphatase and tensin homolog deleted on chromosome 10).
Za normálních okolností PTEN limituje fosforylaci PI[3,4,5]P<sub>3</sub> (fosfatidylinositol 3,4,5-trifosfát). Pokud není PTEN přítomen, PIP<sub>3</sub> aktivuje fosfoinositidin-dependentní kinázu-1 (PDK1), která fosforyluje serin/treoninovou kinázu (Akt) také známou jako protein kináza B (PKB). Fosforylovaná serin/treoninová kináza (P-Akt) řídí buněčné procesy zahrnující migraci, proliferaci a přežití buňky. P-Akt navíc inhibuje komplex genů tuberózní sklerózy (TSC1/TSC2). Za normálních okolností tento komplex genů inhibuje mTOR (mammalian target of rapamycin). Pokud není mTOR inhibován, jeho zvýšený obsah vede k nekontrolovatelnému buněčnému růstu. Ztráta PTEN zvyšuje množství P-Akt, který výrazně inhibuje komplex genů tuberózní sklerózy, což znamená snížení inhibičního vlivu na mTOR a zvýšení jeho množství pak zapříčiňuje nekontrolovaný buněčný růst. CCI-779 je inhibitor mTOR, jedná se o derivát rapamycinu [2].](https://pl-master.mdcdn.cz/media/image/b9d0c4c98a5ee7daf7dab15abb6c5e39.jpeg?version=1537790292)
CS je charakterizován přítomností mnohočetných hamartomů, dermatologickými změnami (faciální papuly, gingivální papilomy a akrální keratóza) a vysokou incidencí systémových malignit, především karcinomu prsu, štítné žlázy a karcinomu endometria (tab. 1).
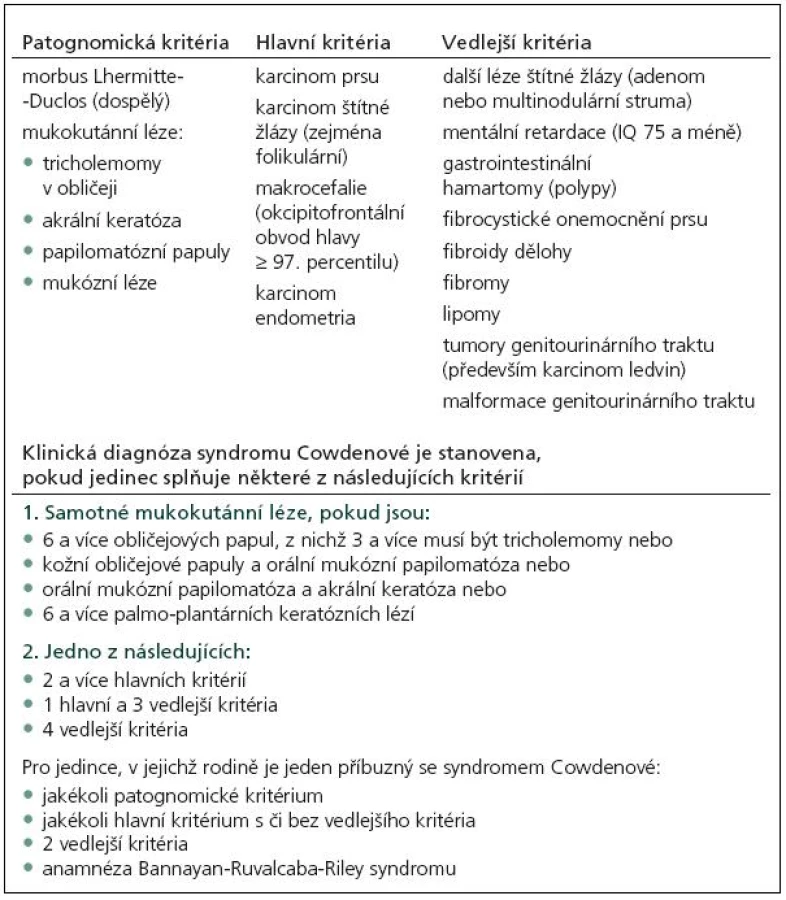
V roce 1991 Padberg zjistil asociaci mezi LDD a CS [14]. Vzhledem k vysokému riziku malignit je nutný adekvátní skríning (tab. 2). Postižení mají 10% riziko vzniku karcinomu štítné žlázy, ženy 25% až 50% riziko vzniku karcinomu prsu během života. U 50–80 % pacientů se nachází makrokranie, přibližně u 10 % zhoršení kognitivních funkcí. V 90 % se onemocnění klinicky manifestuje během prvních 20 let.
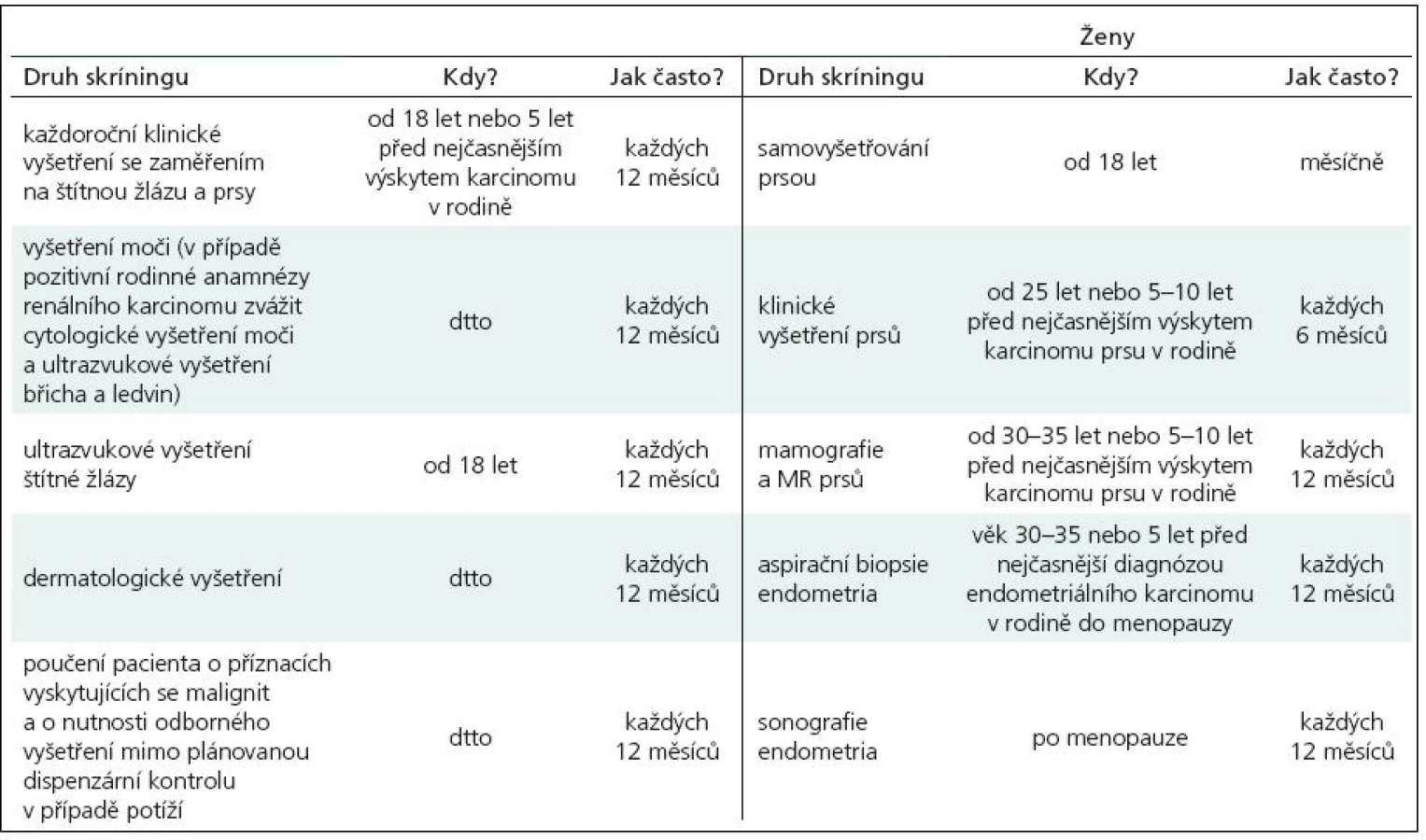
Jedinou efektivní terapií je chirurgická resekce. Vznik recidiv je popisován v rozmezí od 2 měsíců do 20 let, vyskytovat se může i po totálně provedené chirurgické resekci vzhledem k přítomnosti přechodové zóny mezi patologickou a zdravou mozečkovou tkání, která způsobuje výskyt makroskopicky neostré hranice [15]. Vzhledem k těmto skutečnostem se doporučuje provést co nejradikálnější možnou resekci při primooperaci a dále je nutné dlouhodobé sledování pacientů. Přítomnost hydrocefalu si může vyžádat provedení dočasné nebo trvalé drenážní operace. Malá rezidua či recidivy je možno observovat, u větších způsobujících klinické potíže či výrazný mass efekt je nutné přistoupit k reoperaci. Kvůli povaze onemocnění není radioterapie doporučována.
Závěr
V diferenciální diagnostice mozečkových tumorů je třeba zvažovat i možnost vzácných nádorových či nádorům podobných lézí. Nejvýznamnější léčebnou metodou je co nejradikálnější resekce, která však může být ztížena jednak velkým rozsahem léze, jednak také přítomností přechodové zóny způsobující neostrou hranici mezi patologickou a zdravou tkání. Subtotální resekce riziko recidivy významně zvyšuje. Po ní připadá v úvahu sledování nebo reoperace, radioterapie není indikována.
Nezbytné je provedení genetického vyšetření na syndrom Cowdenové. Při pozitivním nálezu je nutný celoživotní pravidelný skríning pacientů stran systémových malignit. U dysplastického gangliocytomu mozečku je nutné dlouhodobé sledování pacientů.
MUDr. Václav Vybíhal
Neurochirurgická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: vvybihal@fnbrno.cz
Přijato k recenzi: 4. 11. 2009
Přijato do tisku: 3. 5. 2010
Zdroje
1. Lhermitte J, Duclos P. Sur un ganglionneurome diffus du cortex du cervelet. Bull Assoc Fr Etud Cancer 1920; 9: 99–107.
2. Robinson S, Cohen AR. Cowden disease and Lhermitte-Duclos disease: an update. Case report and review of the literature. Neurosurg Focus 2006; 20(1): E6.
3. Awwad EE, Levy E, Martin DS, Merenda GO. Atypical MR appearance of Lhermitte-Duclos disease with contrast enhancement. Am J Neuroradiol 1995; 16(8): 1719–1720.
4. Moonis G, Ibrahim M, Melhem ER. Diffusion--weighted MRI in Lhermitte-Duclos disease: report of two cases. Neuroradiology 2004; 46(5): 351–354.
5. Nagaraja S, Powell T, Griffiths P, Wilkonson ID. MR imaging and spectroscopy in Lhermitte-Duclos disease. Neuroradiology 2004; 46(5): 355–358.
6. Douglas-Akinwande AC, Payner TD, Hattab EM. Medulloblastoma mimicking Lhermitte-Duclos disease on MRI and CT. Clin Neurol Neurosurg 2009; 111(6): 536–539.
7. Pirotte B, Goldman S, Baleriaux D, Brotchi J. Fluorodeoxyglucose and methionine uptake in Lhermitte-Duclos disease: case report. Neurosurgery 2002; 50(2): 404–408.
8. Ogasawara K, Beppu T, Yasuda S, Kobayashi M, Yukawa H, Ogawa A. Blood flow and oxygen metabolism in a case of Lhermitte-Duclos disease: results of positron emission tomography. J Neurooncol 2001; 55(1): 59–61.
9. Ogasawara K, Yasuda S, Beppu T, Kobayashi M, Doi M, Kuroda K et al. Brain PET and technetium-99m-ECD SPECT imaging in Lhermitte-Duclos disease. Neuroradiology 2001; 43(11): 993–996.
10. Nowak DA, Trost HA. Lhermitte-Duclos disease (dysplastic cerebellar gangliocytoma): a malformation, hamartoma or neoplasm? Acta Neurol Scand 2002; 105(3): 137–145.
11. Lloyd KK II, Dennis M. Cowden’s disease. A possible new symptom complex with multiple system involvement. Ann Intern Med 1963; 58: 136–142.
12. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997; 16(1): 64–67.
13. Abel TW, Baker SJ, Fraser MM, Tihan T, Nelson JS, Yachnis AT et al. Lhermitte-Duclos Disease: a report of 31 cases with immunohistochemical analysis of the PTEN/AKT/mTOR pathway. J Neuropath Exp Neurol 2005; 64(4): 341–349.
14. Padberg GW, Schot JD, Vielvoye GJ, Bots GT, de Beer FC. Lhermitte-Duclos disease and Cowden disease: a single phakomatosis. Ann Neurol. 1991; 29(5): 517–523.
15. Hashimoto H, Iida J, Masui K, Nishi N, Sakaki T. Recurrent Lhermite-Duclos disease – case report. Neurol Med Chir (Tokyo) 1997; 37(9): 692–696.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2010 Číslo 5
Nejčtenější v tomto čísle
- Neuralgie nervus pudendalis – kazuistika
- Vývoj technik PLIF a TLIF
- Syndrom útlaku ulnárního nervu v oblasti lokte – přehled operačních technik a srovnání jejich výsledků
- Střelná poranění hlavy a mozku