
A Bulgarian family with epileptic seizures as a first manifestation of familial cerebral cavernous malformations
Bulharská rodina s epileptickými záchvaty jako prvním projevem familiárních cerebrálních kavernózních malformací
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Authors:
M. Peycheva 1; E. Viteva 1; A. Trenova 1; O. Chaneva 1; Z. Zahariev 1; E. Tournier-Lasserve 2; F. Riant 2
Authors‘ workplace:
Department of Neurology, Medical University of Plovdiv, Bulgaria
1; Service de génétique moleculaire neuro-vasculaire, Hôpital Lariboisière, Paris, France
2
Published in:
Cesk Slov Neurol N 2018; 81(6): 709-711
Category:
Letters to Editor
doi:
https://doi.org/10.14735/amcsnn2018709
Overview
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Dear Editor,
Familial cerebral cavernous malformations (FCCMs) represent a rare genetic disease with a prevalence of 1 : 5,400– 1 : 6,200 [1]. It is inherited in autosomal-dominant pattern through three genes – KRIT1, CCM2 and PDCD10, located at chromosomes 7q21.2, 7p13 and 3q26.1, resp. [2]. Multiple FCCMs are typically found in the CNS (brain and spinal cord) and sometimes in the retina and skin. Clinical manifestations depend on the localization and size of the lesions. Epileptic seizures, focal neurological deficit, non-specific headache, cerebral hemorrhage, skin and retinal lesions are common [3,4]. The diagnosis is confirmed by MRI and molecular-genetic analysis of the patients.
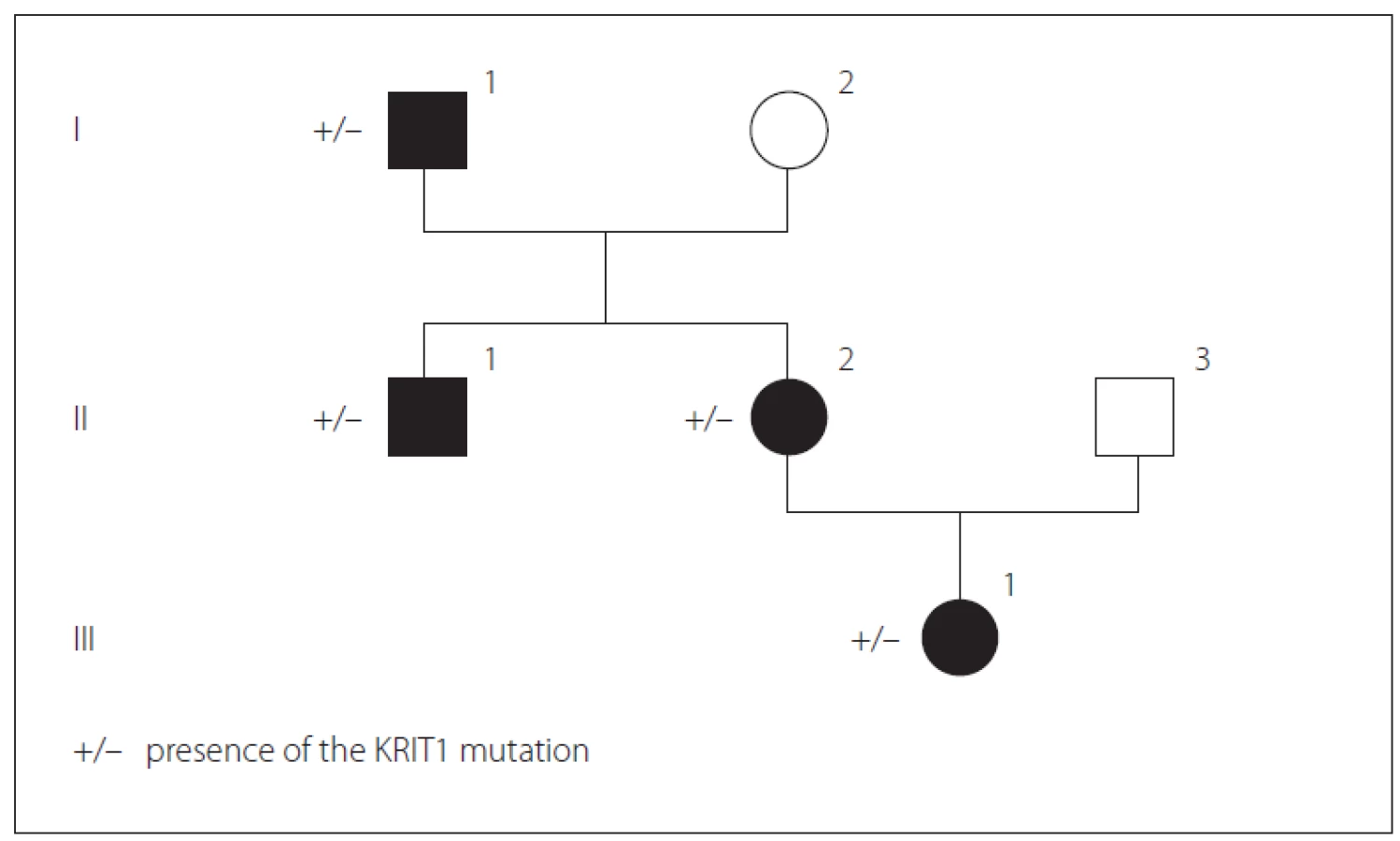
We present a Bulgarian family of Armenian origin with FCCMs, affecting all members from three generations – (Fig. 1). The clinical features and molecular genetic defect were explored.
Obr. 1. Přítomnost mutace KRIT1 v genealogickém stromu rodiny.
In father (I-1), the disease started at a young age (2– 3 decades) with epileptic seizures. At the age of 76, he had double vision and was admitted to the hospital. Upon general examination, a tumor in the neck (about 20 cm in diameter) was evident. Abducens nerve palsy was revealed during neurological assessment. Head MRI showed multiple cavernous lesions with different shapes, localization and size up to 16 mm in diameter. All of them were supra- and infratentorially localized, including the brain stem (Fig. 2A). Some of them had a “popcorn-like” image – specific for a brain cavernoma. The large cystic lesion in the left cervical region, observed also on MRI, was histologically verified as a benign epithelial cyst. Abdominal CT revealed a right suprarenal gland adenoma 1.2 cm in size and two cysts in the left kidney (1.8 and 2.1 cm in size).
Obr. 2. MR cerebrálních kavernózních malformací – otec (A), syn (B), dcera (C, D), vnučka (E).
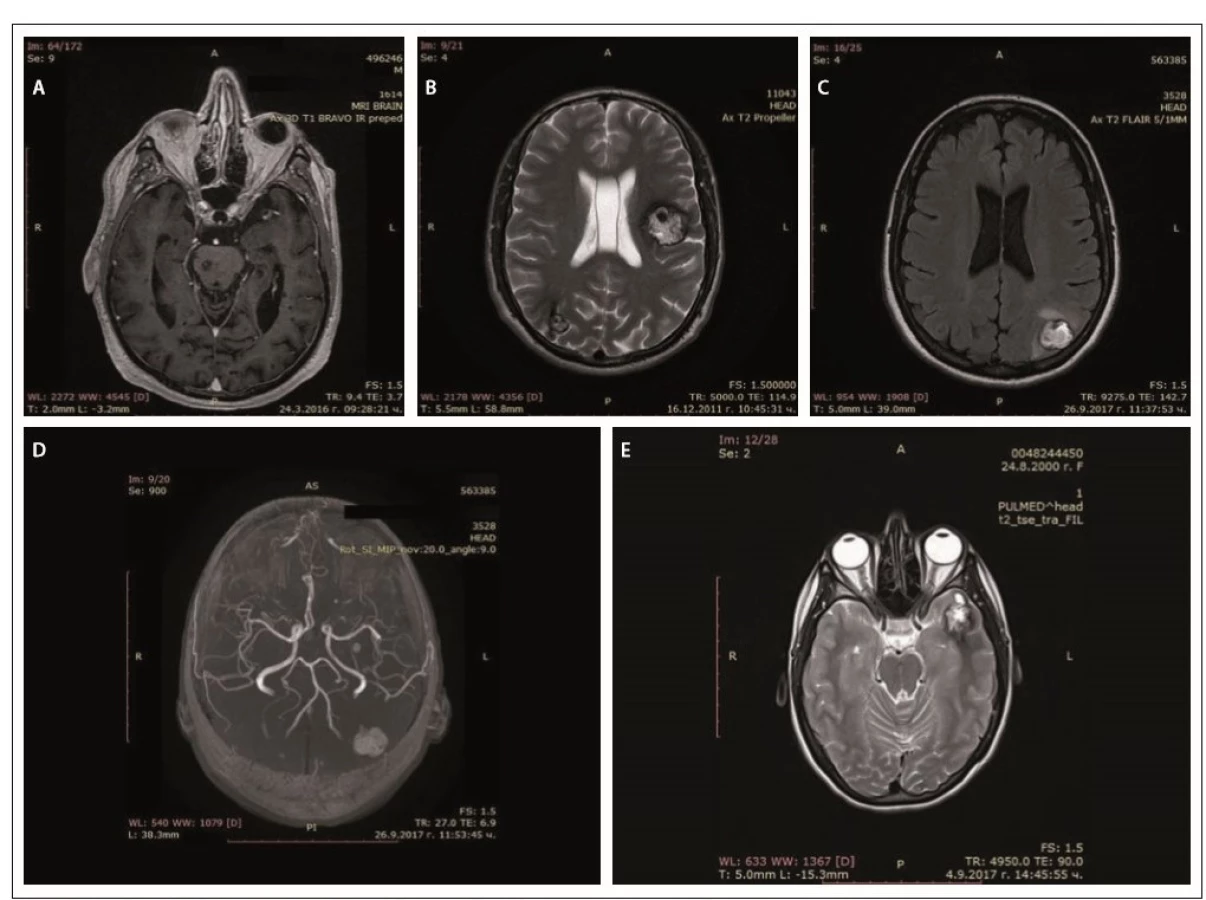
In his son (II-1), the first clinical manifesta-tion was at the age of 28 years of age with two generalized tonic-clonic seizures. Brain concussion was suspected and the patient was admitted to the hospital. No focal neurological signs were found. Four cavernous lesions in supratentorial location were observed on MRI. There were also depositions of hemoglobin degradation products and calcifications. Cavum Vergae was also present (Fig. 2B).
The daughter (II-2) was 44 years old at the time of the assessment, with a history of a cardiac operation for a pulmonary stenosis during childhood. At age 43, she experienced several complex partial epileptic seizures and one generalized tonic-clonic seizure. No focal neurological deficit or cognitive dysfunction were detected upon clinical examination. EEG showed focal slow wave activity in the left frontal and temporal cortex. MRI revealed multiple (at least 13) cavernous malformations of variable sizes. The largest one with a size 22/20 mm was located in the left parieto-occipital cortex and in the subcortical white matter (Fig. 2C). There was also a vascular anomaly in the circle of Willis. The left A1 segment of the anterior cerebral artery was absent and the A2 segment was supplied by the dominant A1 segment of the right anterior cerebral artery and a large anterior communicating artery (Fig. 2D).
In granddaughter (III-1), the first manifestations of the disease were at her age of 17, with focal epileptic seizures, demonstrated by repeating words, inexplicable laughter and impaired consciousness. There were no other complaints or focal neurological signs. EEG showed slow wave activity in the left temporal and occipital areas. Head MRI also revealed multiple cavernous malformations similar to those of the other family members (Fig. 2E).
Examination of all four patients did not find any skin or retinal abnormalities. CT scan of the abdomen of the son, daughter and granddaughter did not show any pathological changes. EEG records of the father and the son were within normal range.
Genetic counseling was proposed for the purpose of diagnostic precision. Genetic screening for FCCMs was done. A typical CCM loss-of-function mutation was identified in exon 11 of the CCM1 gene (NM_004912.3:c.1061_1064dup). It resulted in a stop codon in the open reading frame (p.Ser356Thrfs*2), most probably leading to mRNA degradation. Sanger sequencing of CCM1 exon 11 for all affected relatives detected the same mutation.
All patients were treated with antiepileptic drugs (valproic acid, levetiracetam) with a good therapeutic response with reduction of seizure frequency. Because of the large number and localization of the lesions, surgical excision of the cavernomas was not recommended. Gamma knife surgery of the largest temporal lesion was discussed only for the daughter.
The FCCMs diagnostic criteria include: presence of multiple cerebral cavernous malformations, occurrence of cerebral cavernous malformations in at least two members of a family and presence of a mutation in one of the three genes (KRIT1, CCM2 and PDCD10) causing FCCMs [2,3,5]. Mutations in any of these genes impair the function of protein complexes that are part of the junctions between the vascular endothelial cells, resulting in weakened cell-to-cell junctions and increased leakage from vessels [6,7]. The cavernous malformations histologically present with clustered and enlarged capillaries with a single layer of the epithelium [8]. Neurological manifestations include epileptic seizures (40– 70%), focal neurological deficits (35– 50%), non-specific headache (10– 30%), and cerebral hemorrhages (32%) [3,4]. Non-neurological findings may also be present – vascular skin lesions, retinal vascular lesions, rarely liver cavernoma, renal angioma, atrial myxoma [9] and venous anomalies [10]. The diagnosis is established by MRI, showing multiple cavernous malformations with a reticulated pattern of mixed hyper- and hypointensity on T1- and T2-weighted imaging and a typical hypointense rim on T2-weighted imaging or gradient-echo sequences [8]. Susceptibility-weighted imaging is highly sensitive for small cavernomas. MRI differential diagnosis with other vascular malformations is needed [8]. Molecular-genetic analysis of the patients confirms the genetic defect.
The oldest member of the family has findings without direct relation with the genetic mutation – a rapidly growing epithelial cystic formation on the neck, a suprarenal gland adenoma and two cysts in the left kidney. His daughter has also some atypical signs that might be associated with abnormal angiogenesis – pulmonary stenosis and incomplete circle of Willis. Most members of the family have a late onset of the disease, with the exception of the granddaughter, in whom the first manifestations were at the age of 17.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manu script met the ICMJE “uniform requirements” for biomedical papers.
Accepted for review: 17. 7. 2018
Accepted for print: 29. 10. 2018
Marieta Peycheva
Department of Neurology
Medical University of Plovdiv
Vassil Aprilov 15 A
Plovdiv 4000
Bulgaria
Sources
1. Spiegler S, Rath M, Paperlein C et al. Cerebral cavernous malformations: an update on prevalence, molecular genetic analyses, and genetic counselling. Mol Syndromol 2018; 9(2): 60– 69. doi: 10.1159/ 000486292.
2. Labauge P, Laberge S, Brunereau L et al. Genetics of cavernous angiomas. Lancet Neurol 2007; 6(3): 237– 244. doi: 10.1016/ S1474-4422(07)70053-4.
3. Denier C, Labauge P, Brunereau L et al. Clinical features of cerebral cavernous malformations patients with KRIT1 mutations. Ann Neurol 2004; 55(2): 213– 220. doi: 10.1002/ ana.10804.
4. Battistini S, Rocchi R, Cerase A et al. Clinical, magnetic resonance imaging, and genetic study of 5 Italian families with cerebral cavernous malformation. Arch Neurol 2007; 64(6): 843– 848. doi: 10.1001/ archneur.64.6.843.
5. Verlaan DJ, Davenport WJ, Stefan H et al. Cerebral cavernous malformations: mutations in Krit-1. Neurology 2002; 58(6): 853– 857.
6. Fischer A, Zalvide J, Faurobert E et al. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med 2013; 19(5): 302– 308. doi: 10.1016/ j.molmed.2013.02.004.
7. Spiegler S, Rath M, Hoffjan S et al. First large genomic inversion in familial cerebral cavernous malformation identified by whole genome sequencing. Neurogenetics 2018; 19(1): 55– 59. doi: 10.1007/ s10048-017-0531-7.
8. Mespreuve M, Vanhoenacker F, Lemmerling M. Familial multiple cavernous malformation syndrome: MR features in this uncommon but silent threat. J Belg Soc Radiol 2016; 100(1): 51. doi: 10.5334/ jbr-btr.938.
9. Faure M, Voormolen M, Van der Zijden T et al. Developmental venous anomaly: MR and angiografic features. JBR-BTR 2014; 97(1): 17– 20.
10. Ardeshiri A, Ardeshiri A, Beiras-Fernandez A et al. Multiple cerebral cavernous malformations associated with extracranial mesenchymal anomalies. Neurosurg Rev 2008; 31(1): 11– 17. doi: 10.1007/ s10143-007-0111-7.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2018 Issue 6
Most read in this issue
- Diagnostics, symptomatology and findings in diseases and disorders of the autonomic nervous system in neurology
- New-onset refractory status epilepticus and considered spectrum disorders (NORSE/ FIRES)
- Clinical results of cervical discectomy and fusion with anchored cage – prospective study with a 24-month follow-up
- Pragnancy and multiple sclerosis from a neurologist’s point of view