
Bickerstaff brainstem encephalitis and Guillain-Barré syndrome overlap
Authors:
J. Husárová-Cepková 1; E. Ehler 1,2; L. Ungermann 3; I. Štětkářová 4
Authors‘ workplace:
Department of Neurology, Pardubice, Regional Hospital, Czech Republic
1; Department of Neurology, Faculty of, Health Studies, University of Pardubice, Czech Republic
2; Radiodiagnostic Department, Pardubice, Regional Hospital, Czech Republic
3; Department of Neurology, Third Faculty, of Medicine, Královské Vinohrady University, Hospital, Prague, Czech Republic
4
Published in:
Cesk Slov Neurol N 2022; 85(5): 415-416
Category:
Letters to Editor
doi:
https://doi.org/10.48095/cccsnn2022415
Dear editors,
Bickerstaff brainstem encephalitis (BBE) is a rare acquired autoimmune inflammatory disorder of the nervous system related to Guillain-Barré syndrome (GBS) with ophthalmoplegia and ataxia. Patients present with hyperreflexia and altered consciousness. Miller Fisher syndrome (MFS) also associated with GBS is clinically characterized by ophthalmoplegia, ataxia and areflexia. Both MFS and BBE are observed after preceding infection and have some similarities with GBS. Since the discovery of GQ1b antibodies in early 1990, both disorders provided evidence that are of the same spectrum – GQ1b syndrome. MFS patients who developed limb weakness are overlapping with GBS or those who developed drowsiness are overlapping with BBE [1].
We report a patient, who developed bilateral ophthalmoplegia and ataxia, with weakness and numbness of upper extremities and later also weakness of lower extremities, accompanied by drowsiness with hyperreflexia. Based on clinical and EMG findings, high positivity of anti-GQ1b antibodies and prompt treatment response to intravenous immunoglobulins, we conclude that the final diagnosis of our patients is BBE overlapping with GBS.
On the January 27, 2022, pain on the right side of the face appeared in a 69-year-old woman who was diagnosed with recurrent maxillary sinusitis. The patient has been treated for mild bronchial asthma since childhood (salbutamol and ciclesonide were given for discomfort). She suffered from recurrent maxillary sinusitis on the right side and has undergone carpal tunnel surgery on the right side in 2016 with good outcomes. The patient was originally a civil servant and is now retired.
After 2 days, clarithromycin per os was administered on an outpatient basis. The patient was also examined in cardiology for subcompensation of arterial hypertension with a blood pressure of 149/104 mm Hg and was given amlodipine with a blood pressure adjustment. A neurological examination revealed gait uncertainty and a mild hypoesthesia of the hands and feet and a brain CT revealed sinusitis maxillaris on the right side but otherwise was normal. On the following day, the patient presented with diplopia in the upward gaze and worsened stability gradually making walking impossible. Tingling and dullness of the hands and feet were also observed. A follow-up neurological examination and brain CT with angiography were performed and findings on the blood vessels were normal. On the next day (February 2), the patient was admitted to the department of neurology already displaying oculomotor disorder with diplopia, mild dysphagia and accentuation of trunk ataxia while sitting. Ischemic vascular etiology was considered, and acetylsalicylic acid (ASA) and a prophylactic dose of low molecular weight heparin (LMWH) were administred. On the next day, the patient displayed accentuated diplopia, dysphagia and backward and leftward thrusts while sitting. An MRI showed right sided maxillary sinusitis, but other findings were normal (Fig. 1). On February 4, an EMG was performed indicating normal compound muscle action potential (CMAP) and sensory nerve action potential (SNAP) amplitudes which were otherwise within normal limits including repetitive stimulation. H-reflex was unobtainable (Tab. 1). The patient was no longer able to raise her head in a supine position and presented with worsened diplopia, preserved direct and consensual photoreactions as well as severely restricted vertical movements of the eyeballs, horizontal slow lateral movements of the eyeballs with a small range, and no convergence but with evident miosis. Reflexes C5–8 were low, upper extremities sustained for 10 s and impaired scapular fixation was observed with a right-hand grip of 44 and left-hand grip of 42 kPa. Lower extremities were sustained for 10 s and reflexes L2–S2 were brisk with a left-sided predominance, where extensor hypertonia and Babinski occurred. Hautant was observed in a seated position with a backward pull. The patient was somnolent with hypoesthesia on the left side of the face, acral to the hands and in the lower extremities from the mid-tibia distally. Findings were suggestive of an autoimmune etiology (BBE + GBS) and intravenous immunoglobulin G 2 g/kg was administered for 5 days. A lumbar puncture with cerebrospinal fluid examination was performed showing a nearly normal level of protein (0.56 g/L) and normal cytology.
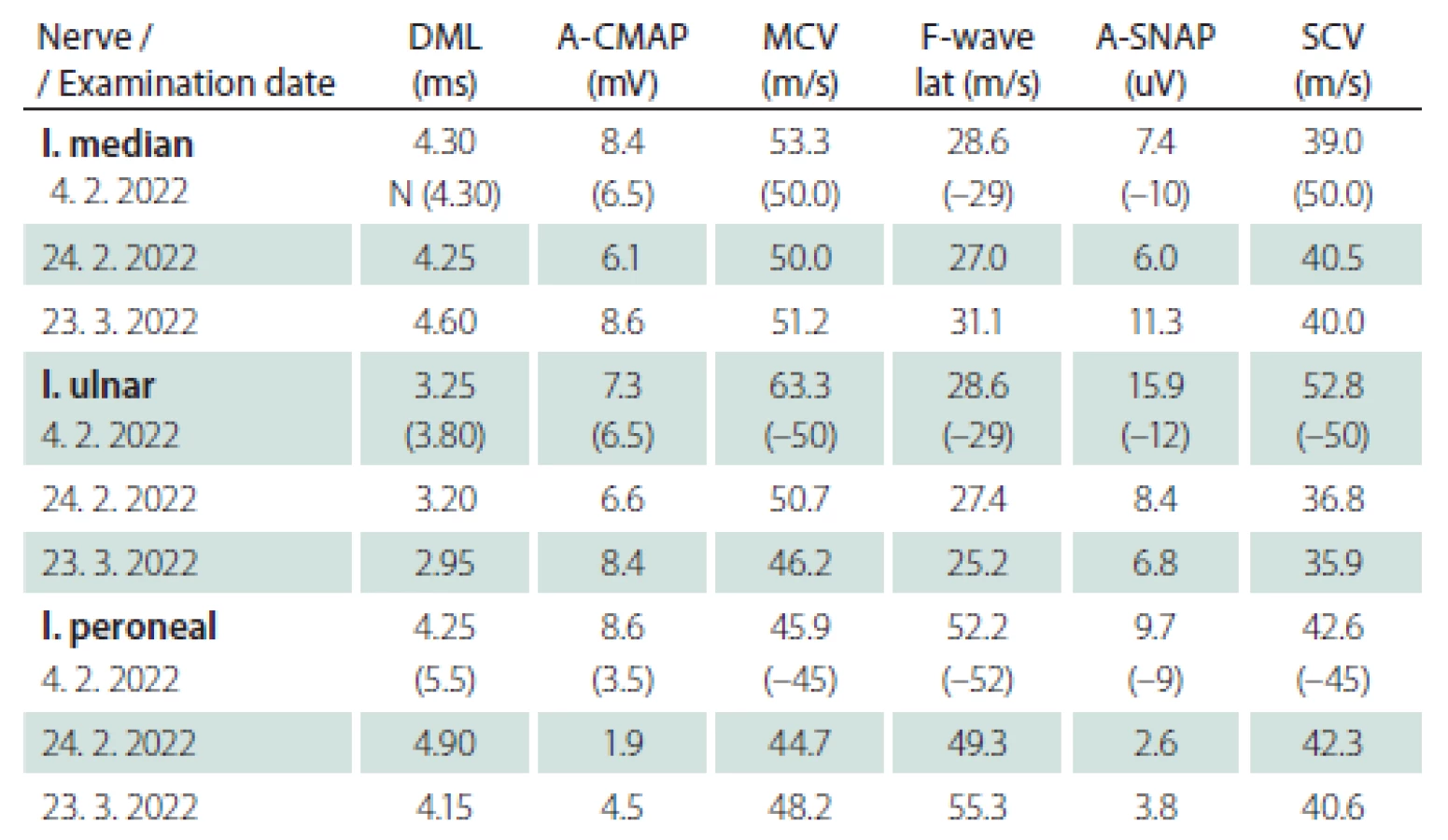
Obr. 1. MR mozku. Axiální T2 vážený snímek – sinusitis maxillaris vpravo s edematózní a zesílenou sliznicí. Normální nález v oblasti mozkového kmene.
During the following days, a progression of fatigue and decline were observed in the patient with further weakening of muscles. The patient became unable to hold out her arms and a gradual progression of respiratory insufficiency ensued. On day 6 of hospitalization, the patient was intubated with full artificial ventilation. On the 9th day of hospitalization, we received laboratory results with an anti-GQ1b level of 419% and no antibodies to other gangliosides were detected.
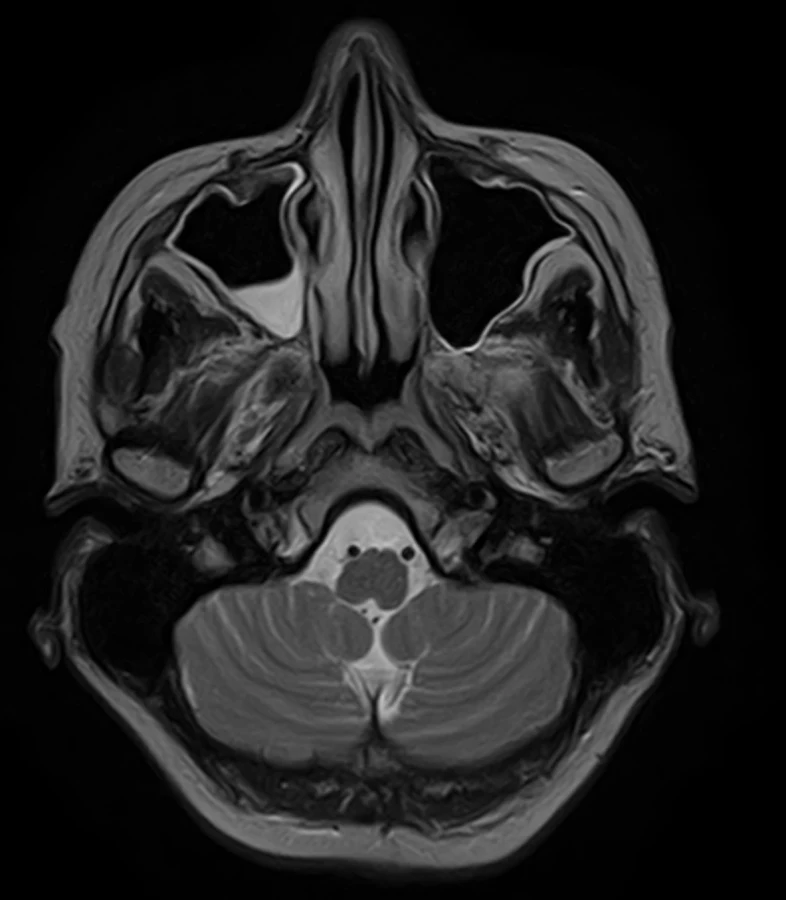
To improve mobility, weaning with intermittent connection to a humidifier began from February 20 and artificial ventilation was stopped on the following day. After two days, food intake per os began. On February 24, an EMG checkup examination was performed revealing lower CMAP amplitudes, a non-elicitable H-reflex, normal blink reflex and a normal brainstem auditory evoked potential (BAEP). An MRI of the brain showed no evidence of a new lesion in the trunk, cerebellum, or corpus callosum (Fig. 2, 3). On March 2, the patient was already alert, lifting and holding her head up and swallowing slowly but safely. The convergence of the eyeballs and the movement of the left eyeball upwards and sideways with the manifestation of diplopia was slightly restricted while walking was already possible with the help of two trekking sticks. A vibratory sensation was not present in the limbs but was adequate in the trunk with a measure of –6/8. A checkup EMG on March 23 showed no evidence of denervation syndrome in the lower extremities.
Obr. 2. MR mozku. Axiální T2 vážený snímek – progrese sinusitis maxillaris. Normální nález v oblasti mozkového kmene.
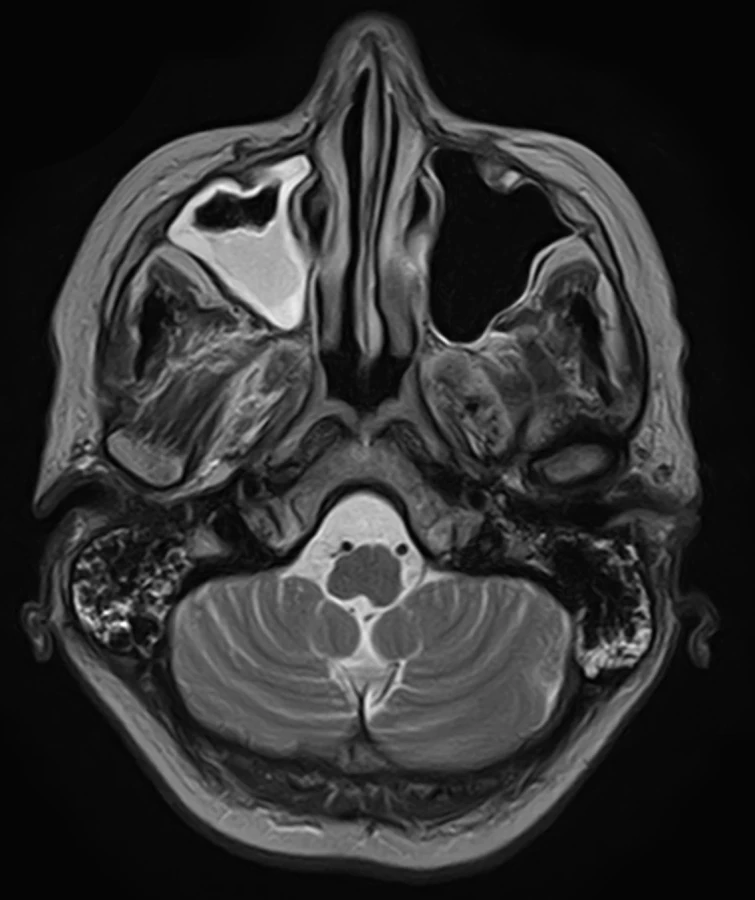
Obr. 3. MR mozku. Sagitální T1 vážený snímek – normální struktury mozkové kmene a corpus callosum.
Discussion
The presence of anti-GQ1b antibodies may explain the complex symptomatology of our patient – GBS and BBE. GQ1b antibodies are expressed in nodal/paranodal region and the neuromuscular junction of the oculomotor nerves, muscle spindle efferents, peripheral nerves and brainstem reticular formation. The neurological effects of anti-GQ1b antibodies are induced by complement-mediated destruction of both perisynaptic Schwann cells and axonal terminals resulting in neuromuscular junction blockade and ultimately muscle weakness [2]. Axonal GBS and MFS are thought to represent acute autoimmune nodopathies with pathogenic mechanism antiganglioside mediated dysfunction of Nav channels clusters at the Ranvier node and by an electrophysiological continuum from conduction failure, reversible in few weeks, to axonal degeneration [1]. Our patient was admitted for quickly developed bilateral ophthalmoplegia, ataxia, with weakness and paresthesia of extremities, weakness of neck flexors and with drowsiness, hyperreflexia, and extensor response of plantar reflex. After Immunoglobulin therapy and artificial ventilation with tracheostomy the improvement of clinical finding began after 14 days. After 25 days the patient began to walk with crutches.
Besides others GQ1b variants differential diagnosis also include Wernicke´s encephalopathy, vascular diseases in vertebrobasilar region, multiple sclerosis, botulism, brainstem tumor [3]. De Castillo et al, 2019 [4] presented a 61-year-old woman admitted with a 14-h history of sudden onset diplopia associated with dizziness, nausea, vomiting, and loss of balance. She had bilateral ophthalmoplegia, left ptosis, bilateral dysmetria and ataxia. She was managed as posterior circulation infarct and given aspirin. Anti-GQ1b antibody was positive and diagnosis MFS in combination with axonal GBS (AMSAN) was made. Anthony et al, 2012 [5] described an 81-year-old man, who developed diplopia, ptosis, shortness of breath. A provisional diagnosis of ocular myasthenia gravis was made. Acetylcholine receptor antibodies were not detected – but 50% patients with ocular myasthenia will be seronegative. After oral pyridostigmine he reported little improvement, which prompted serological testing for the anti-GQ1b antibody, that was positive. On admission to hospital our patient was investigated as an acute ischemic stroke in vertebrobasilar region. CTA was done and treatment with aspirin installed. But after neurophysiological examination the diagnosis was changed to autoimmune neuropathy and BBE, that was later confirmed by finding of high level of GQ1b antibodies.
BBE is an immune-mediated encephalitis, characterized by progressive ophthalmoplegia, ataxia and disturbance of consciousness [6]. Positive anti-GQ1b antibodies are found in 66% and abnormal brain MRI in 30% of patients [3]. Yoshikawa et al [7] presented 73 anti-GQ1b positive with 10 anti-GQ1b negative BBE patients. In BBE patients with positive anti-GQ1b the upper respiratory tract infection and sensory disturbances were more common, abnormal finding on brain MRI were less and consciousness disturbances disappeared earlier. Furthermore, the immunoglobulin G was more frequently administered to anti-GQ1b positive patients MRI abnormalities in form of T2 hyperintensive lesions in brainstem (specially midbrain), cerebellum, corpus callosum, and thalamus have been seen in 30% of BBE patients. Over time, signal characteristics may wax and wane or regress in a descending pattern [3]. The presence of anti-GQ1b IgG antibodies is specific for this disorder but can be absent in 30% of cases. These antibodies can also be seen in an ataxic variant of Guillain-Barré syndrome, known as Miller-Fisher syndrome, characterized by ophthalmoplegia and areflexia. Imaging is usually unremarkable [8]. We have investigated brain MRI of our patient twice during the acute stage. There were no abnormalities of the brain.
Currently, axonal GBS subtypes and MFS are thought to represent acute autoimmune nodopathies with a common pathogenic mechanism characterized by anti-gangliosides mediated dysfunction/disruption of Nav channel clusters and by an electrophysiological continuum from conduction failure, reversible in few weeks, to axonal degeneration [1]. During the stay at the hospital, we found only lower amplitude of CMAP, SNAP, and not obtainable H-reflexes for tibial nerves. There were no signs of acute denervation process (investigated in tibialis anterior muscle).
Alroughani et al, 2015 [2] in their 15-year-old boy with GBS with MFS and BBE elected to treat the patient with a combination of IV methylprednisone (IVMP) and IV Immunoglobulin (IVIg). The rationale was based on the faster effect of IVMP in view of myelitis-like picture, which tends to respond to high dose parental steroids. On the other hand, given that the pathogenesis of anti-GQ1b antibody syndrome is similar to GBS, IVIg was subsequently added as it has been shown to be efficacious in improving outcomes in GBS. Yoshikawa et al, 2020 [7] discussed the therapy. Although a previous study reported that corticosteroids and plasma exchange were mainly administered in BBE, we found that, recently, IVIG or IVIG combined with corticosteroids, were the most prevalent treatments in our cohort. Our patient was treated by IVIG immediately after electrophysiological investigation with established diagnosis of MFS with GBS.
Conclusion
Bickerstaff brainstem encephalitis is a rare diagnosis, but with a clear diagnostic definition. Combination of BBE with a generalized form of GBS is a typical situation in most BBE patients. BBE is very rare in Europe and clinical characteristics are well defined. Combination of BBE and GBS is a presentation of the continuing spectrum of anti-GQ1b syndrome. These patients do not have a poor prognosis.
Financial support
Supported by Cooperation 38 (Neuroscience) research project of Charles University.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Assoc. Prof. Edvard Ehler, MD, CSc.,
FEAN
Department of Neurology
Pardubice Regional Hospital
Kyjevská 44
530 03 Pardubice
Czech Republic
e-mail: eda.ehler@tiscali.cz
Accepted for review: 26. 5. 2022
Accepted for print: 8. 9. 2022
Sources
1. Puma A, Benoit J, Sacconi S et al. Miller Fisher syndrome, Bickerstaff brainstem encephalitis and Guillain-Barré syndrome overlap with persistent non-demyelinating conduction blocks: a case report. BMC Neurol 2018; 18 (1): 101. doi: 10.1186/s12883-018-1104-6.
2. Alroughani R, Thussu A, Guindi RT. Yet another atypical presentation of anti-GQ1b antibody syndrome. Neurol Int 2015; 7 (2): 5770. doi: 10.4081/ni.2015.5770.
3. Bagaria AK, Vyas A, Mathur V et al. Bickerstaff brainstem encephalitis with isolated bilateral ophthalmoplegia: an unusual presentation. Ann Indian Acad Neurol 2021; 24 (4): 624–626. doi: 10.4103/aian.AIAN_823_20.
4. De Castillo LLC, Diestro JDB, Ignacio KHD et al. A rare mimic of acute stroke: rapidly progressing Miller-Fisher syndrome to acute motor and sensory axonal neuropathy variant of Guillain-Barre syndrome. BMJ Case Rep 2019; 12 (3): e228220. doi: 10.1136/bcr-2018-228220.
5. Anthony SA, Thurtell MJ, Leigh RJ. Miller Fisher syndrome mimicking ocular myasthenia gravis. Optom Vis Sci 2012; 89 (12): e118–e123. doi: 10.1097/OPX.0b013e31827717c1.
6. Jing C, Wang Z, Chu C et al. Miller-Fisher syndrome complicated by Bickerstaff brainstem encephalitis: a case report. Medicine 2018; 97 (9): e9824. doi: 10.1097/MD.000 0000000009824.
7. Yoshikawa K, Kuwahara M, Morikawa M et al. Bickerstaff brainstem encephalitis with or without anti-GQ1b antibody. Neurol Neuroimmunol Neuroinflamm 2020; 7 (6): e889. doi: 10.1212/NXI.0000000000000889.
8. Zoghaib R, Sreij A, Maalouf N et al. Autoimmune brainstem encephalitis: an illustrative case and a review of the literature. J Clin Med 2021; 10 (13): 2970. doi: 10.3390/jcm10132970.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2022 Issue 5
Most read in this issue
- Lumbar spine disorder – the new occupational disease
- Cenobamate
- Carotid web
- The dentate gyrus – anatomy, vascular supply, function and neuropathology