
Myasténia gravis
Myasthenia gravis
Myasthenia gravis (MG) is defined as a disorder of the neuromuscular junction with fluctuating weakness of voluntary muscles associated with exhaustibility. The incidence of MG is 7,0 to14,8 cases per million population per year. The prevalence rate ranges from 80,0 to191,6 per million population. The most common age at onset of MG is the second and third decades in women, the sixth and seventh decades in men. MG is a heterogenous disorder with 4 distinct subgroups which differ immunologically, clinically and in responses to various treatments: A. 75 % MG patients have seropositive MG (SPMG) with autoantibodies against nicotinic acetylcholine receptor (AChR). B. 15 % MG patients have SPMG with autoantibodies to AChR associated with thymoma and antibodies against titine. C. 7 % MG patients have seronegative MG (SNMG) to AChR and positive antibodies against MuSK (muscle specific kinase). D. 8 % MG patients have „pure“ SNMG (no autoantibodies to AChR/MuSK). Diagnostic procedures in MG are clinical evaluation, intravenous administration of neostigmin/edrophonium, EMG (repetitive nerve stimulation, single fibre electromyography), autoantibodies to AChR, MuSK, titine; chest CT or chest MR (thymoma diagnosis). Treatment focuses on anticholinesterases, immunosuppressive agents, thymectomy, and short-term interventions such as plasmapheresis, immunoadsorption and intravenous immunoglobulin (IVIg). These treatments, usually in combination, produce remission or improvement in about 90 % of MG patients. Refractory cases to immunosuppressive therapy may improve with plasmapheresis or IVIg on chronic intermittent basis. Treatment should be individualized and there is no single regimen that is appropriate for all patients.
Key words:
myasthenia gravis – neuromuscular junction – pathophysiology – imunopathogenesis – diagnosis – management – prognosis
Autoři:
Peter Špalek
Působiště autorů:
Neurologická klinika SZU FNsP Bratislava-Ružinov
Vyšlo v časopise:
Cesk Slov Neurol N 2008; 71/104(1): 7-24
Kategorie:
Minimonografie
Souhrn
Myasténia gravis (MG) je ochorením nervovosvalového spojenia, ktoré sa prejavuje kolísavou svalovou slabosťou a abnormnou unaviteľnosťou pri telesnej záťaži. Ročná incidencia MG je 7 -14,8 prípadov na 1 milión populácie. Prevalencia MG sa pohybuje v rozmedzí od 80 do 191,6 pacientov s MG na 1 milión populácie. U žien sa MG najčastejšie vyskytuje v druhej a tretej dekáde, u mužov v šiestej a siedmej dekáde. MG je heterogénne ochorenie so 4 podskupinami, ktoré sa odlišujú imunologicky, klinicky a rozdielnou reakciou na rôznu liečbu: A. 75 % pacientov má séropozitívnu MG (SPMG) s autoprotilátkami proti nikotínovým acetylcholínovým receptorom (AChR). B. 15 % pacientov s MG má séropozitívnu MG s autoprotilátkami proti AChR asociovanú s tymómom a autoprotilátkami proti titinu. C. 7 % pacientov má séronegatívnu MG (SNMG) s autoprotilátkami proti MuSK (muscle specific kinase). D. 8 % pacientov má SNMG bez autoprotilátok proti AChR a proti MuSK. V diagnostike MG sa používajú: klinické vyšetrenie (neurologické; myologické), vnútrožilné podanie neostigmínu/edrofónia, EMG (repetitívna stimulácia, single fibre electromyography), vyšetrenie autoprotilátok proti AChR, MuSK, titinu; CT alebo MR vyšetrenie hrudníka (diagnostika tymómov). V terapii sa používajú inhibítory cholínesterázy, imunosupresívne preparáty, tymektómia a procedúry na krátkodobú intervenciu (plazmaferéza, imunoadsorpcia, intravenózny imunoglobulín). Uvedené liečebné metódy, obvykle v kombinácii, docielia remisiu alebo zlepšenie asi u 90 % pacientov s MG. Prípady, ktoré sú refraktérne na imunosupresívnu liečbu, sa zlepšujú pri dlhodobej opakovanej liečbe plazmaferézou alebo IVIg. Liečba pacientov s MG musí byť individualizovaná. V súčasnosti nie je k dispozícii žiadny terapetický postup, ktorý by bol účinný u všetkých pacientov.
Kľúčové slová:
myasténia gravis – nervosvalovéspojenie – patofyziológia – imunopatogenéza– diagnostika – liečba – prognóza
História
Myasténia gravis bola prvýkrát popísaná v r. 1672 známym oxfordským lekárom Thomasom Willisom [1]. Ďalší popis myasténie gravis uskutočnil až po viac ako 200 rokoch londýnsky lekár a vedec Samuel Wilks v roku 1877 [1,2]. V práci „On cerebritis, hysteria and bulbar paralysis“ Wilks klinický obraz myasténie odlíšil od amytrofickej laterálnej sklerózy a hystérie [1]. V roku 1879 popísal Wilhelm Erb v Heidelbergu 3 pacientov s progresívnym priebehom myasténie, s remisiami a exacerbáciami, ktoré viedli k úmrtiu [2]. V roku 1895 nemecký neurológ Friedrich Jolly zhrnul dovtedy 15 známych popisov myasténie a pridal dve vlastné prípady [3]. Jolly podrobne opísal klinickú symtomatológiu ochorenia a navrhol pre ňu názov „Myasthenia gravis pseudo-paralytica“, ktorý bol definitívne prijatý na schôdzi Spoločnosti pre psychiatriu a neurológiu v r. 1899 v Berlíne [3]. V r. 1912 Michael Starr analyzoval 250 dovtedy popísaných prípadov myasténie, z ktorých skončilo 45 % fatálne [3]. V roku 1901 popísal Carl Weigert pri autopsii pacientky s myasténiou tymóm mediastína [1,3]. Prvú tymektómiu uskutočnil v roku 1911 Ernst Sauerbruch v Zurichu u 20 ročnej pacientky s myasténiou gravis a tyreotoxikózou [1]. Moderná história tymektómie je spojená s priekopníckymi prácami Alfreda Blalocka (Baltimore, USA) a Geoffreyho Keynesa (Londýn, UK) v 40. a 50. rokoch minulého storočia [1]. V Československu prvú tymektómiu z indikácie neurológa vykonal v Prahe J. Diviš v r. 1948 [2]. Václav Šmat so spolupracovníkmi na III. Chirurgickej klinike vypracovali v 70. rokoch minulého storočia špeciálnu operačnú techniku – tzv. rozšírenú tymektómiu s odstránením týmusu a celého tukového tkaniva predného mediastína [2]. Do tohto centra boli posielaní na tymektómiu až do roku 1986 aj pacienti zo Slovenska. V roku 1981 založil v Prahe chirurg Jan Vejvalka myastenický dispenzár, čo znamenalo významný prínos v liečbe a sledovaní pacientov s myasténiou gravis [2].
V liečbe myasténie použila v roku 1934 Mary Walker fyzostigmín, o rok neskôr sa začal používať prostigmin, v roku 1954 pyridostigmin bromid (Mestinon) a v roku 1955 ambenonium chlorid (Mytelase) [1, 3]. V roku 1960 publikoval John Simpson autoimunitnú hypotézu patogenézy myasténie gravis [3]. V 70. rokoch viaceré významné objavy potvrdili autoimunitnú patogenézu myasténie gravis, zásadným objavom bol dôkaz cirkulujúcich autoprotilátok proti acetylcholínovým receptorom (viď kapitola patofyziológia a imunopatogenéza MG).
Zásadný prelom v prognóze pacientov s MG nastal po zavedení dlhodobej liečby prednizonom v alternujúcich jednorazových dávkach začiatkom 70. rokov a po zavedení dlhodobej imunosupresívnej liečby azatioprínom alebo cyklofosfamidom
[5,6,7]. Významným medzníkom bolo aj prvé použitie plazmaferézy v roku 1976 [8]. Po plazmaferéze došlo k významnému zlepšeniu myasténie so súčasným poklesom hladiny protilátok proti acetylcholínovým receptorom [8]. V Bratislave bolo v roku 1978 pri Neurologickej klinike ILF konštituované Centrum pre myasténiu gravis s celoslovenskou pôsobnosťou, ktoré sa snaží o čo najrýchlejšiu aplikáciu nových liečebných metód do klinickej praxe [9, 10].
Posledným kľúčovým objavom už v 21. storočí bolo nedávne zistenie Hocha et al (2001), že niektorí pacienti so séronegatívnou myasténiou proti acetylcholínovým receptorom (cca 40 %) majú v sére prítomné autoprotilátky proti MuSK (muscle specific kinase) [11]. Tento nález, ale aj ďalšie poznatky dokazujú, že myasténia gravis je imunopatogeneticky, klinicky a reakciou na liečbu heterogénne ochorenie [12].
Fyziológia nervosvalového prenosu
Myasténia gravis (MG) je prototypom autoimunitného protilátkami sprostredkovaného ochorenia, pri ktorom sú autoprotilátky zamerané na postsynaptickú časť nervosvalového spojenia. Nervosvalové spojenie je pritom prototypom synapsy, o ktorej máme podrobné morfologické, neurofyziologické, neurochemické a neuroimunologické poznatky [4,10,12,13,14,15,18]. Mediátorom nervosvalového prenosu je acetylcholín, ktorý je uložený v tzv. vezikulách (resp. kvantách) v presynaptických zakončeniach motorických nervových vlákien. Jedno presynaptické zakončenie obsahuje približne 300 000 acetylcholínových (ACh) vezikúl, pričom v každej vezikule sa nachádza približne 10 000 molekúl ACh [3,13,15,16].
Postsynaptická časť nervosvalového spojenia je tvorená motorickou platničkou. Povrch platničky je mnohonásobne invaginovaný do tzv. junkčných záhybov, resp. tzv. sekundárnych synaptických záhybov. V ústiach záhybov sa nachádzajú postsynaptické ACh receptory [4,13,14,16,18]. V najväčšej koncentrácii sú lokalizované oproti tzv. miestam uvoľňovania („release sites“) molekúl ACh v membráne presynaptického zakončenia [4,13,16]. U zdravého jedinca sa pohybuje počet ACh postsynaptických receptorov na jednej motorickej platničke od 30 do 40 miliónov [4,9,13,17,19].
Postsynaptický ACh receptor má molekulovú váhu 250 000 a molekulovú veľkosť 360 nm [9,11,14,19]. Štruktúrne je ACh receptor zložený z niekoľkých podjednotiek, chemicky ide o kyslý glykoproteín. Tvorí ho 5 podjednotiek – 2 heterodiméry α, ďalej β, δ a potom buď podjednotka ε, ktorá je prítomná u dospelého typu receptoru alebo podjednotka γ, ktorá je prítomná u fetálneho typu receptoru, a tiež u denervovaných svalových vlákien [4,13,14]. Väzobné miesta pre ACh sú medzi heterodimérmi α 1 a δ alebo α 2 a ε (γ). Bielkovina ACh receptoru je vysoko imunogénna, a hlavná imunogénna oblasť sa nachádza extracelulárne medzi dvomi α podjednotkami [4,14].
Veľmi významnou fyziologickou vlastnosťou postsynaptických ACh receptorov je, že neustále podliehajú spontánnym procesom degenerácie a regenerácie [9,13,14,19,20,21]. Práve tieto vlastnosti ACh receptorov sa významne uplatňujú pri patogenetickej liečbe myasténie gravis. Pri rôznych formách imunoterapie sa spontánne regeneračné procesy receptorov podieľajú na zlepšovaní klinického stavu [4,17,19,21].
Jeden nervový vzruch (akčný potenciál) po dosiahnutí presynaptického zakončenia nervového vlákna aktivuje (otvára) kalciové kanály [4,13,15]. Cez tieto kanály intenzívne prenikajú kalciové ióny z extracelulárneho prostredia (synaptická štrbina) intracelulárne do presynaptického zakončenia. Kalciové ióny potom iniciujú zložitý proces mobilizácie, transportu a uvoľňovania vezikúl ACh do synaptickej štrbiny. Jeden nervový impulz uvoľní mechanizmom exocytózy 150-200 ACh vezikúl [4,11,19,21]. Molekuly ACh vstupujú do interakcie s postsynaptickými ACh receptormi, počas ktorej vznikajú konformačné (tvarové) zmeny receptorov a otvárajú sa iónové kanály asi na 1 msec – ide o tzv. aktívne štádium komplexu ACh – receptor [3,4,13,14,20]. Otvorený iónový kanál umožňuje presun sodíkových a draslíkových iónov podľa ich elektrochemických gradientov, čo vedie k vzniku akčného potenciálu svalového vlákna s následnou kontrakciou.
Jeden nervový impulz uvoľní z presynaptického zakončenia asi 150-200 vezikúl ACh z celkového počtu 300 000, ktoré sa nachádzajú v jednom presynaptickom zakončení. Napriek tomu počet interakcií medzi uvoľnenými molekulami ACh a postsynaptickými ACh receptoromi je 3-4x väčší, ako je potrebné na vyvolanie akčného potenciálu svalového vlákna, resp. vyvolanie svalovej kontrakcie [4,13,18,19,21]. To znamená, že zdravý jedinec má vysokú funkčnú rezervu (safety factor) nervosvalového prenosu. Všetky vplyvy, ktoré znižujú počet interakcií medzi molekulami ACh a postsynaptickými receptormi, môžu viesť k zlyhaniu neuromuskulárnej transmisie s klinicky manifestnou symptomatológiou [4,16-21].
Patofyziológia a imunopatogenéza myasténie gravis
1. Séropozitívna myasténia gravis (autoprotilátky proti ACh receptorom)
Hoci MG má nízku incidenciu a prevalenciu, predstavuje najčastejšie ochorenie, ktoré postihuje nervovosvalové spojenie. Fambrough a Drachman v r. 1973 použitím alfa-bungaro-toxínu (hadí toxín s ireverzibilnou väzbou na postsynaptické ACh receptory) označeným rádioaktívnym izotopom zistili, že množstvo receptorov na postsynaptickej platničke je u pacientov s MG znížené o 70 až 80 % oproti normálnemu počtu 30 až 40 miliónov receptorov u zdravých jedincov [22]. Počet postsynaptických ACh receptorov u pacientov s MG kolíše od 6 do 12 miliónov [16-20]. Použitím hadích toxínov (alfa-cobra-toxín) s pomaly reverzibilnou väzbou na ACh receptory sa umožnilo izolovať a purifikovať ACh receptory [23]. V r. 1973 Patrick a Lindstrom imunizáciou králikov purifikovanými heterológnymi ACh receptormi vytvorili experimentálny autoimunitný model myasténie gravis (EAMG) s typickými klinickými, elektrofyziologickými a farmakologickými vlastnosťami humánneho ochorenia [24]. V ďalších experimentoch sa potvrdilo, že imunizácia heterológnymi receptoromi indukuje autoimunitný proces nielen proti nim, ale aj proti autológnym receptorom [25].
U 80-85 % pacientov s MG sa zistili IgG autoprotilátky proti ACh receptorom. Táto forma myasténie sa označuje ako séropozitívna myasténia gravis (SPMG) – [16-20]. Autoprotilátky proti ACh receptorom sú polyklonálne a hlavne podskupín IgG1 a IgG3 (komplement aktivujúce). Veľkosť titrov protilátok medzi jednotlivými pacientami výrazne kolíše a nekoreluje s klinickou závažnosťou myasténie. Autoprotilátky spôsobujú významnú redukciu funkčných ACh receptorov prostredníctvom komplementom sprostredkovanej lýzy, modulačného vplyvu na degradáciu a regeneráciu ACh receptorov a formou funkčnej (imunofarmakologickej) blokády ACh receptorov [4, 12, 16-18, 20]. K deštrukcii ACh receptoru dochádza väzbou dvoch susedných receptorov protilátkou. Aktivuje sa kaskáda komplementu s produkciou membrány atakujúceho komplexu, zvyšuje sa aktivita tzv. prozápalových cytokinínov a oxidu dusnatého [25,26]. Elektrónmikroskopicky sa na nervovosvalovom spojení zisťuje rozšírenie synaptickej štrbiny, simplifikácia postsynaptických (junkčných) záhybov, výrazne je znížené množstvo ACh receptorov a znížená resyntéza nových receptorov [3,4,16,20,26]. Redukcia ACh receptorov vedie k zlyhaniu neuromuskulárnej transmisie, čo sa klinicky prejaví svalovou slabosťou a abnormnou svalovou unaviteľnosťou. Autoprotilátky je možné merať veľmi senzitívnou imunoprecipitačnou metódou, ktorá používa ACh receptory s naviazaným hadím toxínom, ktorý je označený rádioizotopom. Test prvýkrát popísali Jon Lindstrom so spolupracovníkmi v r. 1976 [27]. Je pomerne jednoduchý a v súčasnosti sa rutinne využíva v diagnostike SPMG.
SPMG spĺňa všetky kritéria autoimunitného protilátkami sprostredkovaného ochorenia [3,4,12,16,18,20,28,29]:
- prítomnosť cirkulujúcej protilátky.
- protilátka vstupuje do interakcie s cieľovým antigénom (ACh receptor)
- pasívny transfer séra, IgG od pacientov so SPMG vyvolá u zvierat model MG
- imunizácia zvierat antigénom (ACh receptor) vyvolá model MG
- zníženie hladiny autoprotilátok (plazmaferéza) spôsobuje zlepšenie MG
Príčina a mechanizmus vzniku autoagresívnej reakcie pri MG, podobne ako pri iných autoimunitných ochoreniach, nie sú známe. V imunopatogenéze SPMG sa významne uplatňuje týmus. Za fyziologických okolností týmus eliminuje niektoré potenciálne autoreaktívne T-bunkové klony. Týmus má veľký význam pri pozitívnej selekcii T-buniek, ktoré rozpoznávajú vlastné a cudzie antigény [4,20,28,29]. Asi 50 % pacientov so SPMG má folikulárnu hyperpláziu týmusu [3,4,9, 7,20,28,29,30,31]. Na rozhraní kôry a drene týmusu v perivaskulárnych priestoroch sa nachádzajú tzv. germinatívne centrá. V nich sú nahromadené autoreaktívne T a B lymfocyty, najmä fenotypy CD4+/CD8- a CD4-/CD8+. Dreňové epiteliálne štruktúry obsahujú myoidné bunky. Ide o nezrelé formy svalových buniek, na ktorých sú lokalizované ACh receptory [4,14,26,27,28]. Pri vzniku MG sa uplatňujú najmä regulačné T lymfocyty. Predpokladá sa, že autoagresívna reakcia proti ACh receptorom môže byť iniciovaná priamo v týmuse [3,4,9,20,28,29]. MHC II pozitívne bunky exprimujú antigénne epitopy ACh receptorov a vytvárajú s receptormi autoreaktívnych T-buniek trojmolekulárny komplex. Aktivácia cytokinínov spôsobuje stimuláciu špecifických B-lymfocytov, ktoré produkujú protilátky proti ACh receptorom.
MG je polygénne ochorenie. Piťha a Matějková (1998) zistili u českých pacientov s myasténiou gravis, najmä u mladých a bez tymómu, zvýšenú frekvenciu výskytu HLA antigénov A1, B8, DR3 a DQ 2 [31]. Starší pacienti sú často nositeľmi HLA haplotypov B7, DR7. MG indukovaná penicilamínom je asociovaná s HLA DR1 [31].
Hoci MG je v súčasnosti asi najlepšie objasneným autoimunitným ochorením, nepodarilo sa u geneticky predisponovaných jedincov odhaliť konkrétny spúšťajúci mechanizmus ochorenia.Významne sa tu zrejme uplatňujú vplyvy vonkajšieho prostredia.
Pacienti vo veku nad 45-50 rokov majú SPMG asociovanú s involvovaným, atrofickým týmusom [12,17,20,28,29,30]. V týchto atrofických týmusoch sa len zriedka zisťujú ojedinelé germinatívne centrá. Pri nálezoch atrofického týmusu býva tymektómia obvykle neúčinná [14,16,17,24,26,28]. Tieto fakty svedčia pre rozhodujúcu účasť extratýmusových autoimunitných mechanizmov v imunopatogenéze SPMG u pacientov vo vyššom veku [17,26].
2. Séronegatívna myasténia gravis (SNMG) s autoprotilátkami proti MuSK
Niektorí pacienti s MG (15-20 %) nemajú autoprotilátky proti ACh receptorom. U pacientov so SNMG je evidentné, že tiež sa jedná o cirkulujúcimi autoprotilátkami sporstredkované ochorenie. Potvrdením tejto skutočnosti je [16,17,18,20,26]:
- U pacientov so SNMG nastáva významné zlepšenie klinického stavu po plazmaferéze.
- Aplikácia sér, IgG od pacientov so SNMG vyvolá u myší experimentálny model myasténie s detekovateľnou poruchou neuromuskulárnej transmisie.
Hoch et al v roku 2001 zistili, že časť pacientov so SNMG má autoprotilátky proti MuSK - muscle specific kinase – najmä triedy IgG4 [11]. MuSK je povrchový receptorický proteín, je súčasťou membrány svalového vlákna a má kľúčovú úlohu pri vývoji neuromuskulárneho spojenia [12,18,32]. Akým mechanizmom spôsobujú autoprotilátky proti MuSK postsynaptickú poruchu neuromuskulárnej transmisie nie je presne jasné. Vo svaloch pacientov s anti-MuSK protilátkami sa nezisťuje signifikantná redukcia ACh receptorov alebo expresie MuSK, junkčné postsynaptické záhyby sú dobre zachované, ich denzita len mierne redukovaná, depozitá imunoglobulínu a komplementu nepatrné [32,33]. V súčasnosti prevláda názor, že autoprotilátky sú namierené proti extracelulárnej časti MuSK a spôsobujú inhibíciu agrínom indukovanej aktivácie MuSK, t.zn. poruchu agregácie, clusteringu ACh receptorov, čím spôsobujú postsynaptické zlyhanie neuromuskulárnej transmisie.
Pacienti so SNMG a autoprotilátkami proti MuSK nemajú hyperpláziu týmusu a ani tymómy, ich týmus býva involvovaný [12,17,32,34,35]. Tymektómia je u tejto skupiny pacientov neefektívna [12,17,32,34,35]. V patogenéze SNMG s autoprotilátkami proti MuSK sa jednoznačne uplatňujú extratýmusové autoimunitné mechanizmy. Pacienti so SNMG a autoprotilátkami proti MuSK tvoria približne 7 % z celkového počtu všetkých pacientov s MG, pričom ženy sú 4x častejšie postihnuté ako muži [17,34,35]. Myastenická symptomatológia býva u pacientov s touto formou MG výraznejšia ako u pacientov so SPMG [12,34,35]. Možno konštatovať, že existujú tri klinické typy generalizovanej SNMG s autoprotilátkami proti MuSK [12,34,35]: 1. typ: generalizovaná MG s výraznou okulobulbárnou slabosťou. Niektorí z týchto pacientov majú atrofiu svalstva jazyka a faciálnych svalov. Končatinové svalstva býva postihnuté, ale nie výrazne. 2. typ: generalizované postihnutie s dominantnýmm postihnutím šijového, ramenného a respiračného svalstva. 3. typ: generalizované postihnutie – neodlíšiteľné od klinických príznakov pri SPMG. Diagnostický význam týchto troch klinických fenotypov však nemožno preceňovať.
SNMG s anti-MuSK protilátkami bola popísaná aj u čisto okulárnej formy myasténie, u izolovaného postihnutia extenzorov šije a aj pri ďalších klinických prejavoch [12]. SPMG s autoprotilátkami proti AchR sa môže vyskytovať v kombinácii so SNMG s anti-MuSK autoprotilátkami, podobne ako sa vyskytuje SPMG v kombinácii s inými autoimunitnými ochoreniami. Kombinovaný výskyt SPMG s autoprotilátkami proti AchR a SNMG s autoprotiltkami proti MuSK bol popísaný v Anglicku a Španielsku [12].
3. Séronegatívna myasténia gravis s negatívnymi autoprotilátkami proti MuSK
Ide o pacientov s okulárnou alebo generalizovanou MG, ktorí nemajú v sére detekovateľné autoprotilátky proti ACh receptorom ani proti MuSK 12, 17, 32). Klinická symptomatológia, elektrofyziologické nálezy, patológia týmusu, reakcia na tymektómiu a ďalšie formy imunoterapie sú pri tejto forme SNMG rovnaké ako pri
SPMG s autoprotilátkami proti ACh receptorom [12,36,37]. Týmus býva u pacientov s touto formou SNMG hyperplastický, s germinatívnymi centrami, s populáciami lymfocytov, ktoré majú rovnaké parametre ako v lymfocyty v týmusoch pacientov so SPMG s autoprotilátkami proti ACh receptorom [37]. Tento nález, spolu s efektívnosťou tymektómie, svedčí pre účasť týmusu v patogenéze SNMG
s negativitou proti MuSK. Hypoteticky sa predpokladá, že táto forma SNMG môže byť podmienená veľmi nízkymi hladinami cirkulujúcich autoprotilátok proti ACh receptorom [12,16,36]. Je však tiež možné, že SNMG s negativitou proti MuSK je klinicky a imunologicky heterogénne ochorenie [12,36]. Z celkového počtu pacientov s MG tvoria pacienti s touto formou SNMG (negativita proti ACh receptorom aj proti MuSK) približne 8 % [17,36].
4. MG asociovaná s tymómom, autoprotilátkami proti ACh receptoru, titinu a ryanodinovému receptoru
Približne 10-15 % pacientov s MG má tymóm [3,4,9,17,20,21,26,38-40]. Títo pacienti majú v 100 % prítomné autoprotilátky proti acetylcholínovým receptorom a nikdy nemajú autoprotilátky proti MuSK. Tymómy neprodukujú myoidné bunky, preto intratýmusová patogenéza tejto myasténie je nepravdepodobná, pri jej vzniku sa uplatňujú extratýmusové imunopatologické mechanizmy. Tymómy však exprimujú celý rad antigénov, ide najmä o titin a ryanodinový receptor. Autoprotilátky proti titinu sú obligátnym nálezom u pacientov s myasténiou a tymómom, častá je aj prítomnosť autoprotilátok proti ryanodinovému receptoru [3,4,17,18,20,26,28,38,39,40]. Protilátky proti titinu aleb ryanodinovému receptoru sa príležitostne zisťujú aj u starších jedincov s netymomatóznou MG. Napriek tomu vyšetrenie autoprotilátok proti titinu má aj diagnostický význam, ich pozitivita nasvedčuje pre myasténiu gravis asociovanú s tymómom [39,40,41]. Po chirurgickom odstránení tymómu dochádza k zreteľnému poklesu autoprotilátok proti titinu [40]. Kalifornskí neurológovia okolo M. Agiusa a nórski neurológovia okolo prezidenta svetovej neurologickej federácie J. Aarliho považujú túto formu myasténie za samostatnú nozologickú jednotku [41].
Epidemiológia
Myasténia gravis postihuje jedincov oboch pohlaví v ktoromkoľvek veku, bez rasovej, etnickej a socioprofesijnej predilekcie [3,9,20,42,43,44]. Výnimkou sú údaje epidemiologickej štúdie z roku 1992 z Virgínie v USA, podľa ktorej sa myasténia gravis vyskytuje častejšie u Afroameričanoch ako u bielej populácie [45]. Prevalencia myasténie sa podľa posledných epidemiologických štúdií pohybuje v hodnotách od 80 do 140 na 1 milión populácie [3,20,44-48]. Prevalencia myasténie gravis v Slovenskej republike bola k 1. januáru 2007 191,6 na 1 milión populácie. Incidencia sa pohybuje od 7 do 10 nových prípadov myasténie na 1 milión populácie ročne [3,20,44-47]. Priemerná ročná incidencia myasténie gravis na Slovensku za obdobie 1997-2006 je 14,8 nových prípadov na 1 milión obyteľstva. Údaje o incidencii a prevalencii myasténie majú stúpajúcu tendenciu, sú podmienené významným zlepšením diagnostiky a liečby MG, a tiež narastajúcim výskytom autoimunitných ochorení [18-21].
Myasténia gravis vzniká častejšie u žien ako u mužov. V štúdiach z 50. až 70. rokoch minulého storočia bol tento pomer 2-3: 1 v prospech žien [3,9,21]. U žien vzniká MG najčastejšie v 2. a 3. dekáde, u mužov v 6. a 7. dekáde [20,48] Pomer postihnutia žien a mužov sa postupne vyrovnáva, na Slovensku bol k 1. januáru 2007 1,4:1. Priemerný vek pri vzniku MG sa u mužov aj žien posúva do vyšších skupín. Priemerný vek žien pri vzniku MG bol na Slovensku podľa štúdie z r. 1983 28,4 rokov, k 1. januáru 2007 sa zvýšil na 45,4 rokov [9]. Priemerný vek slovenských mužov pri vzniku MG bol v r. 1983 45,2 rokov, k 1.1.2007 stúpol na 56,7 rokov [9]. Asi u 10 % pacientov vzniká MG v detskom veku pred 15 rokom života.
Prvá epidemiologická štúdia o SNMG s anti-MuSK protilátkami bola realizovaná roku 2007 v Holandsku [49]. Anti-MuSK protilátky zistili u 35 pacientov z celkového počtu 97 pacientov so SNMG, čo zodpovedá prevalencii 1,9 na 1 milión a incidencii 0,1 na milión [49]. Holandskí neurológovia zistili, že priemerný vek pacientov pri vzniku SNMG s anti-MuSK pozitivitou (35 r.) je o 12 rokov nižší ako u pacientov so SPMG (47 r.) s autoprotilátkami proti ACh receptorom [49]. SNMG s autoprotilátkami proti MuSK postihuje ženy 4x častejšie ako mužov [17,49].
Klinický obraz
Klinické prejavy MG sú veľmi variabilné, čo je podmienené značnými interindividuálnym rozdielmi v distribúcii, intenzite a vývoji myastenickej symptomatológie. Tieto rozdiely sa vysvetľujú rôznym časovým uplatnením heterogénnych autoimunitných mechanizmov v imunopatogenéze a patofyziológii MG [3,4,9,17,19-21,26,28,35,36,39,41,43,47]. Správne určenie diagnózy MG pri jej nízkom výskyte je pre neurológa vždy veľkou výzvou. Všeobecne možno konštatovať, že MG sa manifestuje patologickou svalovou slabosťou, ktorá je združená s abnormnou unaviteľnosťou pri fyzickej záťaži. Tieto ťažkosti sa upravujú, často len čiastočne, v kľude a po podaní inhibítorov cholínesterázy.
Iniciálne príznaky myasténie gravis
V iniciálnom štádiu ochorenia môže byť postihnutý ktorýkoľvek sval, ale určité svalové skupiny bývajú predilekčne postihnuté:
1. Postihnutie vonkajších okohybných svalov (diplopia) a/alebo postihnutie m. levator palpabrae superior (ptóza) sú najčastejším iniciálnym príznakom myasténie asi u 50-60 % pac. [3,9,20,26,50,51,52,53,54,55]. MG sa môže manifestovať aj izolovanou slabosťou musculus orbicularis oculi, ktorá sa prejaví zníženou frekvenciou žmurkania a neschopnosťou dovrieť oko [56]. Asi u 10 % pacientov s okohybnou symptomatológiou nedôjde ku generalizácii myastenických symptómov ani po 3-4 rokoch trvania, vtedy ide o okulárnu formu MG [3,9,20,26,50,51,53,54,55]. Generalizácia okulárnej formy MG však môže nastať aj po viacročnom trvaní ochorenia. Okohybné príznaky sa v priebehu trvania myasténie manifestujú celkove až u 85-90 % pacientov [3,9,20,50,51,53,55,56]. Určité vlastnosti vonkajších okohybných svalov znamenajú predispozíciu k ich častému symptomatickému postihnutiu pri MG [9,56]. Patria k nim: a) Už ľahká slabosť niektorého z externých okohybných svalov spôsobí diplopiu vychýlením vizuálnych osí. b) Okohybné svaly majú vyššiu frekvenciu nervovosvalových spojení (resp. menšie motorické jednotky) c) Okohybné svaly majú nižšiu denzitu ACh receptorov a tým aj nižšiu funkčnú rezervu („safety factor“) neuromuskulárnej transmisie [56].
Pacienti s MG sa subjektívne sťažujú na rozmazané videnie alebo diplopiu kolísavej intenzity a na ptózu, ktorá je často asymetrická. Udávajú, že príznaky sa zhoršujú oslnením, osvitom, prolongovanou vizuálnou záťažou (čítanie, sledovanie TV, šoférovanie). Viacerí pacienti pozorujú, že v prítmí alebo pri nosení tmavých okuliarí diplopia ustupuje.
2. Slabosť svalstva inervovaného bulbárnymi nervami býva iniciálnym príznakom myasténie v 15-20 % [3,9,19,20,21,26,42,50,51,53,58-60]. Prejavuje sa poruchami artikulácie (dysartria), fonácie (zmeny hlasu, rhinolalia), slabosťou mäkkého podnebia (pri pití regurgitácia tekutín nosom), slabosťou jazyka (dysartria, ťažkosti s posúvaním potravy v ústnej dutine) a poruchami prehĺtania (riziko aspirácie častí potravy).
3. Slabosť mimického svalstva alebo žuvacieho svalstva sa vyskytuje ako iniciálny príznak MG u 5 až 10 % pacientov [3,19,20,26,42,50,51,53]. Slabosť mimického svalstva sa prejavuje „ospalým (smutným)“ výrazom tváre, neschopnosťou našpúliť pery, neschopnosťou vyslovovať perové hlásky a neschopnosťou pískať. Prejavom oslabenia mimického svalstva býva aj neschopnosť dovrieť očné štrbiny (slabosť m. orbicularis oculi). Slabosť žuvacieho svalstva sa prejavuje poruchami hryzenia a žuvania potravy. Slabosť žuvacích svalov sa prejaví aj poklesom dolnej čeluste, ktorú si pacienti počas jedenia charakteristicky pritláčajú rukou.
4. Postihnutie končatinového svalstva v proximálnej distribúcii býva iniciálnym prejavom MG asi u 5-10 % pacientov s MG [3,9,19,20,42,50-53]. Pri prirodzenej progresii a generalizácii myasténie sú postihnuté končatinové svaly až u 70-80 % pacientov. Proximálne svalstvo na horných končatinách býva častejšie postihnuté ako na dolných, ale aj z charakteru používania končatín vyplýva, že pacienti si viac uvedomujú postihnutie svalov HK ako DK. Udávajú ťažkosti pri česaní, umývaní vlasov, sprchovaní, pri vešaní prádla, pri chôdzi do schodov, prípadne pri chôdzi mávajú pády.
5. Slabosť niektorej izolovanej svalovej skupiny býva iniciálnym príznakom MG cca u 10 % pacientov. Napr. slabosť extenzorov šije sa prejavuje poklesom, prepadávaním hlavy do anteflexie a pacienti si hlavu typicky podopierajú rukou [20,50,51,54,55]. Postihnutie extenzorov prstov sa prejavuje flekčným držaním prstov a neschopnosťou vystrieť prsty [50,51,55,60]. MG sa môže inicálne manifestovať aj izolovaným postihnutím flexorov bedrového kĺbu alebo extenzorov nohy [3,51,54,55].
6. Slabosť diafragmy, interkostálnych svalov a akcesórnych svalov zapríčiňuje dyspnoické ťažkosti a môže pacienta vitálne ohroziť. Pri prevažnom postihnutí diafragmy má dyspnoe insipiračný charakter, pri oslabení interkostálnych a abdominálnych svalov viac expiračný charakter. Výraznejšie respiračné ťažkosti sa vyskytujú predovšetkým v rámci progresívnej generalizovanej myastenickej symptomatológie. V ojedinelých prípadoch sa MG iniciálne manifestuje aj pod obrazom selektívneho postihnutia dýchacieho svalstva, čo môže spôsobovať značné diagnostické ťažkosti [9,20,50,51,54].
Priebeh myasténie
Intenzita manifestácie prvých myastenických príznakov je podobne variabilná ako uvedená rôznorodosť ich distribúcie. U väčšiny pacientov sa myasténia manifestuje plíživo s kolísajúcou intenzitou príznakov [9,20,50,51,55]. Iniciálne príznaky bývajú ohraničené na určité svalové skupiny, prípadne len na jeden sval. Neskôr, po rôznom časovom odstupe a s variabilnou progresiou, dochádza ku generalizácii myastenickej symptomatológie. Vznik myastenických ťažkostí alebo ich zvýraznenie môže byť spôsobené pod vplyvom viacerých exogénnych nox [3,9,20,21,26,42,43,50,51,52,55,56,57,61,62]. Najčastejšie ide o febrilné ochorenia, najmä infekty horných dýchacích ciest, operácie, lieky s inhibičným vplyvom na neuromuskulárnu transmisiu (aminoglyzidové a polypetidové antibiotiká, tetracyklíny, beta-blokátory, antiarytmiká, neuroleptiká) a tiež psychický stres. K závažným zhoršeniam MG aj s vitálnym ohrozením pacientov úlmom respiračného svalstva môže dôjsť najmä po kurariformných myorelaxanciách, celkových anestetikách a po intravenóznom podaní magnézia [9,61,62]. Pacienti s MG majú absolútnu kontraindikáciu k užívaniu d-penicilamínu. Je dobre známe, že užívanie d-penicilamínu, najmä u pacientov s reumatoidnou artritídou, môže indukovať myasténiu [18,20,26,63,64,65].
U žien dochádza niekedy k zvýrazneniu myastenickej symptomatológie počas menštruácie [43,66]. Počas tehotenstva máva MG tendenciu k spontánnemu zlepšovaniu a vznik myasténie v tehotenstve je vzácny [67,68]. V puerpériu sú popisované zhoršenia myastenickej symptomatológie a môže tiež dochádzať k prvomanifestáciam MG [67,69]. Rozdiely medzi tehotenstvom a puerpériom sa vysvetľujú hormonálnymi vplyvmi a imunosupresívnym pôsobením alfa-fetoproteínu plodu na matku [70]. Po pôrode dochádza k rýchlemu poklesu alfa-fetoproteínu v sére matiek, čím sa vysvetľujú zhoršenia myasténie v puerpériu [70].
Tranzitórna neonatálna myasténia vzniká asi u 7-15 % novorodencov matiek s MG v dôsledku transplacentárneho prieniku materských autoprotilátok do tela plodu počas tehotenstva [3,18,19,20,26,67,68,69]. Nejedná sa o aktívne ochorenie, ide vlastne o prejav „pasívnej imunizácie“. Prognóza tranzitórnej neonatálnej myasténie je výborná. Novorodenci reagujú veľmi priaznivo na adekvátne dávky inhibítorov cholínesterázy a antireceptorické autoprotilátky vymiznú do 2 až 5 týždňov po pôrode.
Prirodzeným priebehom MG, provokáciou exogénnymi noxami, ale aj nedostatočnou imunosupresívnou liečbou (oneskorený začiatok liečby; nízke dávky imunosupresív), môže prísť relatívne náhle k vzniku myastenickej krízy [3,9,20,21,42,50,51,55,71,72,73,74].
V nedávnej minulosti relatívne časté cholinergné krízy alebo zmiešané krízy sú v súčasnosti pri včasnej diagnostickej záchytnosti myasténie a adekvátnom terapeutickom postupe už veľkou vzácnosťou [20,22,72-74]. Varovným signálom hroziacej myastenickej krízy je zvýraznenie slabosti faciobulbárnych svalov, neschopnosť prehĺtať, nemožnosť odkašľať, nekľud pacienta a nástup dušnosti. Liečba myastenickej krízy vyžaduje úzku spoluprácu neurológa (riadi imunopatogenetickú liečbu – kombinovaná imunosupresívna liečba, podávaná v prípade potreby nazogastrickou sondou, plazmaferéza a/alebo vysoké dávky intravenózne podávaných imunoglobulínov) a lekára intenzívnej medicíny [71-74].
Klasifikácia myasténie gravis
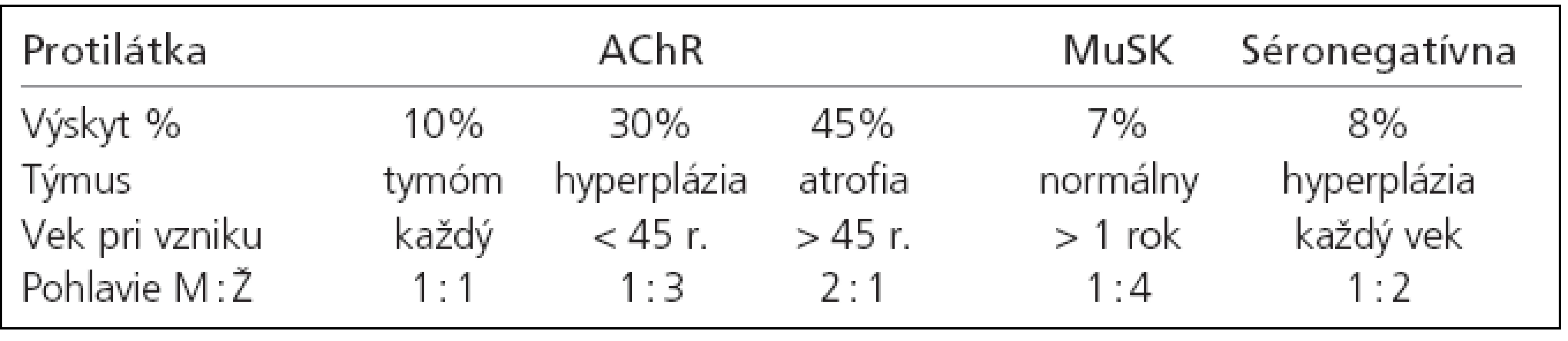
Imunopatogenetická klasifikácia podľa Newsom-Davisa z r. 2007 je v tab. 1. [17]. Diskutovaná je v časti o patofyziológii a imunopatogenéze myasténie gravis. Pôvodná klinická Ossermanova klasifikácia sa mierne modifikovala do podoby, ktorá lepšie zohľadňuje klinické kritéria a klinický priebeh myasténie - tab. 2. [52].

Pre lepšie objektívne posudzovanie klinických nálezov a pre účely klinických štúdií boli navrhnuté viacerými autormi škály s kvantifikovaným myastenickým skóre [75-78]. Podľa autora tohto textu tieto klasifikačné schémy pre potreby bežnej klinickej praxe nemajú podstatnejší význam.
Myasténia gravis a pridružené ochorenia
MG a týmus
MG je obligátne spojená s abnormitami týmusu. U mladých jedincov cca do veku 45 až 50 rokov býva prítomná hyperplázia týmusu, nad touto vekovou hranicou je obligátnym nálezom involúcia, resp. atrofia týmusu [3,4,9,17,20,26,28,42,43,51].
10 až 15 % pacientov pacientov má MG asociovaná s tymómom [17,20,26,38-41,79,80,81]. 2/3 tymómov majú benígny charakter. 1/3 tvoria malígne tymómy, ktoré majú invazívny rast, vzdialené metastázy sú veľmi vzácne [79–81]. Pacienti s tymómom majú v 100 % pozitívne autoprotilátky proti ACh receptorom a nemajú nikdy autoprotilátky proti MuSK [12,17,38,39,41]. Obligátnym markerom asociácie MG s tymómom sú autoprotilátky proti titinu [38,40].
MG a autoimunitné ochorenia
15 až 20 % pacientov s MG má pridružené jedno alebo aj viac autoimunitných ochorení [3,9,20,21,26,38,43,51,82-85]. Najčastejšie ide o autoimunitné ochorenia štítnej žľazy, autoimunitné hematologické ochorenia, reumatoidnú artritídu a Sjögrenov syndróm. Z autoimunitných neurologických ochorení bývajú s MG asociované polymyozitída, Lambert-Eaton myastenický syndróm, neuromyotónia a sclerosis multiplex [3,20,38, 82-85]. Asociácie MG s týmito ochoreniami môžu spôsobovať značné diagnostické problémy a vyžadujú obvykle intenzívny a veľmi dôsledný terapeutický manažment.
Diagnostika
Určenie diagnózy MG u pacientov s rozvinutou generalizovanou symptomatológiou je pomerne jednoduché. U ostatných prípadov správne určenie diagnózy MG spôsobuje často značné problémy. Autoimunitný proces môže postihnúť akýkoľvek sval a svaly v najrôznejšej kombinácii a tak myasténia gravis pri kolísavej intenzite svojich príznakov môže imitovať rôzne neurologické a iné ochorenia [3,21,54,55]. Z katamnestických štúdií v Holandsku, Anglicku, Dánsku, USA, Maďarsku a na Slovensku vyplýva, že u 45 až 80 % pacientov býva MG najprv mylne diagnostikovaná [9,20,21,50,51,54,55,79,86,87]. Z mylných diagnóz pripadá 1/3 na nesprávne neurologické diagnózy , 1/3 na psychiatrické diagnózy a 1/3 na ostatné diagnózy.
K vyšetrovacím metódam pri podozrení na myasténiu gravis patria:
1. Detailná anamnéza a analýza subjektívnych ťažkostí, vplyvu provokačných (zhoršujúcich) faktorov na obvykle kolísavú intenzitu subj ťažkostí, atď.
2. Klasické neurologické vyšetrenie a podrobné myologické vyšetrenie [3,9,20,21,51, 52,55]. Patria sem najmä statické (výdrž) a dynamické (opakované pohyby) záťažové testy, ktoré umožnia demaskovať latentnú svalovú slabosť alebo zvýrazniť už prítomnú svalovú slabosť. Najčastejšie používané testy sú:
Simpsonov test - pri miernej ptóze sa pacient 1 min. pozerá nahor, aktivuje sa aj m. frontalis a dochádza k zvýrazneniu ptózy.
Gorelickov príznak – pri bilaterálnej asymetrickej ptóze: pacient pozerá nahor a prstom pasívne elevujeme viečko na strane výraznejšej ptózy, na kontralaterálnej strane dochádza v priebehu niekoľkých sekúnd k úplnemu poklesu, k uzavretiu očnej štrbiny. Tento fenomén je patognomický pre MG, i keď jeho patofyziologická podstata nie je objasnená. Pri asymetrickej ptóze iného pôvodu (endokrinné oftalmopatie, oftalmoplégia plus syndróm, okulárna myozitída, atď) Gorelickov príznak nie je nikdy prítomný [9,21,87].
Seemanova skúška - čítanie nahlas, demaskujú sa poruchy výslovnosti, fonácie.
Trendelenburgov príznak (statický záťažový test) – stoj na jednej nohe. Pri slabosti v oblasti lumbosakrálneho svalstva dôjde na kontralaterálnej strane k postupnému poklesu pánve. Trendelenburgov príznak je pozitívny napr. aj u pletencových foriem svalových dystrofií, pokles pánve je pri nich okamžitý, pri MG je obvykle postupný.
Dynamické záťažové testy – možno nimi testovať šijové svaly a hlavne akékoľvek končatinové svaly.
3. Elektrodiagnostika:
A. Repetitívna stimulácia periférnych nervov a plexus brachialis elektrickými impulzmi o frekvencii 2 alebo 3 H [89,90,91]: V pozitívnom prípade sa v príslušnom svale zisťuje dekrement - pokles amplitúdy evokovanej odpovede o viac ako 10 % (posudzuje sa zníženie amplitúdy a arey sumačného svalového potenciálu medzi 1. a 4. alebo 5. odpoveďou). Nález je značne špecifický pre poruchu neuromuskulárnej transmisie pri myasténii gravis. Inak môže byť dekrement prítomný pri kongenitálnych myasténiách a hraničné hodnoty dekrementu sa môžu zistiť aj u pacientov s amyotrofickou laterálnou sklerózou.
Diagnostické možnosti repetitívnej stimulácie pri MG sú obmedzené 2 faktormi:
- a) Nervosvalový prenos možno vyšetriť len v niektorých svaloch, ktoré sú inervované periférnymi nervovým štruktúrami prístupnými povrchovej stimulácii elektrickými impulzami.
- b) Pri známych interindividuálnych rozdieloch v lokalizácii myastenickej symptomatológie nález závisí od toho, či vyšetrovaný sval je alebo nie je postihnutý myastenickým procesom.
U nás podrobné práce o elektrodiagnostike pri myasténii gravis publikoval Z. Ambler [89,90,92]. U diagnosticky hraničných prípadov Ambler (1990) s úspechom používa aktivačné testy (1-minútová maximálna izometrická kontrakcia a ischemický test), ktoré zvyšujú senzitivitu (veľkosť dekrementu) pri nízkofrekvenčnej repetitívnej stimulácii [92].
B. Single fibre electromyography (SFEMG )pri vôľovej aktivite a stimulovaná SFEMG – sú vysoko senzitívne, ale žiaľ menej špecifické metodiky [89,90]. Patologické hodnoty hodnotených parametrov (jitter) alebo blokovanie neuromuskulárnej transmisie u pacientov s MG až v 95 % [90,91]. SFEMG umožňuje kvantitatívne študovať neuromuskulárnu transmisiu v jednotlivých nervosvalových spojeniach. Špeciálna intramuskulárna elektróda umožňuje registrovať akčné potenciály 2 susedných svalových vlákien, ktoré patria tej istej motorickej jednotke. Akčné potenciály svalových vlákien, ktoré vznikajú pri aktivácii motorickej jednotky nie sú úplne synchrónne. Časový interval medzi akčnými potenciálmi (tzv. interpotenciálový interval) z 2 susedných vlákien tej istej motorickej jednotky sa do určitej miery mení pri opakovaných za sebou idúcich impulzoch. Tento fenomén sa nazýva jitter a je spôsobený variabilitou synaptického zdržania pri nervosvalovom prenose. V zdravom svale sa priemerná hodnota interpotenciálneho intervalu (jitteru) pohybuje od 10 do 50 usek. Pri MG jitter často presahuje 100 usek. Čím výraznejšia je porucha nervosvalového prenosu, tým je jitter dlhší. Pri výraznej poruche sa objavuje blokáda impulzu, resp. určitá časť impulzov nevyvolá akčný potenciál na 1 z 2 vyšetrovaných svalových vlákien. Progresívne zvyšovanie jitteru poukazuje na progresívny pokles amplitúdy excitačných postsynaptických potenciálov v dôsledku úbytku ACh receptorov, prinajmenšom v 1 z 2 vyšetrovaných svalových vlákien. V svaloch postihnutých myastenickým procesom je typickým nálezom, že niektoré svalové vlákna, resp. ich nervosvalové spojenia, majú normálne hodnoty jitteru, iné predĺžené hodnoty jitteru a v ďalších môže byť prítomný aj blok. Za hraničné kritérium pre diagnózu MG sa považuje ak z 20 registrovaných hodnôt jitteru sú viac ako 2 patologické alebo ak priemerná hodnota jitteru je väčšia ako 34 usek [91].
4. Reparačný farmakologický test. Inhibítory cholínesterázy aplikované intravenózne (Tensilon - edrophonium chlorid; Syntostigmin – neostigminum monomethylsulfuricum) zlepšujú nervosvalový prenos zvýšením počtu interakcií medzi ACh a postsynaptickými ACh receptormi [3,9,42,52,54,55]. Efektívnosť testu sa hodnotí najmä klinicky, ale môže sa aj repetitívnou stimuláciou. Test je diagnosticky pozitívny ak sa klinický nález alebo EMG dekrement pri repetitívnej stimulácii zlepšia alebo úplne upravia. Účinok Tensilonu je krátkodobý (cca 15 min.), po jeho aplikácii možno pozorovať nielen zlepšenie/ústup myastenickej symptomatológie, ale cca po 15 min. manifestáciu pôvodných príznakov. Tensilon však nie je registrovaný v Českej ani v Slovenskej republike.
5. Dôkaz protilátok proti acetylcholínovým receptorom býva pozitívny asi u 85 % pacientov s MG [4,12,16,17,18,20,26]. Potvrdzuje diagnózu SPMG. Veľmi zriedkavo sa vyskytujú u starších jedincov prípady s nízkymi titrami autoprotilátok (cca 0,3-1nmol/l) bez manifestných myastenických prejavov („subklinická forma MG“). Tieto nálezy sa niekedy nesprávne označujú ako falošne pozitívne.
6. Dôkaz protilátok proti MuSK býva pozitívny u 7 % pacientov s MG [12,17,18,32, 34, 35]. Potvrdzuje diagnózu SNMG s autoprotilátkami proti MuSK.
7. Dôkaz protilátok proti titinu. Ich dôkaz nepotvrdzuje samotnú MG, ale svedčí pre asociáciu MG s tymómom [39,40,41,42].
8. Stapediová reflexometria. Pri 30 sekundovej kontinuálnej (alebo prerušovanej) aplikácii nadprahového tónu sa u 85 % pacientov s MG zisťuje pokles veľkosti reflexnej kontrakcie stapediového svalu o 25 až 80 % [9,21,93]. Po intravenóznej aplikácii neostigminu sa tento nález zlepší alebo upraví. Stapediová reflexometria sa v súčasnosti v diagnostike MG používa len zriedkavo. Pred stapediovou reflexometriou je potrebné otoskopické a audiometrické vyšetrenie, a aj pri samotnom vyšetrení je potrebná spolupráca s audiológom.
10. CT (alebo MR) vyšetrenie hrudníka musia absolvovať všetci pacienti s MG za účelom potvrdenia alebo vylúčenia tymómu. Tymóm je absolútnou indikáciou k tymektómii, jediné kontraindikácie sú vysoký vek a závažné pridružené ochorenia [2,3,9,20,21,38,39,55,79,80,84,94,95,114]. CT a MR sú v detekcii abnormít týmusu ekvivalentné, včítane tymómov mediastína [95]. Dôležitý je fakt, že na bežnom roentgenograme sa 25 % tumorov týmusu vôbec nezachytí.
Liečba
Ciele liečby MG sú nasledovné:
- a) zlepšiť čo najlepšie funkcie pacienta s MG
- b) dosiahnúť zlepšenie čo najrýchlejšie
- c) s minimom vedľajších prejavov
- d) pri použití, pokiaľ možno, najjednoduchšej terapie.
Liečba myasténie gravis vyžaduje individuálny prístup ku každému pacientovi. U každého pacienta je nutné osobitne zohľadňovať závažnosť, distribúciu a rýchlosť progresie myastenickej symptomatológie, stupeň funkčného postihnutia, pohlavie, vek a prítomnosť pridružených ochorení. Pacienta je potrebné informovať o prognóze, o bezprostredných a dlhodobých cieľoch liečby. Cieľom liečby je navodiť klinickú alebo farmakologickú remisiu, prípadne stabilizovať ochorenie tak, aby sa dosiahla maximálne uspokojivá kvalita života. Zásadný a veľmi priaznivý obrat v liečbe a prognóze chorých aj s veľmi ťažkými formami MG nastal po zavedení moderných metód imunologickej liečby v 70. a 80. rokoch 20. storočia – dlhodobé podávanie prednizonu v jednorazových alternujúcich dávkach, imunosupresia azatioprínom a cyclofosfamidom, racionálne indikácie na tymektómiu, plazmaferéza, vysoké dávky vnútrožilne podávaného imunoglobulínu a rôzne kombinácie týchto liečebných foriem [3,9,19,20,21,26,52,94].
1. Inhibítory cholínesterázy
Inhibítory cholínesterázy (ICHE) brzdia enzymatickú hydrolýzu acetylcholínu na nervovosvalovom spojení, takže sa zväčšuje množstvo ACh a jeho efekt sa prolonguje [3,19,20,21,26,52,94,95,96]. Tým sa zvýši počet interakcií medzi molekulami ACh a postsynaptickými ACh receptormi, resp. sa zlepší neuromuskulárna transmisia. ICHE vedú u veľkej väčšiny pacientov s MG k výraznému zlepšeniu myastenickej symptomatológie.
Pacienti so SNMG s autoprotilátkami proti MuSK reagujú v 30-60 % na ICHE len nevýrazne alebo vôbec [12,34,35]. U niektorých sa dokonca manifestovala intolerancia s nežiadúcimi muskarínovými a nikotínovými vedľajšími účinkami [34, 35]. Popísaná bola aj hypersenzitivita so zvýraznením myastenických príznakov po užití ICHE [35]. Príčina tejto rozdielnej reaktibility pacientov so SNMG pozitívnou proti MuSK na ICHE nie je známa.
Najpoužívanejším preparátom je pyridostigmin bromid (Mestinon), zriedka sa používa neostigmin bromid (Syntostigmin). Ambenonium (Mytelase) a distigmin (Ubretid) sa používajú len výnimočne. Mestinon je preferovaný kvôli prolongovanému účinku (4-6 hod.) a minimálnym vedľajším gastrointestinálnym účinkom. Terapia sa u dospelých začína dávkami 60 mg každých 4-5 hodín. Najobvyklejšia je počiatočná dávka 4x1 podávaná o 7–11–15–19 hod. V noci Mestinon obvykle nie je potrebné podávať. Hlavným cieľom liečby je priaznivo ovplyvniť najviac postihnuté svaly. Napr. u pacientov s orofaryngeálnou slabosťou podávať Mestinon tak, aby v čase jedla mohli potravu dobre pohrýzť, prežuť a prehltnúť. Nežiadúce muskarínové a nikotínové cholinergné účinky – zvýšená salivácia, bronchiálna sekrécia, hnačky, bradykardia, svalové zášklby – sa vyskytujú zriedkavo, obvykle sú prejavom neuváženého zvyšovania dávok, ktoré by mohlo vyústiť do cholinergnej krízy.
Uspokojiť sa pri MG dobrým terapeutickým efektom ICHE môže byť veľkou chybou neurológa. ICHE síce ostávajú liekmi prvej línie, ale ochorenie ovplyvňujú len symptomaticky, vôbec nemajú vplyv na imunopatogenézu MG. Podľa autora tohto textu je potrebné temer u všetkých pacientov s MG súčasne s podávaním ICHE aplikovať niektorý imunoterapeutický postup (najčastejšie konvenčnú imunosupresívnu liečbu – prednizon a/alebo azatioprín).
2. Imunosupresívna liečba
Kortikosteroidy (prednizon, methylprednizolon)
Prednizon pôsobí imunosupresívne na viaceré humorálne a celulárne zložky imunitného systému, ktoré sa uplatňujú v imunopatogenéze MG. Prednizon má výborný efekt, u 70 % pacientov s MG vedie k úplnemu vymiznutiu myastenických symptómov a u väčšiny ostatných pacientov nastáva aspoň čiastočné zlepšenie [3,9,20,21,94,95,98,99,100]. K zlepšovaniu svalovej sily dochádza k prvých 6-8 týždňoch liečby, úplná remisia nastúpi obvykle až neskôr. Na liečbu prednizonom reagujú najlepšie pacienti s krátkou anamnézou MG, ale aj pacienti s chronickým ochorením – napr. formy MG refraktérne na tymektómiu [9,19,20,21,26,98]. Výbornú odpoveď majú na prednizon myastenici s tymómom, či pred alebo po chirurgickom odstránení tymómu [9,19,20,21,95,98].
Rôzni autori obhajujú rozdielne terapeutické schémy pri nasadzovaní Prednizonu. Najvýhodnejšia a najúčinnejšia je liečba s úvodnou dávkou 1,5–2 mg na kg telesnej hmotnosti denne [3,20, 5]. Niektorí autori doporučujú postupné zvyšovanie úvodných dávok prednizonu so snahou vyhnúť sa prechodnému 7 až 10 dňovému zhoršeniu myasténie, ktoré sa vyskytuje asi u 1/3 pacientov [9,10,94].
Zlepšenie myastenickej symptomatológie sa dostavuje zhruba po 2 až 8 týždňoch podávania maximálnej dávky Prednizonu [20,94,99]. Potom sa prechádza na režim alternujúcich jednorazových dávok prednizonu. Táto schéma podávania prednizonu zabezpečuje jeho optimálny terapeutický efekt pri nízkom riziku nežiadúcich vedľajších prejavov [9,10,94,95,97,100,101]. Základnou podmienkou pre postupné znižovanie prednizonu je stálosť optimálneho klinického zlepšenia MG. Preto je u každého pacienta s MG nutný prísne individuálny prístup k liečbe [3,9,10,19,20,21,94]. Najčastejšie príčiny zlyhania kortikoterapie v liečbe MG sú práve v nesprávnej liečbe:
- nedostatočne vysoká úvodná dávka prednizonu
- nedostatočné trvanie kortikoterapie
- predčasný začiatok znižovania prednizonu
- rýchla redukcia dávok prenizonu
Napr. rýchle alebo „programované“ znižovanie dávok prednizonu a predčasné ukončenie liečby spôsobujú závažné klinické exacerbácie MG, ktoré môžu vyústiť až do myastenickej krízy [3,9,20,21,95].
Vedľajšie prejavy kortikoterapie:
Najčastejšími prejavmi kortikoterapie sú cushingoidná facies a nadváda, ktoré pri znižovaní a najmä po vysadení prednizonu ustúpia. Z ostatných prejavov sa vyskytujú osteoporóza, steroidný diabetes, akcelerácia katarakty, glaukóm, vzácne steroidná myopatia [3,9,20,21,97]. Pre riziko vedľajších prejavov pri dlhodobej kortikoterapii je vhodná strava s nízkym obsahom sodíka a glycidov, vyšším prísunom bielkovín. Profylakticky sa podávajú blokátory H2 receptorov alebo inhibítory protónovej pumpy, preparáty vápníka a vitamín D, podľa potreby sa substituuje draslík. Vhodné je sledovanie kostnej denzitometrie.
Azatioprín (Imuran)
Doba podávania prednizonu k zaisteniu trvalého liečebného efektu je individuálne rôzne dlhá. U väčšiny pacientov presahuje dva roky. V týchto prípadoch, najmä ak je výška udržovacej dávky prednizonu pomerne vysoká, je indikovaná aj imunosupresia azatioprínom. Táto skutočnosť sa v úvode liečby nedá predvídať. Preto je u väčšiny pacientov s MG súčasne indikovaná kombinovaná imunosupresívna liečba prednizonom a azathioprínom [9,21,101]. Palace et al (1998) v randomizovanej dvojito zaslepenej štúdii zistili, že u viac ako 60 % pacientov bolo možné úplne vysadiť Prednizon v priebehu prvých 3 rokov a zvyšok vyžadoval len nízku udržovaciu dávku prednizonu (5-20 mg) [103]. Okrem toho pri komplementárnom efekte oboch medikamentov sa dosahujú lepšie výsledky ako len pri samotnej liečbe Prednizonom [97,103,104]. Prednizon umožňuje rýchly nástup terapeutického efektu a azatioprín je veľmi výhodný u všetkých pacientov, ktorí vyžadujú dlhodobú alebo trvalú udržovaciu imunosupresívnu liečbu. Azatioprín je purínový analóg, ktorý sa v organizme konvertuje na merkaptopurin. Azatioprín pôsobí na syntézu a utilizáciu prekurzorov RNA a DNA [5,21]. Imunosupresívny účinok je dôsledkom jeho interferencie s metabolizmom nukleových kyselín počas proliferácie T-buniek po antigénnej stimulácii. Výsledkom je imunosupresia humorálnych aj celulárne sprostredkovaných autoimuných reakcií. V úvode kombinovanej liečby s prednizonom je azatioprín indikovaný v dávke 2-4 mg/kg telesnej váhy denne [3,9,21,94,94,95,101,102].
Samotné podávanie azatioprínu má veľkú nevýhodu v neskorom časovom nástupe jeho imunosupresívneho účinku, ktorý sa môže prejaviť až po 3-6 mesiacoch liečby [94,95,101,102,103,104]. Azatioprín je však ideálny liek pre dlhodobú udržovaciu imunosupresívnu liečbu [101,102,103,104,105]. Udržovacia imunosupresívna dávka azatioprínu je 1-2 mg/kg telesnej hmotnosti a pacientami je aj dlhé roky veľmi dobre tolerovaná [101,102].
Nežiadúce príznaky azatioprínu sú zriedkavé [21,101,102,104]. Patria k nim leukopénia a/alebo trombocytopénia, hepatotoxicita, intrahepatálna cholestáza a výnimočne alopécia. Potrebné sú pravidelné kontroly krvného obrazu a hepatálnych testov. Celková reakcia na azatioprín (hypersenzitívna reakcia) sa vyskytuje vzácne. Prejavuje sa horúčkou, bolesťami brucha, zvracaním, artralgiami hneď v prvé dni užívania azatioprínu, ktorý je nutné okamžite vysadiť. Výnimočne sa ako prejav hypersenzitívnej reakcie môže vyskytnúť pankreatitída. Po vysadení azatioprínu hypersenzitívne príznaky do 3 dňoch odoznejú.
Cyklofosfamid (Endoxan)
Používa sa v liečbe myasténie gravis len výnimočne [6,9,20,26,94,97,106]. Má síce veľmi dobrý imunosupresívny účinok, ale pomerne časté a závažné nežiadúce účinky. Drachman et al (2003) ho používajú v intravenóznej aplikácii 0,5-1g intravenózne na m2 v opakovaných cykloch po dobu 6-12 mesiacov u pacientov s MG, ktorí sú refraktérni na liečbu prednizonom, azatioprínom, mykofenolát mofetilom a IVIg [106].
Tacrolimus (FK 506)
Preparát používajú japonskí neurológovia [107]. V Európe a USA sú s ním minimálne skúsenosti.
Cyklosporin (Consupren,Sandimun,Equoral)
Má efektívne imunosupresívne účinky, jeho používanie je však spojené s rizikom vedľajších prejavov [20,94,95,97,108]. Cyklosporin blokuje aktiváciu T-buniek, inhibuje produkciu interleukínu-2, redukuje produkciu a uvoľňovanie cytokinínov T helper bunkami [3,94,108]. Obyklá dávka cyklosporinu je 5mg/kg telesnej hmotnosti/deň, optimálna hladina v sére sa pohybuje od 300 do 500 ng/ml [94]. Ak sa docieli optimálne zlepšenie MG, cyklosporin možno opatrne znižovať o 0,5 mg/kg telesnej hmotnosti/deň v 2-3 mesačných intervaloch. Často je nutná dlhodobá udržovacia dávka cyklosporinu 2-3 mg/kg telesnej hmotnosti/deň, lebo pri vysadení cyklosporínu dochádza k exacerbácii MG. Niektorí autori považujú cyklosporín po azatiopríne za imunosupresívum druhej voľby [94,108]. Nevýhodou cyklosporínu sú nežiadúce účinky, pre ktoré sa musí vysadiť cca u 30 % až 55 % pacientov [3,94]. K nežiadúcim prejavom patria toxické poškodenie obličiek (10 %), infekcie (5 %), gastrointestinálne príznaky (10 %), psychiatrické symptómy (5 %), bolesti hlavy (5 %), ale aj arteriálna hypertenzia, poruchy hepatálnych funkcií, hirsutizmus [97].
Mykofenolát mofetil (CellCept)
Mykofenolát mofetil je najnovší sľubný imunosupresívny preparát. Bol použitý v liečbe MG kazuisticky, ale aj v klinických štúdiach [95,109,110]. Účinnou látkou je kyselina mykofenolová, ktorá reverzibilne inhibuje monofosfát dehydrogenázy a tým blokuje syntézu guanozínu a jeho inkorporáciu do DNA [95]. Blokuje proliferáciu T-lymfocytov a aj protilátkovú odpoveď. Podáva sa v dávke 2x1 g. Má pomerne rýchly nástup účinku a dokázal navodiť klinické zlepšenie aj u pacientov s farmakorezistentnou MG [95,110]. Vzhľadom na pomerne rýchly nástup efektu a neveľké spektrum nežiadúcich prejavov (gastrointestinálne, leukopénia, trombocytopénia) sa v niektorých myastenických centrách začal mykofenolát mofetil používať namiesto azatioprínu v kombinovanej imunosupresívnej liečbe s prednizonom [95].
3. Tymektómia
Moderná éra liečby MG tymektómia začala v 40. a 50. rokoch 20. storočia prácami A. Blalocka v Baltimore a G. Keynesa v Londýne [1,2,95]. V Československu bol priekopníkom modernej liečby MG tymektómiou v 60. a 70. rokoch V. Šmat [2,83,112,117,121]. Na Slovensku sa chirurgická liečba MG centralizovala v 80. rokoch minulého storočia [113,114,115,116].
Napriek tomu, že odborných prác o tymektómii pri MG je veľké množstvo, doteraz nebola realizovaná ani jedna randomizovaná štúdia na hodnotenie účinnosti tymektómie pri MG. V súčasnosti je tymektómia akceptovaná v liečbe myasténie gravis asociovanej buď s hyperpláziou týmusu alebo tymómom [17,21,26,28,75,79,114,115,117,118,119]. Tento postoj podporil aj nález tzv. myoidných buniek v hyperplastickom týmuse, na ktorých sa nachádzajú acetylcholínové receptory [3,4,9,21,28,29]. Predpokladá sa, že porucha imunotolerancia a iniciácia autoimunitného procesu môže pri hyperplázii týmusu vzniknúť priamo v ňom.
V prvých rokoch 21. storočia sa indikácie na tymektómiu (TE) podstatne upresnili, k čomu prispelo najmä vyčlenenie nových foriem MG [12,17,35,36,37,41,95]:
A. Na tymektómiu sú indikovaní pacienti s formami MG, u ktorých sa týmus zúčastňuje na imunopatogenéze MG a pacienti s asociáciou MG a tymómu:
- MG + hyperplázia týmusu + autoprotilátky proti AChR. Vek do 45-50 rokov. Ide cca o 30 % chorých z celkového počtu pacientov s MG.
- MG + hyperplázia + žiadne autoprotilátky proti AChR ani proti MuSK. Vek do 50 rokov. Ide cca o 8 % chorých z celkového počtu pacientov s MG.
- MG + tymóm + autoprotilátky proti AChR a titinu. Vek - akýkoľvek. Ide asi o 10-15 % chorých z celkového počtu pacientov s MG. Tymóm je absolútnou indikáciou k tymektómii, s výnimkou pacientov vo vysokom veku a so závažnými pridruženými ochoreniami.
B. Tymektómia nie je indikovaná u tých foriem MG, kde sa týmus nezúčastňuje v imunopatogenéze MG:
- MG + involvovaný týmus + autoprotilátky proti AChR. Vek nad 45-50 rokov. Ide asi o 45 % chorých z celkového počtu pacientov s MG.
- MG + normálny/involvovaný týmus + SNMG s autoprotilátkami proti MuSK. Vek – akýkoľvek. Ide cca o 7 % chorých z celkového počtu pacientov s MG. Významné prognostické faktory pre priaznivý výsledok TE [3,9,19,20,21,26,76,94,115,116,120,121]:
- hyperplázia týmusu
- čo najkratšie trvanie MG od vzniku ochorenia k určeniu dg. a k TE
- detský vek (do 15 rokov)
- SPMG s autoprotilátkami proti AChR
- vek pacientov pod 40 rokov
Významné prognostické faktory pre nepriaznivý výsledok TE sú [3,9,19,20,21,26,76,115,116,120]:
- SNMG s autoprotilátkami proti MuSK
- involvovaný/atrofický týmus
- dlhé trvanie MG (nad 2 roky) od jej vzniku do určenia diagnózy/TE
- vyšší vek (nad 40 - 50 rokov)
Osobitný problém predstavuje problematika indikácií tymektómie v detskom veku. Všeobecne prevládajú váhavé alebo odmietavé postoje k tymektómiam v prepubertálnom veku [20,21,95,97,118]. Cavanagh v r. 1980 zhrnul všetky dovtedy publikované práce o tymektómii a zistil, že do veku 15 rokov bolo operovaných len 38 detí [121]. V slovenskom centre pre myasténiu gravis boli od konca 70. rokov do roku 1989 evidovaní 33 pacienti, u ktorých vznikla MG v detskom veku U 22 detí sa realizovali tymektómie na III. Chirurgickej klinike FVL v Prahe a po r. 1986 na Klinike kardiovaskulárnej chirurgie v Bratislave [30,122]. U 17 detí došlo ku klinickej remisii alebo výraznému klinickému zlepšeniu. U 5 detí tymektómia neovplyvnila MG, zlepšenie nastalo až po imunosupresívnej liečbe. Histologicky sa až u 18 detí zistila hyperplázia týmusu. Obdobné výsledky sme získali u 27 detí, ktoré boli operované v období 1990-2006 v Bratislave [116,120]. Podľa štatistického spracovania je detský vek významný prognostický faktor pre veľmi priaznivý výsledok tymektómie pri MG [21,116,120].
Ďalší problémový okruh predstavujú rozdielne názory na terapeutickú efektívnosť pri rôznych operačných prístupoch k tymektómii. Na väčšine špecializovaných chirurgických pracovísk, včítane pražského a bratislavského pracoviska, je štandardom tzv. rozšírená tymektómia z transsternálneho prístupu, ktorý umožňuje odstránenie týmusu a celého tukového tkaniva predného mediastína [2,3,9,21,79,94,95,114,155,117]. Operácia z cervikálneho prístupu je síce šetrnejšou metódou, ale neumožňuje dostatočnú orientáciu v mediastíne a obmedzuje možnosť úplneho odstránenia týmusu. Na niektorých pracoviskách, najmä v USA, sa používajú transcervikálne a infraaxilárne video-asistované thorakoskopické prístupy. Porovnávanie rôznych operačných prístupov vyznieva v prospech rozšírenej transsternálnej tymektómie [95, 123].
Pacienti s MG sú na tymektómiu indikovaní len v dobrom klinickom stave (farmakologická remisia, minimálna symptomatológia), čo sa u väčšiny dosahuje imunosupresívnou liečbou, plazmaferézou a v prípade potreby aj vysokými dávkami intravenózne aplikovaného imunoglobulínu [3,9,20,21,26,97,115,120,124,125].
4. Plazmaferéza
V centrách pre myasténiu gravis je plazmaferéza (PE; výmena plazmy) štandardnou liečebnou metódou. Efekt PE spočíva v eliminácii autoprotilátok proti ACh receptorom alebo v eliminácii autoprotilátok proti MuSK [8,20,94,95,124,125,126].
Režim PE pozostáva z 2 až 6 výmien plazmy, čo je určované klinickým stavom pacienta. Počas 1 PE sa odstráni cca 50 ml/kg plazmy. Cievny prístup k PE sa najčastejšie získava cez predné kubitálne vény. V prípade potreby je možno zaviesť subklaviálny alebo femorálny katéter. Zlepšenie sa obvykle dostavuje v priebehu 48 hodín po prvej alebo druhej PE. Plazmaferézy sa obvykle opakujú každý druhý deň, v prípade potreby sa môžu opakovať denne.
Cieľom PE je navodiť rýchle zlepšenie klinického stavu pacienta v nasledovných indikáciach [8,20,94,124,125,126]:
- akútne, fulminantné generalizované formy MG
- akútne exacerbácie MG
- hroziace myastenické krízy
- príprava na tymektómiu s cieľom redukovať perioperačnú morbiditu
Účinok PE je najmä v krízových situáciach spravidla veľmi dobrý, ale jeho trvanie je krátke. Vzhľadom ku krátkodobému efektu PE je vždy nutná súčasná kombinovaná imunosupresívna liečba (obvykle prednizon a azatioprín). Pre možnosť skorej reaktivácie MG po ukončení plazmaferetických kúr je u niektorých pacientov potrebné vnútrožilné podanie imunoglobulínu vo vysokých dávkach [20,94,95,97,124,125,126].
PE sa môže aplikovať aj v pravidelných 1 alebo 2 mesačných intervaloch u cielene selektovaných pacientov, ktorí nedostatočne reagujú na kombinovanú imunosupresívnu liečbu [20,94,95,125,126]. Pri plazmaferézach s prístupom z oblasti predných kubitálnych vén nebývajú závažnejšie nežiadúce účinky. Tie (pneumotorax, sepsa, trombóza, pľúcny embolizmus)sa môžu vyskytnúť pri centrálnom venóznom katetre. Nežiadúce prejavy sa však môžu vyskytnúť aj ako dôsledok eliminácie krvných bielkovín (koagulačné faktory, imunoglobulíny). Môže dôjsť k aktivácii koagulácie, komplementu, fibrinolytickej kaskády, agregácii trombocytov. V prípade náhrad plazmy je prítomné riziko hypersenzitívnej reakcie a prenosu infekcie.
Imunoadsorpcia je alternatívou PE. Pri jej aplikácii dochádza k poklesu hladiny autoprotilátok proti AChR, ktoré sa adsorbujú na imunoadsorpčných kolonách [127]. Očistená plazma sa vracia späť pacientovi. Imunoadsorpcia je účinná ako plazmaferéza a má podstatne menej nežiadúcich účinkov. Určitými negatívami sú veľmi vysoká cena a potrebnou technikou disponujú len vysoko špecializované hematologické pracoviská.
5. Intravenózny humány imunoglobulín
Vysoké dávky intravenózne podávaných imunoglobulínov (IVIg), izolovaných z plazmy zdravých darcov, sa ukázali pri MG ako účinné na základe viacerých otvorených nekontrolovaných štúdií, ale aj na základe randomizovaných kontrolovaných štúdií [94,95,97,128,129,130]. IVIg sa uplatňuje viacpočetným mechanizmom účinku. IVIg vytvára väzbu na antiidiotypové protilátky, znižuje tvorbu autoprotilátok, ovplyvňuje kaskádu komplementu s obmedzením produkcie „membrány atakujúceho komplexu“ , blokuje Fc-receptory na fagocytujúcich bunkách, moduluje T-bunkové funkcie s antigénnym rospoznávaním, znižuje produkciu prozápalových cytokinínov, akceleruje katabolizmus IgG a reguluje mechanizmy apoptózy [94,95,97,125,128,129,130]. Používaná dávka je 0,4 g/kg telesnej hmotnosti/deň po dobu 5 dní alebo dávka 2,0 g/kg telesnej hmotnosti/deň po dobu 3 až 5 dní. Podľa klinických štúdií je účinnosť IVIg rovnaká ako efektívnosť plazmaferézy [95,125]. Klinické zlepšenie pri IVIg sa dostavuje cca u 80 % pacientov na 4.-5. deň liečby. V porovnaní s PE je výhodou IVIg, že zlepšenia trvajú dlhšie, od 3-4 týždňov až po niekoľko mesiacov. Indikácie k IVIg sú obdobné ako pri PE: akútne fulminantné formy MG, akútne excerbácie MG, hroziace myastenické krízy a podanie IVIg pred tymektómiou za účelom redukcie perioperačnej morbidity [94,95,125,128,129,130]. V slovenskom Centre pre myasténiu gravis postupujeme pri závažných stavoch myasténie, a to najmä pri hroziacich myastenických krízach tak, že najprv indikujeme kúry plazmaferézy a hneď po nich IVIg, pričom nevyhnutnou a základnou súčasťou terapeutického režimu je intenzívna imunosupresívna liečba (obvykle prednizon a azatioprín). Dosiahnuté výsledky pri tomto postupe sú výborné. Nežiadúce vedľajšie prejavy sú pri IVIg menej časté a menej intenzívne ako pri plazmaferéze. Patria k nim: subfebrílie, erytém, bolesti hlavy, myalgie, len výnimočne môže vzniknúť anafylaktická reakcia alebo aseptická meningitída [95,125,128,129,130].
6. Iná terapia
Rituximab – je monoklónová protilátka proti povrchovému znaku CD 20 T lymfocytov, ktorej pôvodná indikácia je nonhodgkinovský lymfóm. Rituximab sa kazuisticky prejavil ako účinný u pacientov so SPMG a tiež u SNMG s autoprotilátkami proti MuSK, ktorí mali MG refraktérne na konvenčnú imunosupresiu a plazmaferézu [131].
Etanercept (solubilný rekombinantný receptor pre tumor nekrotizujúci faktor alfa) umožňuje u kortikodependentnej MG zníženie dávok Prednizonu, ale podobne ako rituximab, je spojený s rizikom závažných nežiadúcich prejavov [132].
Nedávno sa zistilo, že enzým acetylcholínesteráza, ktorý hydrolyzuje Ach sa vyskytuje v dvoch izoméroch – synaptická (AchE s) a tzv.“readthrough“ (AchE r). Izoméra AchEr sa vyskytuje u zdravých pri strese a prítomná je u pacientov s MG [133]. Okrem iného aktivuje zápalové procesy na postsynaptickej membráne. V experimente tzv. „antisense“ terapia (aplikácia oligodeoxynukleotidu pre mRNA AChE) redukuje na postsynaptickej platničke zápalové zmeny a zvyšuje počet ACh receptorov [132]. V Izraeli prebieha fáza II A klinickej štúdie s preparátom EN 101, od ktorého sa očakáva vyššia účinnosť a menej nežiadúcich prejavov v porovnaní s klasickými inhibítormi acetylcholínesterázy.
Prognóza
Včasné stanovenie diagnózy MG a voľba adekvátneho, optimálneho terapeutického postupu sú rozhodujúcimi faktormi, ktoré ovplyvňujú prognózu pacientov s MG. Pri analýze vplyvu rôznych klinických a laboratórnych ukazovateľov na terapeutickú účinnosť imunosupresívnej liečby a tymektómie sa zistilo, že najvýznamnejší prognostický faktor je čas trvania MG od vzniku do započatia liečby (s výnimkou akútnych fulminantných foriem MG) – [9,19,21,23,51,97,101,113,115,116,120]. S trvaním MG sa rozsah ireverzibilných štrukturálnych zmien postsynaptickej platničky a ACh receptorov zväčšuje a súčasne sa obmedzujú ich prirodzené regeneračné schopnosti. Preto pri nedostatočne účinnej alebo pri oneskorenej liečbe je nádej na úplné vyliečenie alebo podstatné zlepšenie klinického stavu imunosupresívnou liečbou, tymektómiou, PE a IVIg podstatne menšia ako v iniciálnych štádiach ochorenia. Okrem toho zle liečená alebo neliečená MG spravidla progreduje a môže vyústiť do myastenickej krízy. Pre MG ďalej platí, že aj po dlhom období klinickej alebo farmakologickej remisie sú u myastenikov možné exacerbácie ochorenia. Podľa súčasných poznatkov je MG imunopatogeneticky, klinicky aj reakciou na liečbu heterogénne ochorenie. Pre všetky uvedené fakty, a aj mnohé ďalšie, sa osvedčilo riešenie celej problematiky MG v špecializovaných centrách. U každého pacienta s MG je nutná individualizácia liečebného postupu a dlhoročná dispenzarizácia.
V ére pred imunosupresívnou liečbou a pri konzervatívnych postojoch k tymektómii dosahovala letalita pacientov s MG 30-50 % [1,3,12,19,21,50,55,112]. Okrem toho bola diagnostická záchytnosť MG v tomto období nízka. Predpokladá sa, že pomerne značná časť myastenikov zomierala pod mylnými diagnózami. Táto situácia sa významne zlepšila aj zavedením moderných metód do diagnostiky MG. Pri správnom zvolení optimálneho liečebného postupu u každého pacienta s MG by v súčasnosti mala byť letalita nulová, ani jeden pacient by teda nemal zomrieť v kauzálnej súvislosti s MG.
Přijato k recenzi: 12. 12. 2008
Přijato do tisku: 14. 1. 2008
doc. MUDr. Peter Špalek, CSc.
Centrum pre neuromuskulárne ochorenia
Neurologická klinika SZU
FNsP Bratislava – Ružinov
826 06 Bratislava
Tel: 00421 2/48234603
Fax: 00421 2/48234901
e-mail: peter.spalek@seznam.cz
Recenzenti:
prof. MUDr. Zdeněk Ambler, DrSc.
MUDr. Jiří Piťha, CSc.
MUDr. Stanislav Voháňka, CSc., MBA
doc. MUDr. Peter Špalek, PhD

Absolvoval LF Univerzity Komenského v Bratislave (1973). Po absolutóriu pracoval ako sekundárny lekár Oddelenia detskej neurológie DFN, od r. 1974 bol postupne sekundárny lekár, odborný asistent (1977) a primár (1991) Neurologickej kliniky ILF v Bratislave. V r. 1978 založil slovenské centrum pre myasténiu gravis, od roku 1995 je vedúcim Centra pre neuromuskulárne ochorenia. V r. 1977 a r. 1980 získal I. a II. atestáciu z neurológie, r. 1983 titul kandidáta vied (Myasténia gravis: Register a epidemiológia na Slovensku. Diagnostický význam stapediovej reflexometrie. Analýza účinnosti imunologických foriem liečby.) a r. 1991 habilitoval (Diagnostika a liečba ochorení neuromuskulárnej transmisie a primárne myogénnych ochorení). Bol pozvaný na pobyty na zahraničné pracoviská zaoberajúce sa myasténiou gravis: r. 1991 Oxford (John Newsom-Davis, Angela Vincent), r. 1993 Groningen (Hans Oosterhuis, Jan Kuks) a r. 1995 Viedeň (Wolfgang Grisold). Je autorom alebo spoluautorom 187 publikácií, z nich je viac ako 40 pôvodných prác, a 8 kapitol v monografiách. Niekoľko publikácií bolo odmenených cenami Slovenskej neurologickej spoločnosti, Slovenskej chirurgickej spoločnosti a Slovenskej sexuologickej spoločnosti. Jeho hlavnými klinickými a výskumnými záujmami sú myasténia gravis, Lambertov-Eatonov myastenický syndróm, neuromyotónia, tymómy a paraneoplastická autoimunita, polymyozitída, dermatomyozitída a imunogénne neuropatie. Je predsedom Sekcie pre neuromuskulárne ochorenia Slovenskej neurologickej spoločnosti. Organizoval 9 domácich a zahraničných konferencií o neuromuskulárnych ochoreniach.
Vědomostní test
1. Patofyziologickou podstatou pri myasténii gravis je:
- a) znížená degredácia acetylcholínu
- b) kongenitálny deficit acetylcholínesterázy
- c) redukcia počtu postsynaptických acetylcholínových receptorov
- d) redukcia počtu uvoľňovaných acetylcholínových kvánt
2. Ktorá z nasledovných príčin najčastejšie vyvoláva exacerbácie myasténie gravis:
- a) hypertyreóza
- b) infekcie horných dýchacích ciest
- c) steroidný diabetes
- d) hypersenzitívna reakcia na azatioprín
3. Ktorá z nasledujúcich foriem myasténie nie je indikovaná na tymektómiu:
- a) SPMG s autoprotilátkami proti ACh receptorom
- b) SNMG s negativitou proti AChR aj MuSK
- c) SPMG asociovaná s tymómom
- d) SNMG proti AChR a pozitívna proti MuSK
4. Ktorý liečebný postup je najúčinnejší pri riešení ťažkej akútnej myastenickej symptomatológie?
- a) tymektómia
- b) plazmaferéza
- c) cyklosporín
- d) azatioprín
5. 26ročná pacientka so SPMG – forma II A. Ktorý liečebný postup je najsprávnejší:
- a) tymektómia bez inej liečby
- b) cyclosporín
- c) plazmaferéza s následnou tymektómiou
- d) tymektómia s následnou plazmaferézou
6. V akom veku vzniká najčastejšie MG u žien:
- a) nad 70 rokov
- b) v 2.–3. dekáde
- c) v detstve
- d) v 5.–6. dekáde
7. Ktorý liečebný postup docieli najrýchlejšie zlepšenie u novodiagnostikovanej MG:
- a) inhibítory cholínesterázy
- b) tymektómia
- c) mykofenolát mofetil
- d) azatioprín
8. Gorelickov príznak sa vyšetruje pri:
- a) rinolalia, dysfónia
- b) asymetrická ptóza
- c) porucha okulomotoriky s diplopiou
- d) obojstranná slabosť musculus orbicularis oculi
9. Ako často sa vyskytuje tymóm u pacientov s MG:
- a) 1 až 5%
- b) 10–15%
- c) 20–30%
- d) 30–40%
10. U ktorej formy myasténie sa tymóm nevyskytuje:
- a) ženy nad 60 rokov
- b) pacienti s okulárnou MG
- c) SPMG s autoprotilátkami proti ACh receptorom
- d) SNMG (proti ACh receptorom) s protilátkami proti MuSK
11. Priemerná ročná incidencia myasténie gravis na Slovensku je:
- a) 6,5 prípadov na 1 milión obyvateľov
- b) 11,7 prípadov na 1 milión obyvateľov
- c) 14,8 prípadov na 1 milión obyvateľov
- d) 8,6 prípadov na 1 milión obyvateľov
12. Prítomnosť autoprotilátok proti titinu svedčí pre:
- a) asociáciu myasténie s tymómom
- b) asociáciu myasténie s Lambert- Eatonovým myastenickým syndrómom
- c) diagnózu SNMG s autoprotilátkami proti MuSK
- d) hroziacu myastenickú krízu
13. Myasténia gravis sa stáva klinicky manifestnou ak sa počet funkčných acetylcholínových receptorov zredukuje o:
- a) 10–20%
- b) 25–35%
- c) 40–50%
- d) 70–80%
14. Ktorý z nasledovných liekov môže ohroziť myastenikov útlmom respiračného svalstva:
- a) intravenózne podanie neostigmínu
- b) intravenózne podanie magnézia
- c) intravenózne podanie kalcia
- d) intravenózne podanie kofeínu
15. Seemanova skúška je pri MG záťažový diagnostický test pre:
- a) slabosť artikulačného a fonačného svalstva
- b) obojstrannú ptózu
- c) slabosť šijového svalstva
- d slabosť žuvacieho svalstva
16. Ktorá terapia je pri hodnotení dlhodobého efektu v liečbe MG najúčinnejší:
- a) IVIg
- b) mykofenolát mofetil
- c) prednizon v kombinácii s azatioprínom
- d) plazmaferéza
17. Ktoré z nasledovného odlíši autoimunitnú myasténiu gravis od kongenitálnej?
- a) zlepšenie po inhibítoroch cholínesterázy
- b) slabosť vonkajších okohybných svalov
- c) dekrement pri repetitívnej stimulácii
- d) zlepšenie po plazmaferéze
18. Najčastejšou príčinou exacerbácie MG pri kortikoterapii je:
- a) rýchle alebo „programované“ znižovanie dávok prednizonu
- b) asociácia myasténie s tymómom
- c) asociácia myasténie s hypertyreózou
- d) nedostatočný efekt inhibítorov cholínesterázy
19. Úvodná terapeutická dávka azatioprínu je:
- a) 2–4 mg/kg telesnej hmotnosti/deň
- b) 1 mg/kg telesnej hmotnosti/deň
- c) 4–6 mg/kg telesnej hmotnosti/deň
- d) 6–8 mg/kg telesnej hmotnosti/deň
20. U ktorého vyšetrenia má pozitívny nález najvyššiu špecificitu pre myasténiu gravis?
- a) single fibre electromyography
- b) Trendelenburgov príznak
- c) repetitívna stimulácia periférnych nervov
- d) dôkaz autoprotilátok proti titinu
správná je jedna nebo více odpovědí
Za správné vyřešení testu získá řešitel 5 kreditů ČLK.
Správné odpovědi:
WWW.CSNN.EU
Zdroje
1. Hughes T. The early history of myasthenia gravis. Neuromusc Dis 2005; 15: 878
886.
2. Šmat V, Schutzner J. Historie léčby myasthenia gravis. In: Schutzner J, Šmat V et al. Myasthenia gravis – komplexní pojetí a chirurgická léčba. Praha: Galén 2005: 11-17.
3. Oosterhuis HJGH. Myasthenia gravis. Groningen: Neurological Press 1997.
4. Gooch CL. Myasthenia gravis and Lambert-Eaton myasthenic syndrome. In: Rolak LA, Harati Y. Neuroimmunology for the clinician. Newton: Butterworth-Heinemann 1997: 263-299.
5. Warmolts JR, Engel WK. Benefit from alternate-day prednisone in myasthenia gravis. N Engl J Med 1972; 286: 17-19.
6. Nouza K, Šmat V. The favorable effect of cyclophosphamide in myasthenia gravis.
Rev franc clin biol 1968; 13: 161-163.
7. Mertens HG, Balzereit F, Leipert M. The treatment of severe myasthenia gravis with immunosuppressive agents. Eur Neurol 1969; 2: 323-339.
8. Pinching AJ, Peters DK, Newsom-Davis J. Remission of myasthenia gravis following plasma exchange. Lancet 1976; 2: 1373-1374.
9. Špalek P. Myasthenia gravis – register a epidemiológia na Slovensku. Diagnostický význam stapediovej reflexometrie. Analýza účinnosti imunologických foriem liečby. Kandidátska dizertačná práca. Bratislava: LF Univerzity Komenského 1983.
10. Špalek P, Lisý Ľ. Myasthenia gravis – liečba Prednizonom. Čs Neurol Neurochir 1982; 45/78: 418-424.
11. Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Autoantibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 2001; 7: 365-368.
12. Špalek P. Myasténia gravis a autoprotilátky – dlhý a nekončiaci sa príbeh. Čes Slov Neurol Neurochir (editorial). V tlači 2008.
13. Engel AG. Anatomy and molecular architecture of the neuromuscular junction. In: Engel AG. Myasthenia gravis and myasthenic disorders. Oxford: University Press 1999: 3-39.
14. Kaminski HJ, Ruff RL. Structure and kinetic properties of the acetylcholine receptor. In: Engel AG. Myasthenia gravis and myasthenic disorders. Oxford: University Press 1999: 40-64.
15. Molenaar P. Synthesis, storage and release of acetylcholine. In: Vincent A, Wray D. Neuromuscular transmission – basic and applies aspects. Manchester: University Press 1990; 62-81.
16. Vincent A. Immunology of disorders of neuromuscular transmission. Acta Neurol. Scand. 2006; 183 (Suppl.): 1-7.
17. Newsom-Davis J. The emerging diversity of neuromuscular junction disorders. Acta Myol 2007; 16: 5-10.
18. Špalek P, Vincent A. Autoantibodies at the neuromuscular junction. Neurológia (Bratisl) 2007; 2 (Suppl. 1): 11-12.
19. Špalek P. Ochorenia nervovosvalového prenosu. In: Rovenský J et al. Reumatológia v teórii a praxi. Martin: Osveta 1998: 651-659.
20. Sanders DB, Andrews PI, Barohn RJ, Massey JM, Phillips LH. Myasthenia gravis. Hagerstown, Baltimore: Lippincott Wiliams Wilkins 1999.
21. Špalek P. Diagnostika a liečba ochorení neuromuskulárnej transmisie a primárne myogénnych ochorení. Habilitačná práca. Bratislava: LF University Komenského 1990.
22. Fambrough DM, Drachman DB, Satyamurti S. Neuromuscular junction in myasthenia gravis: decreased acetylcholine receptors. Science 1973; 182: 293-295.
23. Engel AG, Tsujihata M, Sakakibara H, Lindstrom J, Lambert EH. Ultrastucture evidence for acetylcholine receptor dysfunction in myasthenia gravis and its autoimmune model. In: Rowland LM. Pathogenesis of human muscular dystrophies. Amsterdam: Elsevier 1977: 133-142.
24. Patrick J, Lindstrom J. Autoimmunity response to acetylcholine receptor. Science 1973; 180: 871-872.
25. Lindstrom JM. Experimental autoimmune myasthenia gravis: induction and treatment. In: Engel AG. Myasthenia gravis and myasthenic syndromes. Oxford: University Press 1999: 65-86.
26. Newsom-Davis J, Beeson D. Myasthenia gravis and myasthenic syndromes: autoimmune and genetic disorders. In: Karpati G, Hilton-Jones D, Griggs RC. Disorders of voluntary muscles. Cambridge: University Press 2001: 660-672.
27. Lindstrom JM, Seybold ME, Lennon VA, Wittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Neurology 1976; 26: 1054-56.
28. Kuks JBM. The thymus and myasthenia gravis. Doctoral thesis. Groningen: University of Groningen 1992.
29. Hohlfeld R, Wekerle H. The immunopathogenesis of myasthenia gravis. In: Engel AG. Myasthenia gravis and myasthenic syndromes. Oxford: University Press 1999: 87-110.
30. Spalek P, Smat V, Benko J, Vejvalka J. Thymectomy in juvenile myasthenia gravis. Prague: VIth International Symposium of Child Neurology 1985: 77.
31. Piťha J, Matějková E. Asociace HLA antigenu s myasthenia gravis u české populace. Čes Slov Neurol Neurochir 1998; 61/94: 7-12.
32. Vincent A, Leite MI. Neuromuscular junction autoimmune disease: muscle specific kinase antibodies and treatments for myasthenia gravis. Curr Opin Neurol 2005; 18: 519-525.
33. Selcen D, Fukuta T, Shen XM, Engel AG. Are the MuSK antibodies the primary cause of myasthenic symptoms ? Neurology 2004; 62: 1945-1950.
34. Deymeer F, Gungor-Tuncer O, Yilmaz V, Parman P, Serdaroglu P, Ozdemir A et al. Clinical comparison of anti-MuSK – vs – AchR – positive and seronegative myasthenia. Neurology 2007; 68: 609-611.
35. Wolfe GI, Trivedi JR, Oh SJ. Clinical review of muscle-specific tyrosine kinase-antibody positive myasthenia gravis. J Clin Neuromusc Dis 2007; 8: 217-224.
36. Vincent A, Mc Conville J, Farrugia ME, Newsom-Davis J. Seronegative myasthenia gravis. Sem Neurol 2004; 24: 125-133.
37. Hayashi A, Shiono H, Ohta M, Ohta K, Okumura M, Sawa Y. Heterogeneity of immunopathological features of AChR/MuSK autoantibody-negative myasthenia gravis. J Neuroimmunol 2007; 189: 163-168.
38. Spalek P, Hancinova V, Schnorrer M, Vincent A. Thymomas, myasthenia gravis, associated autoimmune diseases and antibodies against antogens at neuromuscular junction. Eur J Neurol 2006; 13 (Suppl.): 255.
39. Špalek P. Tymómy a paraneoplastická autoimunita. Čes Slov Neurol Neurochir 2002; 65/98: 367-373.
40. Chen XJ, Qiao J, Xiao BG, Lu CZ. The significance of titin antibodies in myasthenia gravis. Correlation with thymoma and severity of myasthenia gravis. J Neurol 2004; 251: 1006-1012.
41. Agius MA, Richman DP, Fairclough RH, Aarli J, Gilhus NE, Romi F. Three forms of immune myasthenia. Ann N Y Acad Sci 2003; 998: 454-456.
42. Osserman KE, Genkins G. Studies in myasthenia gravis: review of twenty year experience in over 1200 patients. Mount Sinai J Med, 1971; 38: 497-537.
43. Beekman R, Kuks JBM, Oosterhuis HJGH. Myasthenia gravis: diagnosis and follow-up of 100 consecutive patients. J Neurol Sci 1997; 244: 112-118.
44. Somnier FE, Keiding N, Paulson OB. Epidemiology of myasthenia gravis in Denmark. A longitudinal and comprehensive population survey. Arch Neurol 1991; 48: 733-739.
45. Phillips LH, Torner JC, Anderson MS, Cox GM. The epidemiology of myasthenia gravis in central and western Virginia. Neurology 1992; 42: 1888-1893.
46. Phillips LH, Torner JC. Epidemiologic evidence for a changing natural history of myasthenia gravis. Neurology 1996; 47: 1233-1238.
47. Vincent A, Palace J, Hilton-Jones D. Myasthenia gravis. Lancet 2001; 357: 2122-2128.
48. Špalek P. Epidemiology of myasthenia gravis in Slovak Republic – a 25 years longitudinal study (1978-2002). Lek Obzor 2003; 52 (Supl.): 2.
49. Niks EH, Kuks JBM, Verschuuren JJ. Epidemiology of myasthenia gravis with anti-muscle specific kinase antibodies in the Netherlands. J Neurol Neurosurg Psychiat 2007; 78: 417-418.
50. Oosterhuis HJGH. The natural course of myasthenia gravis: a long-term follow-up study. J Neurol Neurosurg Psychiat 1989; 52: 1121-1127.
51. Grob D. Natural history of myasthenia gravis. In: Engel AG. Myasthenia gravis and myasthenic disorders. Oxford: University Press 1999: 131-145.
52. Piťha J, Ambler Z. Myasthenia gravis. In: Neurologie 2003: Triton 2003: 188-202.
53. Schumm F. Myasthenia gravis – klinische Aspekte. Akt Neurol 1998; 25(Suppl. 2): 22-27.
54. Szobor A. Myasthenia gravis. Budapest: Akadémiai Kiadó 1990.
55. Simpson JA. Myasthenia gravis and myasthenic syndromes. In: Walton J. Disorders of voluntary muscles. Edinburgh: Churchill Livingstone 1981:585-624.
56. Gangadhar DV, Johnson LN, Borchert M. Isolated weakness of the orbicularis oculi muscle from myasthenia gravis. Neuro-ophtalmology 1989; 9: 327-329.
57. Kaminski HJ, Spiegel P, Ruff RL. Why are eye muscles frequently involved in
in myasthenia gravis? Neurology 1990; 40: 1663-1669.
58. Dziewas R, Warnecke T, Ritter M, Dittrich R, Schilling M, Schabitz WR et al. Fatigable swallowing in myasthenia gravis – proposal of a standardized test and report of a case. J Clin Neuromusc Dis 2006; 8: 12-15.
59. Llabrés M, Molina-Martinez FJ, Miralles F. Dysphagia as the sole manifestation of myasthenia gravis. J Neurol Neurosurg Psychiat 2005; 76: 1297-1300.
60. Nicolle MW. Wrist and finger drop in myasthenia gravis. J Clin Neuromusc Dis 2006; 8: 65-69.
61. Špalek P. Antibiotiká a neuromuskulárna transmisia. Prakt Lék 1982; 62: 130-132.
62. Ambler Z. Inhibiční vpliv léku na nervosvalový prenos. Farmakoter Zpravy 1989; 35: 49-53.
63. Mičeková D, Ondrašík M, Špalek P, Švec V. Myasthenia gravis pri liečbe reumatoidnej artritídy D-penicilamínom. Prakt Lék 1986; 66: 649-650.
64. Kuncl RW, Pestronk A, Drachman DB, Rechthand E. The pathophysiology of penicilamine-induced myasthenia gravis. Ann Neurol 1986; 20: 740-744.
65. Drosos AA, Christou L, Galanapoulou V, Tziofa AG, Tsiakou EK. D-penicilamine induced myasthenia gravis: clinical, serological and genetic findings. Clin Exp Rheumatol 1993; 11:387-391.
66. Leker RR, Karni A, Abramsky O. Exacerbation of myasthenia gravis during the menstrual period. J Neurol Sci 1988; 156: 107-111.
67. Špalek P. Myasthenia gravis, tehotnosť a tranzitórna neonatálna myasténia. Prakt Gynekol 1996; 3: 129-132.
68. Hoff JM, Daltveit AK, Gilhus NE. Myasthenia gravis in pregnancy and birth: identifying risk factors, optimising care. Eur J Neurol 2007; 14: 38-43.
69. Spalek P, Soskova M, Oros M. Myasthenia gravis, pregnancy and transient neonatal myasthenia. Neuromusc Dis 2002; 8: 735.
70. Brenner T, Abramsky O, Lisak RP. Influence of alpha-fetoprotein in vitro and in vivo immune responses to acetylcholine receptor. Ann N Y Acad Sci 1981; 377: 208-221.
71. Kalischewski P, Berrouschot J, Baumann I, Wagner A, Schneider D. Krisen bei Myasthenia Gravis, Management und Therapie auf der Neurologischen Intensivsstation. Akt Neurol 1998; 25 (Suppl 2): 62-63.
72. Hughes RAC, McLuckie A. Acute neuromuscular paralysis. In: Hughes RAC. Neurological Emergencies. London: BMJ Books: 332-358.
73. Drábková J. Myasthenia gravis z pohledu intenzívní medicíny. In: Schutzner J, Šmat V. Myasthenia gravis. Komplexní pojetí a chirurgická léčba. Praha: Galén 2005: 93-112.
74. Bedlack RS, Sanders DB. On the concept of myasthenic crisis. J Clin Neuromusc Dis 2002; 4: 40-42.
75. Schroer C, Bucka C, Sokolowski P, Kohler W. Myasthenia gravis – Klassifikationen und Scores. Akt Neurol 1998; 25 (Suppl. 2): 30-32.
76. Oosterhuis HJGH, Limburg PC, Hummel-Tappel E, The TH. Anti-acetylcholine receptor antibodies in myasthenia gravis: clinical and serological follow-up of individual patients. J Neurol Sci 1983; 58: 371-385.
77. Besinger UA, Toyka KV, Heininger K. Long-term correlation of clinical course and acetylcholine receptor antibody in patients with myasthenia gravis. Ann N Y Acad Sci 1981: 371-379.
78. Jaretzki A, Barohn RJ, Ernstoff RM. Myasthenia gravis. Recomendations for clinical research standards. Neurology 2000; 55: 16-23.
79. Schutzner J, Tvrdoň J. Chrurgická léčba thymomu. In: Schutzner J, Šmat V. Myasthenia gravis. Komplexní pojetí a chirugická léčba. Praha: Galén 2005: 63-72.
80. Špalek P, Schnorrer M. Tymómy: nové poznatky a ich nová WHO klasifikácia.
Lek Obzor 2002; 51: 248-250.
81. Koc F, Yerdelen D, Sarica Y. Myasthenia gravis and invasive thymoma with multiple intracranial metastases. J Clin Neuromusc Dis 2003; 4: 171-173.
82. Spalek P, Orolin D, Lisy L. Immunosuppressive drug treatment in overlap myasthenic syndrome: combined myasthenia gravis and Lambert-Eaton myasthenic syndrome. In: Bartko D, Gersterbrand F, Turcani P. Neurology in Europe I. London: John Libbey and Co Ltd 1989: 603-607.
83. Spalek P, Brozman B, Lisy L, Vincent A. Lambert-Eaton myasthenic syndrome – recent developments, diagnostic methods and case report of a patient with concomitant myasthenia gravis. Čes Slov Neurol Neurochir 1999; 62/95: 163-166.
84. Špalek P, Schnorrer M, Cibulčík F. Kombinovaný výskyt akútnej myasténie gravis a akútnej polymyozitídy u troch pacientov, u dvoch v asociácii s tymómom. Rozhl Chir 2000; 79: 468-470.
85. Spalek P, Cibulcik F, Vincent A. Myasthenia gravis and associated neurologic autoimmune disorders. Neuromusc Dis 2004; 14: 618.
86. Sneddon J. Myasthenia gravis – a difficult diagnosis. Brit J Psychiat 1980; 136: 92-93.
87. Sorensen TT, Hol EB: Myasthenia gravis in county of Viborg, Denmark. Eur Neurol 1989; 29:177-179.
88. Gorelick PB, Rosenberh M, Pagano RJ. Enhanced ptosis in myasthenia gravis. Arch Neurol 1981; 38: 531.
89. Ambler Z, Stalberg E. EMG diagnostika myasthenie gravis. Čs Neurol Neurochir 1986; 49/82: 60-65.
90. Ambler Z, Stalberg E. Myasthenický syndróm a jeho EMG diagnostika. Čs Neurol. Neurochir 1986; 49/82: 66-69.
91. Stalberg E, Sanders DB. Electrophysiological tests of neuromuscular transmission. In: Clinical neurophysiology. London: Butterworths 1981: 88-116.
92. Ambler Z. Aktivační testy při diagnostice myasthenie gravis repetititivní stimulací. Čs Neurol Neurochir 1990; 53/86: 78-82.
93. Špalek P, Hupka Š. Diagnostický význam stapediovej reflexometrie pri myasthenia gravis. Čas Lék čes 1982; 45: 1388-1390.
94. Seybold ME. Treatment of myasthenia gravis. In: Engel AG. Myasthenia gravis and myasthenic syndromes. Oxford: Univesity Press 1999:167-201.
95. Wolfe GI, Gross B. Treatment review and update for myasthenia gravis. J Clin Neuromusc Dis 2004; 6: 54-98.
96. Janzen RWC. Basistherapie der Myasthenie – Azethylcholinesterasehemmer (AchE-Hemmer). Akt Neurol 1998; 25 (Suppl. 2): 42-45.
97. Piťha J, Šimková L, Nováková I. Konzervatívní terapie myasthenia gravis. In: Schutzner J, Šmat V. Myasthenia gravis. Komplexní pojetí a chirugická léčba. Praha: Galén 2005: 79-91.
98. Bedlack RS, Sanders DB. Steroid treatment for myasthenia gravis. Steroids have an important role. Muscle Nerve 2002; 25: 117-121.vv
99. Seybold ME, Drachman DB. Gradually increasing doses of prednisone in myasthenia gravis: reducing the hazards of treatment. N Engl J Med 1974; 290: 81-64.
100. Johns TR. Long-term corticosteroid treatment of myasthenia gravis. Ann NY Acad Sci 1987: 505: 568-583.
101. Spalek P, Lisy L. Long-term immunosuppressive therapy in 134 myasthenia gravis patients. J Neurol Sci 1990; 90 (Suppl): 415-416.
102. Kuks JBM, Djojoatmodjo S, Oosterhuis HJGH. Azathioprine in myasthenia gravis: observation in 41 patients and review of literature. Neuromusc Disord 1991; 1:423-431.
103. Myasthenia Gravis Clinical Study Group. A randomized clinical trial comparing prednison and azathioprine in myasthenia gravis: results of the second interim analysis. J Neurol Neurosurg Psychiat 1993; 56: 1157-1163.
104. Palace J, Newsom-Davis J, Lecky B, the Myasthenia Gravis Study Group. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Neurology 1998; 50: 1778-1883.
105. Spalek P. Register of myasthenia gravis and myasthenic syndromes in Slovak Republic. Oxford: Euro-Myasthenia III 1991: 52.
106. Drachman DB, Jones RJ, Brodsky RA. Treatment of refractory myasthenia: „rebooting“ with high-dose cyclophosphamide. Ann Neurol 2003; 53: 29-34.
107. Wakata N, Saito T, Tanaka S. Tacrolimus hydrate (FK 506): therapeutic effects and selection of responders in the treatment of myasthenia gravis. Clin Neurol Neurosurg 2003; 106: 5-8.
108. Piťha J. První zkušenosti s terapií cyklosporinem A u myasthenia gravis. Čes Slov Neurol Neurochir 1994; 57/90: 237-241.
109. Hauser RA, Malek AR, Rosen R. Succesful treatment of a patient with severe refractory myasthenia gravis using mycophenolate mofetil. Neurology 1998; 51: 912-913.
110. Meriggioli MN, Rowin J, Richamn JG. Mycophenolate mofetil for myasthenia gravis: a double blind, placebo controlled pilot study. Ann NY Acad Sci 2003; 998: 494-499.
111. Mee J, Paine M, Byrne E. Immunotherapy of ocular myasthenia gravis reduces conversion to generalized myasthenia gravis. J Neuroophtalmol 2003; 23: 251-255.
112. Šmat V, Vejvalka J. Význam thymektomie v léčbě myasthenia gravis. Čs. Neurol Neurochir 1970: 33/66: 246-251.
113. Špalek P, Orolin D, Siman J. Tymektómia v liečbe myasténie gravis. Lék Obzor 1989; 38: 309-312.
114. Schnorrer M, Spalek P, Belacek J, Carsky S. Thymom und Myasthenia gravis. Viszeralchirurgie 1998; 33: 171-174.
115. Schnorrer M, Hraška V, Špalek P, Čársky S. Stanovenie významu prognostických faktorov pri chirurgickej liečbe myasténie gravis. Rozhl Chir 1999; 78: 223-227.
116. Spalek P, Schnorrer M, Sitarova K. Thymectomy in 347 myasthenia gravis patients (1990-2006). Eur J Neurol 2007; 14 (Suppl 1): 270.
117. Schutzner J, Šmat V. Thymus a chirugická léčba myasthenia gravis. In: Schutzner J, Šmat V. Myasthenia gravis. Komplexní pojetí a chirurgická léčba. Praha: Galén 2005: 31-61.
118. Lanska DJ. Indication for thymectomy in myasthenia gravis. Neurology 1990; 40: 1828-1829.
119. Gronseth GS, Barohn RJ. Thymectomy for nonthymomatous autoimmune myasthenia gravis (an evidence-based review). Neurology 2000; 55: 7-15.
120. Špalek P, Schnorrer M, Sosková M, Sitárová K. Myasthenia gravis – analýza faktorov ovplyvňujúcich terapeutickú efektívnosť tymektómie. Čes Slov Neurol Neurochir 2007; 70/103 (Suppl): 26.
121. Cavanagh NPC. The role of thymectomy in childhood myasthenia. Develop Med Child Neurol 1980; 22: 668-674.
122. Spalek P, Orolin D, Smat V. Myasthenia gravis and myasthenic syndromes in children J Autoim 1989; 2: 947.
123. Jaretzki A III. Thymectomy for myasthenia gravis: analysis of controversies regarding technique and results. Neurology 1997; 48 (Suppl 5): 52-63.
124. Spalek P, Hlavacikova M, Babincova A. Plasmapheresis in therapy of immunopathological neurological diseases. Piestany: Proceedings of International Congress on Haematology 2000: 55-56.
125. Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C. Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis: Myasthenia Gravis Clinical Study Group. Ann Neurol 1997; 41: 789-796.
126. Yeh JH, Chen WH, Chiu HCh, Bai ChH. MuSK antibody clearence during serial sessions of plasmapheresis for myasthenia gravis. J Neurol Sci 2007; 263: 191-193.
127. Pták J. Imunoadsorbce – nová možnost léčby autoimunitních onemocnění. Prakt Lék 2000; 82: 336-339.
128. Bednařík J, Voháňka S, Kadaňka Z. Léčba autoimunitních nervosvalových onemocnění intravenóznym lidským imunoglobulinem – přehled problematiky. Čes Slov Neurol Neurochir 1999; 62/95: 67-74.
129. Evoli A, Palmisani MT, Bartoccioni E, Padua L, Tonali P. High-dose intravenous immunoglobulin in myasthenia gravis. Ital J Neurol Sci 1993; 14: 233-237.
130. Newsom-Davis J. Myasthenia gravis and the Lambert Eaton myasthenic syndrome. In: Said G. Treatment of neurological disorders with intravenous immunoglobulins. London: Martin Dunitz 2000: 93-101.
131. Hain B, Jordan K, Deschauer M, Zierz S. Successful treatment of MuSK antibody-positive myasthenia gravis with rituximab- Muscle Nerve 2006; 33: 575-580.
132. Rowin J, Meriggioli MN, Tuzun E, Leurgans S, Christadoss P. Etanercept treatment in corticosteroid-dependent myasthenia gravis. Neurology 2004; 63: 2390-2392.
133. Nizri E, Hamra-Amitay Y, Sicsic C, Lavon I, Brenner T. Anti-inflammatory properties of cholinergic up-regulation: A new role for acetylcholinesterase inhibitors. Neuropharmacology 2006; 50: 540-547.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2008 Číslo 1
Nejčtenější v tomto čísle
- Bezpečnosť MRI vyšetrenia u pacientov s kovovými implantátmi a implantovanými prístrojmi
- Difuzní gliomy mozkového kmene u dětí. Noční můra dětského onkologa.
- Myasténia gravis
- Klinické využití protilátek u roztroušené sklerózy