
Genetické a neurobiologické aspekty komorbidního výskytu poruch autistického spektra a epilepsie
Genetic and neurobiological aspects of comorbid occurence of autism spectrum disorder and epilepsy
Autism spectrum disorder (ASD) is ranked among neurodevelopmental and neuropsychiatric disorders with clinical onset in childhood. In recent years, this disorder has come to the forefront of scientific interest, mainly due to increasing prevalence of up to 1/ 68 in 2014. The genetic causes of the disorder and the pathophysiological mechanisms that might be involved in the development of ASD are revealed. Comorbid occurrence with epilepsy is quite common, in up to 46% of cases. This article summarizes the current knowledge in this field with a focus on the hypothesis of excitatory-inhibitory imbalance. Some genetic causes of ASD and current diagnostic options are also discussed. The pathophysiology of the co-morbidity of ASD and epilepsy is discussed in terms of possible therapeutic interventions.
Keywords:
Genetics – Autism – Epilepsy – Autism spectrum disorder – ASD
Authors:
P. Danhofer 1; O. Horák 1; Š. Aulická 1; K. Česká 1; J. Pejčochová 1; L. Fajkusová 2; H. Ošlejšková 1
Authors‘ workplace:
Klinika dětské neurologie LF MU a FN Brno, Centrum pro epilepsie Brno
1; Centrum molekulární biologie a genové terapie Interní hematoonkologické kliniky LF MU a FN Brno
2
Published in:
Cesk Slov Neurol N 2019; 82(2): 148-154
Category:
Review Article
doi:
https://doi.org/10.14735/amcsnn2019148
Overview
Poruchy autistického spektra (PAS) se řadí mezi neurovývojové a neuropsychiatrické poruchy s klinickou manifestací v dětském věku. V posledních letech se tato porucha dostává do popředí vědeckého zájmu, a to především z důvodu narůstající prevalence až na 1/ 68 v roce 2014. Odhalují se genetické příčiny poruchy a patofyziologické mechanizmy, které by se na rozvoji PAS mohly podílet. Komorbidní výskyt s epilepsií je poměrně častý, a to až ve 46 % případů. Práce shrnuje dosavadní poznatky v této oblasti se zaměřením na hypotézu excitačně-inhibiční nerovnováhy. Jsou probrány i některé genetické příčiny PAS a současné možnosti diagnostiky. Patofyziologie komorbidního výskytu PAS a epilepsie je diskutována z pohledu možných terapeutických intervencí.
Klíčová slova:
autizmus – epilepsie – poruchy autistického spektra – genetika – PAS
Poruchy autistického spektra
Poruchy autistického spektra (PAS) se řadí mezi neurovývojové a neuropsychiatrické poruchy s klinickou manifestací v dětském věku charakterizované potížemi v sociální interakci a komunikaci, omezenými zájmy a repetitivními prvky v chování. Příznaky přetrvávají celoživotně a děti s PAS proto po 18. roce věku přecházejí do péče lékařů pro dospělé pacienty.
Častěji se PAS vyskytují u mužů v poměru 4 : 1. Odhadovaná prevalence PAS v populaci se zvýšila z 1/ 476– 1/ 323 v 90. letech 20. století [1,2] na 1/ 68 v roce 2014 [3]. Takový značný nárůst prevalence může být dán zčásti zvýšenou informovaností a povědomím populace o této poruše, zčásti i změnami v diagnostických postupech. Svůj podíl mohou mít i faktory ze strany matky a faktory prostředí. Infekce plodu v prenatálním období může negativně zasáhnout do vývoje imunoregulačních mechanizmů [4], podstatný vliv je přikládán febriliím [5]. Dále jsou studovány např. zvýšená koncentrace toxických látek v ovzduší [6] a vliv těžkých kovů, kde výsledky jsou kontroverzní [7,8].
V oblasti klasifikace PAS byla během posledních let provedena celá řada změn. Autizmus lze dělit na vysokofunkční a nízkofunkční (na podkladě kognitivního profilu), autizmus s regresí nebo bez ní (s ohledem na vývojové aspekty), syndromický nebo nesyndromický aj. U nesyndromického autizmu je PAS primární diagnózou a není součástí komplexní poruchy, která je charakterizována vývojovými abnormitami a malformacemi. Naproti tomu u syndromického (atypického) autizmu je znám genetický syndrom, v rámci něhož část pacientů vykazuje PAS (např. Angelmanův syndrom, syndrom fragilního chromozomu X, Rettův syndrom).
Pro klinické účely vycházejí v ČR diagnostická kritéria PAS z mezinárodní klasifikace nemocí MKN-10. Zde se autizmus řadí mezi pervazivní neurovývojové poruchy (F84.0–F84.9). Praktičtější je, a to především z pohledu možnosti srovnání pacientů se soubory publikovanými ve světovém písemnictví, využití klasifikace DSM (diagnostický a statistický manuál). Tato klasifikace nemocí zařadila PAS do svého obsahu ve svém třetím vydání a od té doby prošla celou řadou změn. Kritéria PAS v DSM-III [9] vycházela z původních Kannerových případů [10] a byla poměrně přísná. DSM-IV [11] poté spektrum autizmu rozšířilo o případy méně závažně (pervazivní vývojové poruchy jinak nespecifikované [PDD-NOS] a Aspergerův syndrom). Poslední revize DSM-V z roku 2013 [12] kombinuje všechny podskupiny do jedné diagnózy poruch autistického spektra. PAS se již nedělí do jednotlivých subkategorií, ale diferencují se pouze varianty autizmu [12].
Současným zlatým standardem v diagnostice autizmu je podrobná psychologicko-psychiatrická anamnéza a testování alespoň dvěma škálami – celosvětově uznávaným nástrojem je The Autism Diagnostic Interview – Revised (ADI-R) a The Autism Diagnostic Observation Schedule (ADOS). Testování musí provádět specialista na PAS. ADI-R je velmi podrobná dotazníková škála založená na strukturovaném pohovoru s rodiči. ADOS je standardizovaný diagnostický test, který skóruje na základě přímé observace dítěte a zohledňuje i jeho vývojový stupeň a věk. Je doporučován jako vhodný standardizovaný diagnostický observační nástroj. Vyšetřující nabízí dítěti interaktivní aktivity, které jsou navrženy tak, aby bylo možno hodnotit sociální interakci a komunikaci i repetitivní prvky v chování, jež jsou podkladem diagnostiky PAS [13].
Na Klinice dětské neurologie LF MU a FN Brno byla v roce 2007 provedena retrospektivní studie 204 dětí s PAS s cílem zjistit, zda je diagnóza stanovena časně nebo dochází ke zpoždění v diagnostickém procesu. Závěrem této studie bylo, že diagnostika autizmu je často provedena pozdě, a tím je znemožněno zahájení časných edukačních, behaviorálních a léčebných intervencí [14]. Zvýšené povědomí o PAS, lepší informovanost široké veřejnosti a formování týmů specialistů, kteří se zabývají diagnostikou této poruchy, přináší v tomto ohledu slibné výsledky a v současné době lze konstatovat, že u většiny pacientů je diagnostika PAS dokončena před dosažením 5. roku věku [15].
Epilepsie u pacientů s PAS
Komorbidní výskyt autizmu a epilepsie byl znám již od doby, kdy byl autizmus poprvé popsán Leo Kannerem v roce 1943 [10]. Je všeobecně známo, že u pacientů s autizmem je výskyt epilepsie vyšší než v běžné populaci. Prevalence však kolísá v poměrně širokém rozmezí – 2 [16] až 46 % [17]. Na Klinice dětské neurologie LF MU a FN Brno bylo hodnoceno 205 dětí s PAS ve vztahu ke komorbidnímu výskytu epilepsie nebo epileptiformní aktivity v EEG. PAS byly spojeny s epileptickými záchvaty ve 40 % případů a s epileptiformní aktivitou v EEG bez manifestních záchvatů ve 20 % případů. Nejvyšší výskyt epilepsie byl ve skupině dětí s dětským autizmem (66 %) a atypickým autizmem (30,1 %). Nejnižší výskyt byl zjištěn u dětí s Aspergerovým syndromem (3,9 %) [18]. Výsledky studií jsou značně nekonzistentní [16,17]. Je to dáno především opakovanými změnami v klasifikačních schématech a různými diagnostickými postupy. Velký podíl nese i spektrum vyšetřovaných pacientů. Obecně lze říci, že soubory z terciárních center soustřeďující „komplikovanější“ pacienty (tj. často farmakorezistentní s četnějšími komorbiditami) vykazují vyšší výskyt komorbidního výskytu PAS a epilepsie ve srovnání se soubory „běžných“ pacientů s PAS dispenzarizovaných v sektorovém ambulantním provozu.
Faktory diskutované v souvislosti s výskytem epilepsie u pacientů s PAS
Deficit intelektu
Přítomnost intelektového deficitu u PAS (tj. IQ < 70) je běžně asociována se zvýšenou mírou výskytu komorbidní epilepsie. Metaanalýza z roku 2008, která studovala data z publikovaných studií z let 1963– 2006, ukazuje prevalenci epilepsie u dětí s PAS mladších 12 let 21,4 % v případě přítomného intelektového deficitu ve srovnání s 8 % u dětí bez deficitu v intelektu [19]. Další metaanalýza – z roku 2012, která zahrnula jen studie, kde follow-up byl delší než 12 měsíců, ukázala prevalenci 23,7, resp. 1,8 % [20]. Silná asociace mezi intelektovým deficitem a epilepsií u dětí s PAS vysvětluje nižší výskyt epilepsie u pacientů s Aspergerovým syndromem, kde je intelekt normální.
Pohlaví
Existuje zvýšené riziko epilepsie u dívek s PAS ve srovnání s chlapci [21].
Etiologie onemocnění
Výskyt epilepsie u pacientů s nesyndromickým neboli idiopatickým autizmem je nižší než u pacientů se syndromickým (atypickým) autizmem. Například dle výsledků jedné studie 20 % u nesyndromické formy ve srovnání s 33 % u syndromických PAS [22]. I přesto je ale i u pacientů s idiopatickou formou PAS výskyt epilepsie vyšší než v běžné populaci. Syndromické formy PAS spojené s epilepsií jsou popsány např. u Rettova syndromu, Rett-like syndromů (mutace v genu pro methyl CpG binding protein 2 [MECP2], mutace v genu pro cyclin-dependent kinase-like 5 [CDKL5]), Angelmanova syndromu (mutace v genu pro ubiquitin-protein ligázu E3A [UBE3A]), tuberózní sklerózy (mutace v genu pro tuberous sclerosis complex [TSC] 1 a TSC2), neurofibromatózy typu I (mutace v genu pro neurofibromin 1 [NF1]), Dravetova syndromu (mutace v genu pro sodium voltage-gated channel alpha subunit 1 [SCN1A]), tedy syndromů, se kterými se v praxi setkáváme nejčastěji.
Vývojový regres
Vývojový regres je definován jako ztráta již dříve naučených dovedností. Odhaduje se, že zhruba 30 % pacientů s PAS prochází vývojovým regresem, který se typicky objevuje mezi 18. a 24. měsícem věku [23]. Výskyt epilepsie a/ nebo epileptiformní patologie v EEG je považován za rizikový faktor pro přítomnost vývojového regresu [23,24].
Vliv epilepsie a/ nebo epileptiformní aktivity v EEG na rozvoj PAS
Předpokládá se, že epileptiformní abnormita se může spolupodílet na rozvoji neuropsychologického deficitu u pacientů s PAS. Studie 77 dětí s PAS prokázala, že epileptiformní výboje v EEG jsou signifikantně více spojeny s abnormálním vývojem v 1. roce života [24]. Například u Rettova syndromu, syndromu fragilního X, Angelmanova syndromu nebo Prader-Williho syndromu lze současně nalézt jak patologii v EEG, tak i příznaky PAS. Je však nutné brát v úvahu, že nejsme schopni přesně vystopovat, kdy se epileptiformní aktivita v EEG u daného pacienta objevila poprvé. Je ovšem možné, že epileptiformní výboje v EEG, které se objeví v kritickém období vývoje, jsou částečně odpovědné za rozvoj autistických symptomů [25] nebo se mohou asociovat s autistickým regresem u těchto dětí [24].
Epileptiformní výboje mohou mít negativní dopad na senzorické, paměťové a vyšší kognitivní funkce, a to především pokud se vyskytují ve spánku, který je považován za klíčový v procesech učení a paměti. Toto je patrno zvláště u pacientů s Landau-Kleffnerovým syndromem, kde se ve spánku setkáváme s kontinuálními výboji v EEG. Pacienti mají těžký kognitivní deficit a často i PAS [26]. Potlačení výbojů antiepileptickou medikací však nepřináší vždy jednoznačný a předvídatelný efekt [27].
Vidíme tedy, že epileptiformní výboje v EEG mají dopad na kognitivní funkce pacienta, ale potlačení výbojů nevede k předpokládanému zlepšení neuropsychologického profilu. V takovém případě je nutné zvažovat i jiné faktory, které se dostávají do hry, jak je probíráno dále v textu.
Zvířecí modely ukazují, že pokud je epilepsie provokována epileptickým statem (status epilepticus; SE), lze pozorovat rozvoj aseptického zánětu, objevují se poruchy buněčného metabolizmu a poruchy na úrovni iontových kanálů a receptorů. Rozvíjí se poruchy vedení vzruchu, tvoří se aberantní neuronální sítě. Nakonec může dojít až k apoptóze buněk. Zároveň jsou poškozeny neuronální okruhy citlivé na rozvoj dalších záchvatů a nejsou schopny udržet normální kognitivní funkce [28]. Pravda je, že samotný SE se často objevuje jako důsledek zánětu, infekce nebo traumatu. Pak je obtížné rozlišit, jaký je podíl samotného etiologického procesu a jak se v rozvoji kognitivního deficitu uplatňuje následný SE. Potlačení záchvatů však vede jen k minimálnímu zlepšení kognitivního profilu pacientů. Proto je možné, že samotná příčina epilepsie může vést přímo k poškození neuropsychologických funkcí autistických pacientů [25] více než další epileptický proces.
Etiopatogeneze komorbidního výskytu epilepsie a PAS
Genetické aspekty v patogenezi komorbidního výskytu epilepsie a PAS
Studie s dvojčaty ukazují, že dědičnost PAS je zhruba 85– 92 % [29,30]. I v případě, že se nepodaří identifikovat mutaci odpovědnou za rozvoj PAS, je riziko pro další potomky vyšší [31]. V etiopatogenezi PAS se předpokládá role jak vzácných, tak běžných variant genů. S pokrokem na poli molekulárně-genetických technologií (jako jsou chromozomální microarray technologie nebo celoexomové sekvenování – whole exome sequencing) bylo identifikováno více než 800 chromozomálních lokusů a genů, které by mohly být asociovány s PAS, což poukazuje na vysoce heterogenní genetickou architektoniku této poruchy [3]. Geny a jejich příslušné proteiny se uplatňují v celé řadě biologických procesů, např. remodelaci chromatinu, regulaci genové transkripce, buněčném růstu a proliferaci a v neuronálně specifických procesech, jako jsou např. synaptická organizace a aktivita, dendritická morfologie a axonogeneze [3].
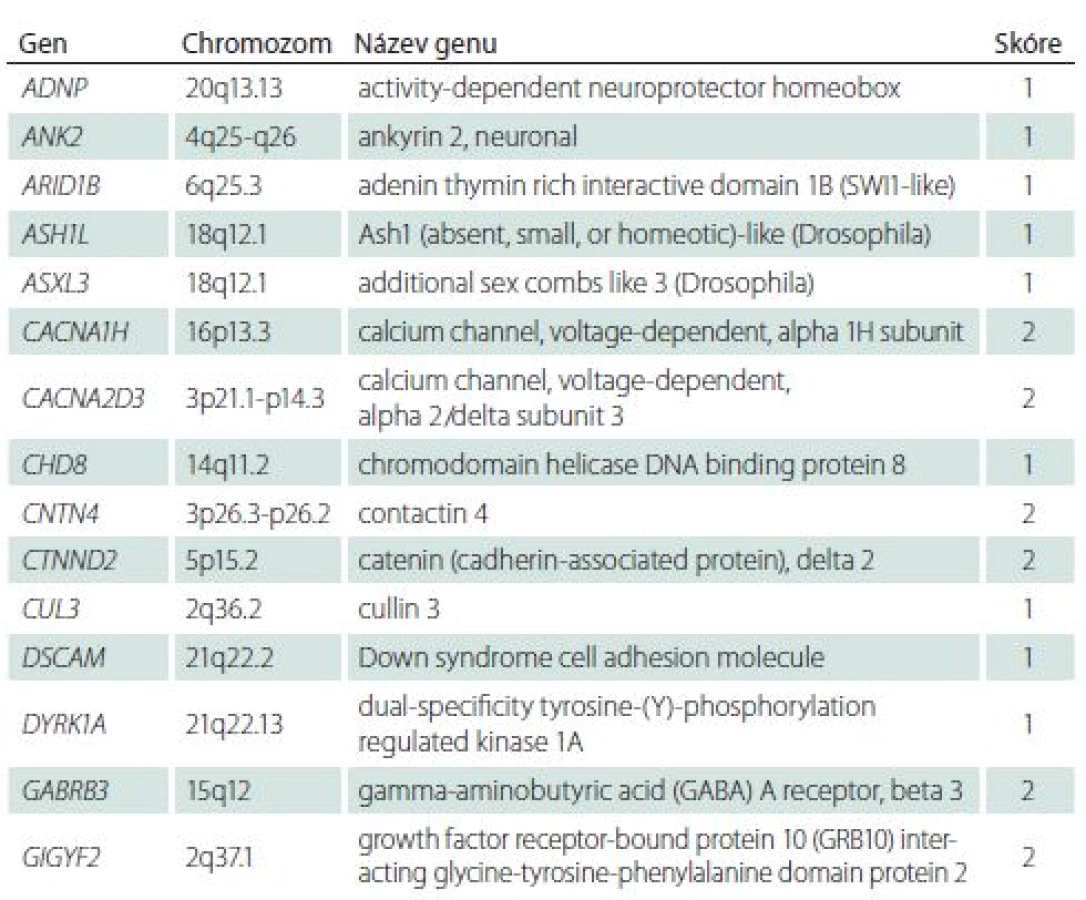
Přehlednou databází genů asociovaných s PAS je Simons Foundation Autism Research Initiative (SFARI) gene [32]. Databáze podává informace o hladině významnosti dané mutace na podkladě výsledků zvířecích a humánních studií. Všechny geny, pro které existuje hladina úrovně důkazu v diagnostice PAS, jsou skórovány v databázi: 1 – vysoce důvěryhodný a 2 – silný kandidátní gen. Je jich 51 (aktuální počet v roce 2018) a jsou uvedeny souhrnně v tab. 1.

Co se týká komorbidního výskytu PAS a epilepsie, riziko rozvoje epilepsie u dětí s PAS je 12,8 %, u jejich sourozenců 2,3 %, což je 2× vyšší riziko než v běžné populaci. Genetické pozadí se tedy týká nejen PAS, ale i epilepsie v rodinách s mnohočetným výskytem autizmu [33,34]. Vztah mezi PAS a epilepsií zůstává nejasný; může zde být kauzální vztah mezi těmito dvěma poruchami (především v případě epileptických encefalopatií či syndromického autizmu) nebo jsou obě výsledkem stejného neuropatofyziologického procesu. V každém případě se předpokládá, že na rozvoj PAS a epilepsie mají vliv nejen dědičné, ale i další faktory [25], jak bude detailněji probráno dále.
Imunitní faktory v patogenezi komorbidního výskytu epilepsie a PAS
Zánětlivé procesy v CNS hrají poměrně významnou roli ve společné patogenezi epilepsie a PAS. Jejich dopadem mohou být změny indukované mediátory zánětu v excitačních neuronálních sítích.
U komorbidního výskytu PAS a epilepsie nacházíme zvýšení HMBG-1 (high mobility group box protein 1), klíčové zánětlivé molekuly, která aktivuje signální cestu přes interleukin (IL)-1 a IL-1ß. U pacientů s PAS toto zvýšení koreluje s postižením v sociální interakci [35]. Stejně tak zvýšení exprese HMBG-1 bylo detekováno v hipokampu pacientů s epilepsií temporálního laloku (TLE) [36] a dětí s febrilními křečemi [37]. Poznatky opět ukazují na aktivaci IL-1 signální kaskády vedoucí k zánětlivé reakci se zvýšenou excitabilitou v neuronálních sítích. Ukazuje se, že antagonisté HMBG-1 jsou v preklinických studiích účinné v kontrole záchvatů [36].
U pacientů s PAS bylo detekováno zvýšení Th1 prozánětlivých cytokinů (např. IL-6, tumor necrosis factor α [TNF-α] a interferon γ [IFN-γ]), hladina protizánětlivých Th2 cytokinů (např. IL-4, IL-5, IL-10) zůstává nezměněna. U pacientů tím dochází ke zvýšení poměru Th1/ Th2 (T-helper lymphocytes 1/ T-helper lymphocytes 2). Periferní lymfocyty u pacientů s PAS vykazují 2× vyšší hladiny prozánětlivých cytokinů ve srovnání s kontrolami. Pokud byl TNF-α vyšetřen u všech nepostižených sourozenců pacientů s PAS, byla zjištěna opět signifikantně vyšší hladina této molekuly, což svědčí pro určitý podíl genetiky v nastavení zánětlivých signálních kaskád [38]. Stejně tak byla zjištěna elevace prozánětlivých cytokinů (TNF-α, IL-1β a IL-6) u pacientů s TLE. Tyto cytokiny mají in vivo i in vitro prokázaný prokonvulzivní efekt [39].
Dalšími společnými patofyziologickými vodítky mezi PAS a epilepsií mohou být porucha integrity hematoencefalické bariéry (HEB), mikrogliální aktivace a zánět indukovaný stresem. Mastocyty lokalizované v hypotalamu mohou být aktivovány stresem, jak bylo pozorováno u pacientů s PAS, a mohou vést k poruše HEB. Aktivace mastocytů vede ke spuštění prozánětlivých kaskád (např. exprese IL-6, vaskulárního endoteliálního růstového faktoru – vascular endothelial growth factor [VEGF]), které mají dopad na úrovni tight junctions HEB, což způsobuje poruchu její integrity [40]. Porucha integrity HEB byla zjištěna i na modelech chronické epilepsie, kde je opět důsledkem aktivace prozánětlivých signálních kaskád [41].
Společným důsledkem aktivace zánětlivého procesu u pacientů s PAS a epilepsií je zvýšení excitability mozku a progrese klinického vyjádření obou syndromů. Následkem zvýšení excitability v mozkových neuronálních okruzích nastane porucha excitačně-inhibiční (E/ I) rovnováhy, která představuje velmi křehké ekvilibrium mezi excitačními a inhibičními vlivy mozku. Dochází pak k nekontrolovatelným neuronálním výbojům a rozvoji záchvatů, jak bude blíže vysvětleno v další kapitole [25]. U pacientů s PAS a epilepsií byla v post mortem studiích zjištěna astroglióza, odpovědná za poruchu zpětného vychytávání glutamátu, což vede ke zvýšení jeho extracelulárních hladin a zvýšení excitability. Aktivace mikroglie (opět pozorována u obou procesů) zvýšení glutamátu ještě podporuje. Stejný efekt má aktivace IL-6, která stimuluje tvorbu excitačních synapsí [42].
Hypotéza společného patofyziologického mechanizmu komorbidní epilepsie a PAS
Shrnutím výše uvedených pozorování a výsledků lze předpokládat, že autizmus a epilepsie mohou mít alespoň v některých případech společný neurobiologický podklad. Jak bylo uvedeno v úvodu, existují syndromy, pro které je společný výskyt těchto dvou diagnóz typický. Právě v těchto případech je patrné a všeobecně přijímané, že jak PAS, tak epilepsie mohou mít společnou genetickou příčinu, která postihuje synaptické funkce a vývoj mozku. Jedna z nejrozšířenějších hypotéz, která popisuje komorbidní výskyt PAS a epilepsie, předpokládá, že neurovývojový defekt různého původu (např. genetického, metabolického, imunitního nebo působením faktorů vnějšího prostředí) vede ke změně struktury excitačních a inhibičních okruhů, což má za následek rozvoj perzistující E/I nerovnováhy [42]. Tento vztah ilustruje obr. 1 [43].
Fig. 1. Schematic representation of the pathophysiological model of the comorbid occurence of autism spectrum disorder and epilepsy – hypothesis of excitatory-inhibitory dysbalance. Taken and modified from [43]. GABA – gamma-aminobutyric acid
![Schematické znázornění patofyziologického modelu komorbidního výskytu poruch autistického spektra a epilepsie – hypotéza excitačně-inhibični nerovnováhy. Převzato a modifikováno z [43]. GABA – gama aminomáselná kyselina<br>
Fig. 1. Schematic representation of the pathophysiological model of the comorbid occurence of autism spectrum disorder and epilepsy – hypothesis of excitatory-inhibitory dysbalance. Taken and modified from [43]. GABA – gamma-aminobutyric acid](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/37bd2254c37987e6d082383f454cff61.jpeg)
Podkladem hypotézy E/ I rovnováhy je předpoklad, že normální mozkové funkce závisí na dokonalé rovnováze mezi excitačními a inhibičními vstupy do klíčových mozkových buněk. Pokud je přítomna nadměrná excitace nebo nedostatečná inhibice, dojde k hyperexcitabilitě neuronální sítě a rozvoji záchvatů. U celé řady epileptických syndromů může být za rozvoj záchvatů odpovědná nedostatečná inhibice, např. alterace receptorů pro gama aminomáselnou kyselinu (GABA) nebo funkce interneuronů. Může také dojít k nadměrné excitaci např. na úrovni excitačních receptorů nebo excitačních neuronálních okruhů. V souladu s teorií E/ I rovnováhy je i princip terapie antiepileptiky, který známe z běžné neurologické praxe: použití GABAergních agonistů ke zvýšení inhibice nebo snižování excitačních vlivů blokátory sodíkových nebo vápníkových kanálů.
V oblasti rozvoje PAS je hypotéza excitačně-inhibiční rovnováhy přijímána již více než 10 let. Pokud E/ I nerovnováha vede k rozvoji záchvatů, pak samotná přítomnost záchvatů může indikovat rozvoj E/ I nerovnováhy i u pacientů s PAS [25]. Ztráta inhibice cestou alterace GABAergní transmise, dysfunkce interneuronů nebo abnormální migrace byla dokumentována i u pacientů s PAS. Bylo zjištěno snížení hladin GABA v kortikálních oblastech mozku u těchto pacientů [44] a snížení GABAA receptorů v oblasti frontálního kortexu [45]. Post mortem studie ukazují redukci parvalbuminových interneuronů v mediálním prefrontálním kortexu [46]. E/ I nerovnováha může být vysvětlením v patogenezi i u celé řady genetických syndromů, které spojují epilepsii a PAS – mutace v genu pro SCN1A, mutace v genu pro podjednotku epsilon-1 glutamátového receptoru (GRIN2A) nebo mutace v genu pro phosphatase and tensin homolog (PTEN) [25].
Logickým důsledkem této hypotézy je úvaha, že antiepileptika by mohla zlepšit kognitivně-behaviorální komorbidity u autistických pacientů. Zvířecí modely nabízí velmi slibné výsledky, např. u SCN1A +/ - myší bylo prokázáno zlepšení sociálních a paměťových funkcí po léčbě klonazepamem [47]. Bohužel antiepileptika v případě podání pacientům s autizmem tento efekt nevykazují [48,49]. Naše zkušenosti ukazují, že podání klonazepamu pacientům se syndromem Dravetové nevede ke zlepšení kognitivních a behaviorálních funkcí (vlastní nepublikované pozorování).
Z předchozího vyplývá, že problematika autistických projevů u pacientů s epilepsií nebude jen důsledkem samotné epilepsie nebo E/ I nerovnováhy. Do hry vstupují další faktory. E/ I rovnováhu nelze chápat jako statické ekvilibrium. Dokonce i v klidovém stavu jsou mozkové struktury aktivní a informace v mozku jsou zpracovávány prostřednictvím specifických rytmů. Právě tyto mozkové rytmy jsou schopny koordinovat neuronální výboje, a tak je umožněn přenos informací. Základní roli v uvedených procesech hraje GABAergní inervace. GABAergní inhibice vytváří rytmickou aktivitu oscilací a „rytmické ticho“ neuronů během oscilací způsobuje okno, ve kterém jsou informace rozmělňovány na „sousta“, která mohou být účinně přenášena a interpretována [50]. Díky alterované GABAergní transmisi u pacientů s PAS jsou pozměněny mozkové rytmy a koordinace oscilací [25]. Objevuje se celá řada důkazů, že u těchto pacientů jsou redukovány γ a α rytmy a fázová synchronizace po zrakovém nebo sluchovém stimulu. Maturační profil oscilací a synchronizací neuronálních sítí během klidového stavu je u dětí a adolescentů s PAS abnormální [51]. E/ I nerovnováha tedy vede ke změně mozkových rytmů a špatné koordinaci v oblasti neuronálních sítí. Funkční konektivita je poškozena a to může mít za následek rozvoj senzorických, percepčních a sociálních potíží u pacientů s PAS [52] (obr. 1).
Terapeutické perspektivy
Pochopení patofyziologického substrátu komorbidního výskytu PAS a epilepsie je stěžejní z pohledu možných terapeutických intervencí. Hypotéza porušené E/ I rovnováhy nabízí hned několik cílových molekul v léčebných možnostech těchto onemocnění.
GABA agonisté
Pokusy na SCN1A myších modelech ukázaly zlepšení chování po léčbě klonazepamem [47]. Chybí však konzistentní data na dalších zvířecích modelech a je nutno také brát v potaz možnou paradoxní reakci léků ovlivňujících GABAA receptory u pacientů s PAS díky perzistující excitační GABA aktivitě. Ostatně příkladem může být zvýšení úzkosti u některých pacientů s PAS po léčbě diazepamem [53]. Naproti tomu GABAB agonista R-baklofen (arbaclofen, STX209) byl účinný v léčbě zvířecích modelů s PAS [54]. Dokonce jsou již výsledky klinických studií na pacientech s fragilním syndromem X. Ukazuje se, že STX209 může zlepšit symptomatiku u pacientů s PAS [55]. Co se týká ostatní GABAergních látek v klinické praxi, jako např. riluzol, tiagabin, vigabatrin, zlepšení symptomatiky PAS je rozporuplné a nepřesvědčivé [56].
Neurosteroidy
Řadí se mezi pozitivní modulátory GABAA receptorů. Příkladem je syntetický derivát progesteronového metabolitu allopregnanolonu, ganaxolon. Ukázal se efektivní v redukci záchvatů na myších modelech epileptických spazmů [57] a chování u myší s PAS [58]. Výzkum ganaxolonu se nachází ve fázi II u pacientů s refrakterní epilepsií a u pacientů s PAS a syndromem fragilního X.
Antagonisté glutamátového receptoru
Předpokládá se jejich efekt na snížení hyperexcitability u pacientů s PAS a komorbidní epilepsií a byla provedena celá řada studií u pacientů s PAS a syndromem fragilního X. Výsledky jsou opět nekonzistentní a rozporuplné. Antagonista N-metyl-D-aspartát receptoru memantin ukázal pozitivní efekt na myších modelech [59] a dokonce i v klinické studii u pacientů s autizmem [60].
Mammalian Target of Rapamycin (mTOR) inhibitory
Zapojení mTOR inhibitorů je klíčové v regulaci řady buněčných procesů – růstu, proliferaci a translaci proteinů. Komponenty mTOR signální cesty v mozku jsou lokalizovány na synapsích, kde kontrolují synaptogenezi. mTOR inhibitor rapamycin je schopen zlepšit neurobehaviorální deficit u myší s PAS [61]. Probíhají klinické studie s mTOR inhibitory u pacientů s tuberózní sklerózou a refrakterní epilepsií, kde měly pozitivní vliv na redukci záchvatů, jak ukazuji výsledky III. fáze klinické studie [62].
Závěr
Komorbidní výskyt PAS a epilepsie není zřejmě pouhou koincidencí, ale jedná se o velmi komplexní a vícesubstrátový proces. Porozumění patofyziologickému substrátu tohoto komorbidního výskytu je stěžejní nejen pro pochopení procesů probíhajících v mozku u pacientů, ale může být zásadní i z pohledu možných terapeutických intervencí. Hypotéza porušené E/ I rovnováhy nabízí hned několik cílových molekul v léčebných možnostech těchto onemocnění (GABA agonisté, neurosteroidy, antagonisté glutamátových receptorů, mTOR inhibitory a další). Výsledky na zvířecích modelech jsou slibné. Jejich aplikace na člověka ale musí projít ještě složitou cestou, která je vzhledem k závažnosti těchto diagnóz a limitovaným terapeutickým možnostem velkou výzvou v celosvětovém vědeckém měřítku.
Tento projekt a publikace byly podpořeny z fondu Lékařské fakulty Masarykovy univerzity pro juniorského výzkumníka MU Dr. Pavlínu Danhofer, Ph.D. a MU Dr. Štefánii Aulickou, Ph.D. (č. projektu: 2726 - IRP 2018-ROZV/ 24/ LF/ 18).
MUDr. Pavlína Danhofer, Ph.D.
Klinika dětské neurologie LF MU a FN Brno
Černopolní 9
613 00 Brno
e-mail: pavlina.cahova@seznam.cz
Přijato k recenzi: 11. 10. 2018
Přijato do tisku: 14. 1. 2019
Sources
1. Arvidsson T, Danielsson B, Forsberg P et al. Autism in 3-6-year-old children in a suburb of Goteborg, Sweden. Autism 1997; 1(2): 163– 173. doi: 10.1177/ 1362361397012004.
2. Baird G, Charman T, Baron-Cohen S et al. A screening instrument for autism in 18 months of age: a 6-year-follow-up study. J Am Acad Child and Adolesc Psychiatry 2000; 39(6): 694– 702. doi: 10.1097/ 00004583-200006000-00007.
3. Yin J, Schaaf CP. Autism genetics – an overview. Prenatal Diagn 2017; 37(1): 14– 30. doi: 10.1002/ pd.4942.
4. Meltzer A, Van der Water J. The role of the immune system in autism spectrum disorder. Neuropsychopharmacology 2017; 42(1): 284– 298. doi: 10.1038/ npp.2016.158.
5. Brucato M, Ladd-Acosta C, Li M et al. Prenatal exposure to fever is associated with autism spectrum disorder in the boston birth cohort. Autism Res 2017; 10(11): 1878– 1890. doi: 10.1002/ aur.1841.
6. Talbott EO, Marshall LP, Rager JR et al. Air toxics and the risk of autism spectrum disorder: the results of a population based case-control study in southwestern Pennsylvania. Environ Health 2015; 14: 80. doi: 10.1186/ s12940-015-0064-1.
7. Golding J, Rai D, Gregory S et al. Prenatal mercury exposure and features of autism: a prospective population study. Mol Autism 2018; 9: 30. doi: 10.1186/ s13229-018-0215-7.
8. Gump BB, Dykas MJ, MacKenzie JA et al. Background lead and mercutry exposures: psychological and behavioral problems in children. Environ Res 2017; 158: 576– 582. doi: 10.1016/ j.envres.2017.06.033.
9. American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 3rd ed. Washington, DC: American Psychiatric Association 1980.
10. Kanner L. Early infantile autism. J Pediatr 1944; 25: 211– 217.
11. American Psychiatric Association. Diagnostic and statistic manual of mental disorders, 4th ed. (DSM-IV). Washington, DC: American Psychiatric Association 1994.
12. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington, VA: American Psychiatric Association 2013.
13. Lord C, Rutter M, DiLavore PC et al. Autism diagnostic observation schedule. Los Angeles: Western Psychological Services 2001.
14. Ošlejšková H, Kontrová I, Foralová R et al. D. The course of diagnosis in autistic patients: the delay between recognition of the first symptoms by parents and correct diagnosis. Neuro Endocrinol Lett 2007; 28(6): 895– 900.
15. Juříková Z, Jambrikovičová M, Ošlejšková H. Vývoj socioekonomického statu u pacientů s poruchou autistického spektra v průběhu let. Neurol praxi 2016; 17(2): 108– 112.
16. Amiet C, Gourfinkel-An I, Laurent C et al. Epilepsy in simplex autism pedigrees is much lower than the rate in multiplex autism pedigrees. Biol Psychiatry 2013; 74(3): e3– e4. doi: 10.1016/ j.biopsych.2013.01.037.
17. Hughes JR, Melyn M. EEG and seizures in autistic children and adolescents: further findings with therapeutic implications. Clin EEG Neurosci 2005; 36(1): 15– 20.
18. Ošlejšková H. Dušek L, Makovská Z et al. The incidence of epileptic seizures and/ or epileptiform EEG abnormalities in children with childhood and atypical autism. Cesk Slov Neurol N 2008; 71/ 104(4): 435– 444.
19. Amiet C, Gourfinkel-An I, Bouzamondo A et al. Epilepsy in autismis associated with intellectual disability and gender: evidence from a meta-analysis. Biol Psychiatry 2008; 64(7): 577– 582. doi: 10.1016/ j.biopsych.2008.04.030.
20. Woolfenden S, Sarkozy V, Ridley G et al. A systematic review of two outcomes in autism spectrum disorder – epilepsy and mortality. Dev Med Child Neurol 2012; 54(4): 306– 312. doi: 10.1111/ j.1469-8749.2012.04223.x.
21. Danielsson S, Gillberg IC, Billstedt E et al. Epilepsy in young adults with autism: a prospective population-based follow-up study of 120 individuals diagnosed in childhood. Epilepsia 2005; 46(6): 918– 923. doi: 10.1111/ j.1528-1167.2005.57504.x.
22. Parmeggiani A, Barcia G, Posar A et al. Epilepsy and EEG paroxysmal abnormalities in autism spectrum disorders. Brain Dev 2010; 32(9): 783– 789. doi: 10.1016/ j.braindev.2010.07.003.
23. Barger BD, Campbell JM, McDonough JD. Prevalence and onset of regression within autism spectrum disorders: a meta-analytic review. J Autism Dev Disord 2013; 43(4): 817– 828. doi: 10.1007/ s10803-012-1621-x.
24. Hrdlicka M, Komarek V, Propper L et al. Not EEG abnormalities but epilepsy is associated with autistic regression and mental functioning in childhood autism. Eur Child Adolesc Psychiatry 2004; 13(4): 209– 213. doi: 10.1007/ s00787-004-0353-7.
25. Velíšková J, Silverman JL, Benson M et al. Autistic traits in epilepsy models: why, when and how? Epilepsy Res 2018; 144: 62– 70. doi: 10.1016/ j.eplepsyres.2018.05.009.
26. Deonna T, Roulet-Perez E. Early-onset acquired epileptic aphasia (Landau-Kleffner syndrome, LKS) and regressive autistic disorders with epileptic EEG abnormalities: the continuing debate. Brain Dev 2010; 32(9): 746– 752. doi: 10.1016/ j.braindev.2010.06.011.
27. Besag FM. Epilepsy in patients with autism: links, risks and treatment challenges. Neuropsychiatr Dis Treat 2017; 14: 1– 10. doi: 10.2147/ NDT.S120509.
28. Lenck-Santini PP, Scott RC, Mechanisms responsible for cognitive impairment in epilepsy. Cold Spring Harb Perpect Med 2015; 5(10): pii: a022772. doi: 10.1101/ cshperspect.a022772.
29. Steffenburg S, Gillberg C, Hellgren L et al. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry 1989; 30(3): 405– 416.
30. Folstein S, Rutter M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry 1977; 18(4): 297– 321.
31. Ozonoff S, Young GS, Carter A et al. Recurrence risk for autism spectrum disorders: a baby siblings research consortium study. Pediatrics 2011; 128(3): e488– e495. doi: 10.1542/ peds.2010-2825.
32. Simons Foundation. Simons Foundation Autism Research Initiative Gene. [online]. Available from URL:http:/ / sfari.org/ .
33. Amiet C, Gourfinkel-An I, Laurent C et al. Does epilepsy in multiplex autism pedigrees define a different subgroup in terms of clinical characteristics and genetic risk? Mol Autism 2013; 4(1): 47. doi: 10.1186/ 2040-2392-4-47.
34. Ekinci O, Arman AR, Isik U et al. EEG abnormalities and epilepsy in autistic spectrum disorders: clinical and familial correlates. Epilepsy Behav 2010; 17(2): 178– 182. doi: 10.1016/ j.yebeh.2009.11.014.
35. Emanuele E, Boso M, Brondino N et al. Increased serum levels of high mobility group Box 1 protein in patients with autistic disorder. Prog Neuropharmacol Biol Psychiatry 2010; 34(4): 681– 683. doi: 10.1016/ j.pnpbp.2010.03.020.
36. Maroso M, Balosso S, Ravizza T et al. Toll-like receptor 4 and high mobility group Box 1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 2010; 16(4): 413– 419. doi: 10.1038/ nm.2127.
37. Choi J, Min HJ, Shin JS. Increased levels of HMGB1 and pro-inflammatory cytokines in chldren with febrile seizures. J Neuroinflammation 2011; 8: 135. doi: 10.1186/ 1742-2094-8-135.
38. Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism. J Neuroimmunol 2001; 120(1– 2): 170– 179.
39. Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 2005; 46(11): 1724– 1743. doi: 10.1111/ j.1528-1167.2005.00298.x.
40. Theoharides TC, Zhang B. Neuroinflammation, blodd.brain barrier, seizures and autism. J Neuroinflammation 2011; 8: 168. doi: 10.1186/ 1742-2094-8-168.
41. Benson MJ, Manzanero S, Borges K. The effects of C5aR1 on leukocyte infiltration following pilokarpine-induced status epilepticus. Epilepsia 2017; 58(4): e54– e58. doi: 10.1111/ epi.13698.
42. Nelson TE, Olde Engberink A, Hernandez R et al. Altered synaptic transmission in the hippocampus of transgenic mice with enhanced central nervous system expression of interleukin-6. Brain Behav Immun 2012; 26(6): 959– 971. doi: 10.1016/ j.bbi.2012.05.005.
43. Bozzi Y, Provenzano G, Casarosa S. Neurobiological bases of autism-epilepsy comorbidity: a focus on excitation/ inhibition imbalance. Eur J Neurosci 2018; 47(6): 534– 548. doi: 10.1111/ ejn.13595.
44. Puts NA, Wodka EI, Harris AD et al. Reduced GABA and somatosensory function in children with autism spectrum disorder. Autism Res 2017; 10(4): 608– 619. doi: 10.1002/ aur.1691.
45. Zurcher NR, Bhanot A, McDougle CJ et al. A systematic review of molecular imaging (PET and SPECT) in autism spectrum disorder: current state and future research opportunities. Neurosci Biobehav 2015; 52: 56– 73. doi: 10.1016/ j.neubiorev.2015.02.002.
46. Ariza J, Rogers H, Hashemi E et al. The number of chandelier and basket cells are differentially decreased in prefrontal cortex in autism. Cereb Cortex 2018; 28(2): 411– 420. doi: 10.1093/ cercor/ bhw349.
47. Han S, Tai C, Westenbroek RE et al. Autistic like behavior in Scn1a +- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012; 489(7416): 385– 390. doi: 10.1038/ nature11356.
48. Frye RE, Rossignol D, Casanova MF et al. A review of traditional and novel treatments for seizures in autism spectrum disorder: findings from a systematic review and expert panel. Front Public Health 2013; 1: 31. doi: 10.3389/ fpubh.2013.00031.
49. Hellings JA, Nickel EJ, Weckbaugh M et al. The overt aggression scale for ratin aggression in outpatient youth with autistic disorder: preliminary findings. J Neuropsychiatry Clin Neurosci 2005; 17(1): 29– 35. doi: 10.1176/ jnp.17.1.29.
50. Buzsaki G. Neural syntax: cell assemblies, synapsemblies and readers. Neuron 2012; 68(3): 362– 385. doi: 10.1016/ j.neuron.2010.09.023.
51. Vakorin VA, Doesburg SM, Leung RC et al. Developmental changes in neuromagnetic rhythms and network synchronyin autism. Ann Neurol 2017; 81(2): 199– 211. doi: 10.1002/ ana.24836.
52. Simon DM, Wallace MT. Dysfunction of sensory oscillations in autism spectrum disorder. Neurosci Biobehav Rev 2016; 68: 848– 861. doi: 10.1016/ j.neubiorev.2016.07.016.
53. Marrosu F, Marrosu G, Rchel MG et al. Paradoxical reactions elicited by diazepam in children with classic autism. Funct Neurol 1987; 2(3): 355– 361.
54. Silverman JL, Pride MC, Hayes JE et al. GABAb receptor agonist R-baclofen reverses social deficits and reduces repetitive behavior in two mouse models of autism. Neuropsychopharmacology 2015; 40(9): 2228– 2239. doi: 10.1038/ npp.2015.66.
55. Veenstra-VanderWeele J, Cook EH, King BH et al. Arbaclofen in children and adolescents with autism spectrum disorder: a randimizes controlled, phase II trial. Neuropsychopharmacology 2016; 42(7): 1390– 1398. doi: 10.1038/ npp.2016.237.
56. Brondino N, Fusar-Poli L, Panisi C et al. Pharmacological modulation of GABA function in autism spectrum disorders: a systematic review of human studies. J Autism Dev Disord 2016; 46(3): 825– 839. doi: 10.1007/ s10803-015-2619-y.
57. Yum MS, Lee M, Ko TS et al. A pottential effect of ganaxolone in an animal model of infantile spasms. Epilepsy Res 2014; 108(9): 1492– 1500. doi: 10.1016/ j.eplepsyres.2014.08.015.
58. Kazdoba TM, Hagerman RJ, Zolkowska D et al. Evaluation of the neuriactive steroid ganaxolone on social and repetitive behaviors in the BTBR mouse model of autism. Psychopharmacology (Berl) 2016; 233(2): 309– 323. doi: 10.1007/ s00213-015-4115-7.
59. Kim KC, Rhee J, Park JE et al. Overexpressiono of telomerase reverse transcriptase induces autism-like excitatory phenotypes in mice. Mol Neurobiol 2016; 53(10): 7312– 7328. doi: 10.1007/ s12035-015-9630-3.
60. Joshi G, Wozniak J, Faraone SV et al. A prospective open-label trial of memantine hydrochloride for the treatment of social deficits in intellectually capable adults with autism spectrum disorder. J Clin Psychopharm 2016; 36(3): 262– 271. doi: 10.1097/ JCP.0000000000000499.
61. Huber KM, Klann E, Costa-Mattioli M et al. Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci 2015; 35(41): 13836– 13842. doi: 10.1523/ JNEUROSCI.2656-15.2015.
62. French JA, Lawson JA, Yapici Z et al. Adjunctive everolimus therapy for treatment resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a pahes 3, randomised, double-blind, placebo conrolled study. Lancet 2016; 388(10056): 2153– 2163. doi: 10.1016/ S0140-6736(16)31419-2
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2019 Issue 2
Most read in this issue
- Intradurálne extramedulárne nádory chrbtice
- Rychlá diagnostika chemokinu CXCL13 v mozkomíšním moku u pacientů s neuroboreliózou
- Genetika nervosvalových onemocnění
- Roztroušená skleróza a těhotenství z pohledu gynekologa – možnosti asistované reprodukce