
Současná kortikoterapie u nádorů mozku
Current Corticosteroid Treatment in Brain Tumours
Considering that there is no curative treatment for the majority of malignant brain tumours, supportive therapy plays a very important role and treatment with corticosteroids is its integral part. The goal of corticosteroid therapy is to maintain adequate quality of life and functional self-sufficiency. Corticosteroids have been used to effectively treat oedema around brain tumours since 1960s. In addition to their antioedematous action, their antiemetic and antilymphocytic effect has been utilised in neurooncology. However, these positive impacts are accompanied by a range of adverse cardiovascular, muscular, and psychiatric effects. Despite the widespread use of corticosteroids in neurooncology, no relevant data have been available so far concerning their optimal and safe administration in this specific case. Although there has been a development in disease-modifying treatment modalities over the recent years, the prognosis of patients with brain tumours remain poor; therefore, maintaining an acceptable quality of life continues to be a priority. Rational corticosteroid prescription to minimise toxicity is one of the major factors affecting the quality of life in patients with brain tumours.
Key words:
brain tumours – corticosteroid treatment – adverse effects – quality of life
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
:
O. Kalita; M. Vaverka; L. Hrabálek
:
Neurochirurgická klinika LF UP a FN Olomouc
:
Cesk Slov Neurol N 2016; 79/112(5): 521-527
:
Review Article
Vzhledem k tomu, že u většiny maligních nádorů mozku neexistuje kurativní forma léčby, hraje podpůrná terapie velmi důležitou roli a kortikoterapie je její nedílnou součástí. Jejím záměrem je udržení dostatečné kvality života a funkční soběstačnosti pacienta. Kortikoidy jsou již od 60. let 20. století využívány k efektivní léčbě edému v okolí mozkových nádorů. Kromě antiedematózního působení se využívá v neuroonkologii i účinek antiemetický a antilymfocytární. Tyto pozitivní dopady jsou však doprovázeny celou řadou nepříznivých vedlejších kardiovaskulárních, muskulárních a psychiatrických projevů. Navzdory tomu, že kortikoidy jsou široce používány v neuroonkologii, neexistují dosud relevantní data, která by se týkala jejich optimálního a bezpečného podávání v tomto specifickém případě. Během posledních let došlo k rozvoji léčebných modalit modifikujících průběh nemoci, avšak prognóza pacientů s nádory mozku zůstává špatná, a proto udržení akceptabilní kvality života je i nadále prioritou. Rozumná preskripce kortikosteroidů za účelem minimalizace toxicity je jedním z významných prvků ovlivňujících kvalitu života těchto pacientů.
Klíčová slova:
nádory mozku – kortikoterapie – nežádoucí účinky – kvalita života
Úvod
Kortikosteroidy tvoří skupinu biologických mediátorů, hormonů, produkovanou kůrou nadledvinek. Tyto hormony můžeme rozdělit na glukokortikoidy (např. kortizol) a mineralokortikoidy (např. aldosteron), které regulují celou řadu procesů, jako je metabolizmus, vodní a iontové hospodářství, imunitní a stresovou reaktivitu.
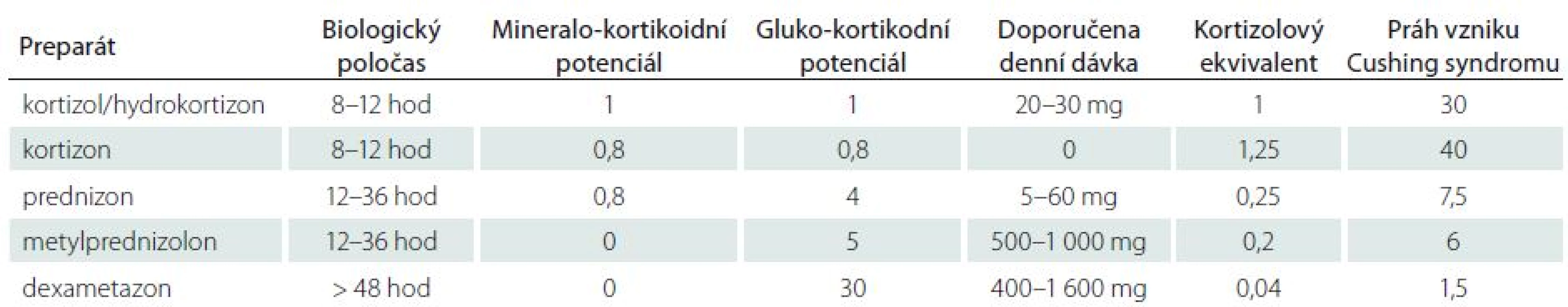
Steroidy jsou metabolizovány v játrech pomocí cytochrom-P450-oxydázových enzymů [1]. Za fyziologických podmínek se 90 % kortikosteroidů váže v plazmě na transportní protein transkortin, zbytek se váže na albumin nebo cirkuluje volně. Kortikosteroidy uplatňují svůj efekt na buněčný metabolizmus po navázání na glukokortikoidní receptory na buněčné membráně v buňkách všech tkání. Následně je informace přenášena intracelulárně na specifické DNA sekvence v buněčném jádře, kde moduluje transkripční aktivitu [2,3]. Ve snaze využít některé ze zmíněných vlastností pro léčebné účely byla v průběhu posledního půlstoletí vytvořena celá řada syntetických kortikosteroidů, kortikoidů, které se liší ve svém glukokortikoidním a mineralokortikoidním působení, biologickým poločasem apod. (tab. 1).
Patofyziologie účinku kortikosteroidů v mozkové tkáni
Kortikoidy jsou relativně neefektivní při cytotoxickém a intracelulárním edému. Naopak jsou velmi účinné při řešení vazogenního edému, který vzniká poškozením hematoencefalické bariéry [4]. Dle analýzy z roku 1994 [5] po sedmi dnech podávání dexametazonu je transport přes hematoencefalickou bariéru již konstantní, což je podstatou stabilizace hematoencefalické bariéry a redukce mozkového edému. Je nutné poznamenat, že stabilizace hematoencefalické membrány se týká i extravazálního úniku kontrastní látky během MR vyšetření mozku [6]. Proto pokud během terapie mozkových nádorů dojde na kontrolním MR zobrazení ke snížení postkontrastního sycení patologické léze, je pro hodnocení vhodné znát i recentní dávku kortikoterapie.
Obecně patologický vzestup permeability cév a hematoencefalické bariéry je většinou spojen se vzestupem paracelulární permeability [7]. Mezi základní mechanizmy vzniku vazogenního mozkového edému v okolí nádoru patří:
- Pokles exprese funkčních proteinů tigh junction; např. maligní gliomy neexprimují funkční protein okludin, exprimují jen nízkou hladinu klaudinu-1 v nádorových cévách a zvýšeně exprimují akvaporin-4 v endoteliálních výběžcích, což vše vede k poškození hematoencefalické bariéry [8–10].
- Poškození již exprimovaných proteinů tigh junction; typickým příkladem je VEGF zprostředkované poškozením hematoencefalické bariéry [11]. Tento protein má velmi silnou permeabilní aktivitu, která je 1 000krát silnější než histamin [12]. U gliomů, VEGF m RNA je až 50krát zvýšeně exprimované ve srovnání s normální mozkovou tkání. Tato zvýšená exprese koreluje s kapilární permeabilitou u lidských gliomů [13].
Dalšími spouštěči poškození hematoencefalické bariéry jsou prozánětlivé cytokiny a jiné mediátory, jako např. leukotrieny C4, které jsou nalézány ve vysoké koncentraci u glioblastomů, přičemž koncentrace koreluje s velikostí peritumorózního edému [14,15].
Jak bylo řečeno, kortikoidy jsou schopny dočasně hematoencefalickou bariéru stabilizovat, což se děje několika mechanizmy:
- Ovlivněním sekrece cytokinů, jako je např. VEGF, kdy kortikosteroidy na úrovni DNA redukují expresi VEGFR v buňkách nádoru mozku a v buňkách hematoencefalické bariéry [16–19].
- Protizánětlivým mechanizmem, který je zprostředkován přímou aktivací apoptózy bílých krvinek [20] nebo např. regulací procesu genové exprese interleukinů (IL-1, IL-6, IL-8, IFN), granulocyty-makrofágy kolony stimulujícího faktoru a tumornekrotizující faktoru [16].
- Expresí genů a molekul jako jsou např. klaudin, okludin, cadherin, které modifikují permeabilitu hematoencefalické bariéry [21,22].
Indikace kortikoterapie u nádorů mozku
Kortikoidy jsou podávány u nádorů mozku jak samostatně, tak jako podpůrná léčba jedné ze základních léčebných modalit (chirurgie, radioterapie, chemoterapie). Cílem je podpora dosažení klinické a radiologické léčebné odpovědi. Objem a doba podávání kortikoidů závisí primárně na odpovědi tumoru na základní léčbu. Pokud je dosaženo kompletní odpovědi na základní terapeutickou modalitu, není většinou již nutné v kortikoterapii pokračovat. Existuje-li částečná nebo stabilní odpověď na základní léčebnou modalitu, je podávání kortikoidů prodlužováno z důvodů symptomatické kontroly perzistujícího perilezionálního, mozkového edému. Vliv kortikoterapie na klinickou a radiologickou odpověď byl zahrnut i do dvou posledních systémů hodnotících efekt léčby u gliomů – McDonaldova kritéria a RANO kritéria [23,24]. V souladu s těmito kritérii radiologická a klinická odpověď na terapii není hodnocena jako kompletní, není-li u pacienta ukončeno i podávání kortikoidů.
Skupinou mozkových nádorů s atypickou reakcí na kortikoterapii jsou primární a sekundární lymfomy [25,26]. U těchto nádorů dochází často k promptní reakci spojené se zastavením buněčného dělení a indukcí buněčné smrti. Zmíněná apoptóza u B a T lymfocytů je většinou zprostředkována p38 mitogen-aktivizovanou proteinkinázou. Tento tlumivý efekt je však jen přechodný [27].
V současnosti nejsou dostupné informace o tom, že by kortikoterapie blokovala růst gliomů nebo mozkových metastáz. Naopak některé práce uvádí, že kortikoidy mohou částečně stimulovat růst gliomových buněk, nebo dokonce zvyšovat odolnost vůči chemoterapii [28–32]. I toto by měl být důvod pro velmi kritické hodnocení aplikace kortikoidů během léčby nádorů mozku.
Antiemetický efekt dexametazonu a metylprednizolonu je využíván již řadu let, ale přesný mechanizmus účinku opět není znám [33]. Některé práce ukázaly, že vlivem kortikoterapie dochází k sníženému uvolňování 5-HT3 z krevních elementů či je přímo snížena exprese 5-HT3 receptorů [34,35]. Další práce prokazovaly přímý, centrální entiemetický efekt kortikoidů na prodlouženou míchu [36].
Vedlejší účinky kortikoterapie a kortikosteroidní toxicita
Dlouhodobé podávání glukokortikoidů vede k funkčnímu hypokortikalizmu jako důsledku zpětnovazebného útlumu produkce kortikoliberinu v hypothalamických neuronech, adrenokortikotropního hormonu v kortikotrofních buňkách hypofýzy a následně i kortizolu v kůře nadledvin. Pravděpodobnost vzniku hypokortikalizmu je výrazně individuální a je obecně závislá na dávce a době aplikace steroidů. Na tento fenomén je nutno myslet při snaze o snížení či vysazení kortikoterapie, protože ten není možný bez opětovné fyziologické aktivace osy hypothalamus–hypofýza–nadledviny [37].
Fyziologickým účinkem steroidů je k retenci Na+ iontů a k exkreci K+ iontů. Při kortikoterapii vzniká výrazná hypokalemie a hypokalemická alkalóza s následnou nutností substituce kalia [38,39].
Druhým problémem je vlastní efekt exogenních steroidů na lidské tkáně. Obecným problémem u nádorů mozku je odlišení toxicity vyvolané kortikoidy od vlastní progrese nemoci [40,41]. Jedna ze starších retrospektivních studií zaměřená na neuroonkologické pacienty objevila, že až 51 % pacientů mělo nejméně jednu z forem steroidní toxicity [42]. Jiná retrospektivní studie zaměřená na radioterapii mozkových metastáz ukázala, že nežádoucí vedlejší účinky kortikoterapie byly typické pro pacienty s úvodní dávkou dexametazonu 16 mg/den. V této skupině mělo 91 % pacientů v průběhu léčby nejméně jeden z vedlejších nežádoucích projevů. Naopak pacienti s nižší úvodní dávkou měli i nižší toxické projevy a pouze 65 % pacientů registrovalo alespoň jeden vedlejší nežádoucí účinek [43]. V tab. 2 je přehled základních vedlejších negativních účinků kortikoterapie u neuroonkologických pacientů.

V textu bychom chtěli rozvézt ty nejzávažnější. Jedním z nejčastějších vedlejších účinků kortikoterapie je sekundární hypertenze vyskytující se v cca 20 % případů. Byl potvrzen její vznik v závislosti na dávce kortikoterapie [44–46].
Hyperglykemie je velmi závažný vedlejší efekt kortikoterapie u primárních nádorů mozku vyskytující se u cca 50 % pacientů [47]. Samotný steroidní diabetes mellitus je nejčastějším sekundárním typem tohoto onemocnění. Přetrvávající hyperglykemie u pacientů tři měsíce po operaci se ukázala být signifikantním nezávislým prognostickým faktorem korelujícím se špatným celkovým přežíváním [48].
Dalším vedlejším projevem kortikoterapie je imunosuprese spojená především s vysokým rizikem vzniku kvasinkových a mykotických infekcí [49,50].
Kortikoidy indukovaná myopatie se vyskytuje u 60 % pacientů s kortikoterapií a je zapříčiněna poklesem syntézy proteinů a katabolizmem svalových proteinů [51]. Existují dvě formy steroidy indukované myopatie: generalizovaná myopatie spojená s rabdomyolýzou nastupující ihned po započetí vysokodávkové kortikoterapie a klasická forma vznikající postupně, během dlouhodobé kortikoterapie [52–54]. Dle některých údajů myopatie častěji vzniká u fluorosteroidů (dexametazon), ale jednoznačné závěry komparativních studií chybí [55–57].
Psychiatrické vedlejší účinky kortikoterapie (insomnie, emoční labilita, hypomanické a manické ataky) jsou během léčby zaznamenány až u 60 % neuroonkologických pacientů [58–60]. Obtíže většinou mizí po ukončení kortikoterapie.
Kortikoidy indukovaná osteoporóza nebyla u pacientů s nádory mozku dříve studována, ale s prodlužující se dobou přežívání pacientů se i ona dostává do popředí. Kortikoterapií indukovaná osteoporóza je nejčastější formou iatrogenní osteoporózy [61,62]. U pacientů s kortikoidy dosahuje riziko vzniku jakékoliv osteoporotické fraktury až 50 %, přičemž riziko zlomeniny stehenní kosti či obratle je až pětinásobné oproti běžné populaci [63,64]. Protože mnoho pacientů s nádorem mozku je na trvalé antiepileptické medikaci, je nutno zdůraznit, že i léky na bázi kyseliny valproátové a fenytoinu mohou také vznik osteoporózy podporovat [65–67].
Atypická situace je u primárních či sekundárních lymfomů mozku vyplývajících z kortikoidy indukované apoptózy T a B [25–27]. Dle jedné z prací měli pacienti s lymfomy po kortikoterapii v 59 % radiologickou odpověď na MR a v 84 % klinickou odpověď [68]. Radiologická odpověď na kortikoterapii projevující se redukcí objemu či úplným vymizením („ghost tumour“) postkontrastně se sytícího nádoru na MR [69] je jedním z klinicko-diagnostických testů u těchto nádorů. V případě podezření na lymfomy mozku je nutná histologická verifikace pomocí biopsie. Aby byl odběr vzorku maximálně výtěžný, je nutné cca 1–2 dny před výkonem vysadit kortikoidy, což vede k opětovné radiologické transparentnosti nádoru.
Lékové interakce s kortikoidy
Jak bylo řečeno, steroidy jsou odbourávány v játrech pomocí cytochrom-P450-oxydázových enzymů [1], a proto všechny léky, které jsou eliminovány stejným mechanizmem, mohou kompetitivně ovlivňovat hladinu svou i kortikoidů. Dále sama kortikoterapie vede k hypokalemii, a tak je vždy nutné zvažovat aplikaci diuretik zvyšujících exkreci kalia [39]. Uvedená hypokalemie naopak zvyšuje toxicitu glykosidových kardiotonik [70]. Kortikoidy na jedné straně podporují glukoneogenezi a na straně druhé vedou ke snížené periferní utilizaci glukózy, což vede k hyperglykemii. Exogenní steroidy také snižují účinek inzulinu a perorálních antidiabetik cestou zvýšené periferní rezistence na inzulin [71–73]. U perorálních antikoagulancí (Warfarin) kortikoidy snižují účinnost a u nesteroidních antiflogistik antirevmatik zvyšují riziko krvácení např. do gastrointestinálního traktu [74]. Kortikosteroidy samy o sobě nejspíše nejsou přímo odpovědné za vznik peptických vředů během terapie; nicméně je zřejmé, že podávání glukokortikoidů kompromituje hojení slizničních defektů a může i maskovat symptomy při evoluci peptického vředu. Také při kombinaci s nesteroidními antiflogistiky existuje zvýšené riziko rozvoje gastrointestinálních vředů. Naopak snížený účinek kortikosteroidů způsobují léky jako rifampicin, fenytoin, barbituráty [37,74].
Prediktory toxicity léčby
Existuje konsenzus, že kortikoidní toxicita odpovídá jak kumulativní dávce kortikosteroidů, tak době podávání kortikoterapie [75,76]. Při hodnocení kumulativní dávky se ukázalo, že pacienti, kteří obdrželi celkovou dávku dexametazonu větší než 4 g/den, měli incidenci toxicity 27 % oproti 13 % u pacientů s dávkou menší než 4 g/den [77]. Dalším potenciálním prediktorem může být hladina sérového albuminu, kdy nejvyšší toxicity dosahovali pacienti se sérovým albuminem pod 0,25 g/l [78].
Variabilita toxicity kortikoterapie
U některých podskupin pacientů s v průběhu kortikoterapií setkáváme s toxicitou mnohem dříve než u jiných. Starší práce zabývající neuroonkologickými pacienty popisovaly, že u 1/3 pacientů se objevily první známky toxicity během prvních tří týdnů léčby [77]. Kortikoidy reagují s glukortikoidním receptorem, jehož gen se nachází na 5. chromozomu. Gen glukokortikoidního receptoru obsahuje nejméně tři promotory, které zajišťují pro tkáň specifickou expresi receptoru [79]. Právě potranskripční a potranslační modifikace zprostředkovaná glukokortikoidními receptory na extracelulární, intracelulární a intranukleární úrovni je základem velké variability klinické odpovědi [80–85].
Na uvedené variabilitě se může podílet i odbourávání steroidů, kdy enzymaticky (cytochrom-P450) kompromitovaní pacienti mohou mít sklon k projevům toxicity kortikoterapie [86].
Praktická kortikoterapie v neuroonkologii
Existuje obrovská variabilita v kortikoidní preskripci v neuroonkologii, s velmi malým konsenzem v literatuře [43].
Výběr kortikosteroidu
Dexametazon je nejčastěji předepisovaný kortikoid pro léčbu nádorem indukovaného mozkového edému [87]. Byl to první kortikoid použitý v předoperační přípravě před operací mozkového nádoru [88]. Dexametazon má lepší průnik do CNS než prednizolon [89]. Dále má nejmenší mineralokortikoidní aktivitu ze všech steroidů, což se projevuje mnohem menší retencí tekutin v porovnání s prednizolonem [90]. Dexametazon má dlouhý biologický poločas a vysokou účinnost, takže není třeba frekventní aplikace. Naopak jeho dlouhý biologický poločas může při dlouhodobém používání vést k vyššímu riziku nástupu útlumu fyziologické sekrece nadledvinek [91].
Startovací dávka kortikoterapie
U dexametazonu je obecně akceptováno 16 mg/den jako úvodní dávka kortikoterapie, a to jak pro primární, tak pro sekundární nádory mozku. Tato startovací dávka se datuje od roku 1961, kdy byla vytvořena první dose-response křivka použití dexametazonu v předoperační přípravě u primárního nádoru mozku [88,92]. Novější studie a klinické zkušenosti předpokládají, že nižší startovací dávka může být v mnoha případech dostatečná. Jedna z randomizovaných studií ukázala, že skupiny pacientů se startovací dávkou dexametazonu 4, 8 a 16 mg/den dosáhly během jednoho týdne stejného zlepšení Karnofsky performance scale [93]. Musíme mít na mysli, že jak frekvence, tak tíže steroidní vedlejších efektů odpovídá dávce a délce kortikoterapie a startovací dávka může podpořit oba tyto aspekty.
Pokračovací kortikoterapie
V klinické praxi se v délce podávání kortikoidů odráží individuální odpověď pacienta na léčbu a/nebo zvyklosti lékaře či pracoviště. Jedna prospektivní studie sledovala dynamiku podávání kortikoidů u pacientů s gliomy po radioterapii. I po třech měsících po ukončení radioterapie stále vyžadovalo kortikoterapii 71 % pacientů s maligními gliomy [94]. Dle jiné, retrospektivní studie pacienti s primárními nádory mozku vyžadovali kortikoidy průměrně 23 týdnů po radioterapii, kdežto sekundární nádory mozku jen průměrně sedm týdnů [95].
Přestože se prakticky všem pacienti s nádorem mozku během léčby aplikují kortikoidy, optimální dávkování během chirurgické a onkologické terapie a po ní není přesně určeno [87]. Starší studie ukázala, že postupné snižování denní dávky dexametazonu podávané dvakrát denně (8 mg/den, 4 mg/den, 2 mg/den) bylo možné u 14 z 20 pa- cientů, z toho 13 pacientů ze 14 bylo bez symptomů steroidní léčby 30. den po ukončení terapie [95]. Dle další práce o metastatických nádorech mozku je doporučováno, aby dávky kortikoidů byly snižovány pomalu během doby dvou týdnů nebo u symptomatických pacientů i déle, a to vše s ohledem na individuální léčebný režim a při respektování všech dlouhotrvajících následků kortikoterapie [96]. Existují dva aspekty týkající se snižování dávky kortikoidů. Zaprvé je to riziko znovuobjevení se symptomů nitrolební hypertenze a zadruhé snaha o opětovné obnovení, utlumené fyziologické sekrece nadledvin. Signifikantní útlum fyziologické sekrece kortikosteroidů se objevuje za cca dva týdny, což značí, že při ukončování terapie vyžaduje naši pozornost každý pacient, který přijímá kortikoidy dále než 10 dní [91]. Pokusy vypočítat přesnou dávku kortikoidů u neuroonkologických pacientů s použitím tělesné hmotnosti, povrchu těla, věku zatím stále selhávají [97]. Otázkou zůstává nejen celková denní dávka, ale i rozložení dávky steroidů během dne. Endogenní steroidy jsou vyplavovány v organizmu v cirkadiálním rytmu s ranním maximem. Dle posledních prací rozložení denní dávky kortikoterapie kopírující cirkadiální rytmus hormonální sekrece nadledvin výrazně snižuje riziko vzniku nežádoucích vedlejších účinků [98].
Jak vyplývá z předchozích řádků, u každé déletrvající kortikoterapie je nutné diagnostikovat, sledovat a léčit vedlejší nežádoucí účinky. Frekvence a typ laboratorních, zobrazovacích a klinických vyšetření u těchto sekundárních symptomů a syndromů odpovídá strategii léčby primárních chorob. U rozkolísaných kalemií a glykemií, které by mohly mít závažné následky, jsou vhodné denní laboratorní kontroly, a to až do stabilizace stavu. Kontroly krevního tlaku, laboratorní vyšetření krevního obrazu (vč. diferenciálního rozpočtu bílých krvinek) se řídí tíží nežádoucích účinků, s frekvencí vyšetření cca 1–3krát/měsíc. U osteoporózy je indikována denzitometrie, a to 1–2krát/rok. Po steroidy indukované myopatii či psychóze pátráme při každé klinické kontrole.
Stran terapie u hypokalemie je nutná substituce kalia, u arteriální hypertenze je vhodná aplikace antihypertenziv a kalium šetřících diuretik a u osteoporózy je vhodný zvýšený přísun potravin s vysokým obsahem vápníku a substituce vitaminu D. V případě steroidního diabetu nebo poruch glukózové tolerance jsou doporučována režimová opatření (dieta, pohyb) či medikamentózní terapie (perorální antidiabetika, inzulin). Opět je nutné zdůraznit, že základní profylaxí vzniku nežádoucích, vedlejších účinků kortikoidů je minimalizování dávky a doby podávání.
Kortikoterapie v terminální fázi nemoci
V posledních několika týdnech života je celková dávka kortikoidů výrazně navýšena jako reakce na nárůst edému mozku při progresi nádoru. Z důvodu dlouhodobé kortikoterapie dochází k projevům mnoha nežádoucích vedlejších efektů [99]. Pacienti velmi nepříjemně vnímají především kožní projevy a otoky vznikající na podkladě retence tekutin. Vysoké dávky kortikoidů mohou zapříčinit vznik psychiatrických projevů vč. tzv. terminální agitovanosti, což je multifaktoriální, ireverzibilní, deliriu podobný stav. Obecně je lepší postupné navyšování kortikoterapie než náhlá změna.
Terapie nahrazující kortikoidy – budoucnost
Potřeba dalších látek s obdobnými účinky jako kortikosteroidy je nasnadě. Během doby byla vytvořena řada agens s obecně imunosupresním efektem, ale také s výraznými, třebaže kvalitativně jinými, vedlejšími efekty. V terapii edému mozku vyvolaného nádorem však nebyl žádný jiný medikament použit. Výjimkou je bevucizumab, který cestou blokace VEGF receptoru zabraňuje vaskulogenezi a angiogenezi u nádorů a zároveň stabilizuje hematoencefalickou bariéru, čímž redukuje perilezionální edém v okolí nádoru mozku [100,101].
Závěr
Kortikosteroidy zůstávají nejefektivnější agens pro léčbu peritumorózního otoku mozku. Vedlejší efekty kortikoterapie redukují jasný benefit této terapie. Skutečná frekvence toxicity kortikoterpie u neuroonkologických pacientů není zcela přesně známa. Při znalosti prediktivních faktorů vzniku vedlejších efektu je nutné začít s nejnižší efektivní dávkou na nejkratší možnou dobu trvání terapie. Při terapii vedlejších projevů kortikoterapie jsou využívány stejné přístupy jako u primárních onemocnění (tab. 2). Základem pro rozvahu jsou potřeby pa- cienta s využitím konceptu personalizované medicíny [102–104].
Doporučení standardní kortikoterapie
- Výběr kortikoidu – dexametazon je kortikoid první volby pro kontrolu nádorem indukovaného edému mozku. Změna na prednizolon může být provedena v případě těžké formy proxymální myopatie vyvolané dexametazonem nebo v případě podpory postupného vysazování kortikoterapie.
- Startovací dávka dexametazonu – optimální dávka je určena maximálním benefitem a minimální toxicitou, ale není přesně známa; jestliže je použita startovací dávka dexametazonu 16 mg/den, měla by být snižována co nejdříve, jakmile je to možné, za předpokladu udržení benefitu tak, aby byla rychle nalezena nejnižší účinná dávka. Prvních 10 dnů kortikoterapie bychom měli považovat za úzké okno k nalezení optimální dávky s tím, že snižování dávky je dáno i rychlostí obnovy funkce nadledvinek, pacientovou tolerancí a jeho odpovědí na další léčebné modality.
- Pokračovací dávka dexametazonu – pacient by měl být ponechán na kortikoterapii minimální potřebnou dobu. Časné snížení je jediným známým prostředkem k identifikaci potřeby a minimalizace zbytečného prodlužování terapie. Pokus o snížení dávky by mělo být proveden u všech pacientů a obava ze znovuobjevení symptomů by neměla lékaře odradit od redukce dávek. Redukce dávky by měla být rychlá v prvních 10 dnech kortikoterapie (cca každý 1.–3. den), ale pomalejší po 10. dnu (cca každý 4.–7. den) s cílem dosažení fyziologické dávky. Není ospravedlnitelné ponechat fixní dávku kortikoterapií s klinickým hodnocením každé 1–2 měsíce. Při redukci dávky by měli být pacient a jeho okolí poučeni o příznacích znovuobjevení se symptomů. Vzhledem k znalostem o fyziologickém, cirkadiálním rytmu sekrece hormonů nadledvin by se i podávání syntetických preparátů mělo řídit tímto pravidlem, takže největší část denní dávky kortikoidů by měl být podána v ranních hodinách.
Doporučení kortikoterapie v terminálním stadiu nemoci
- V úvodu je vhodné podstatně navýšit dávku dexametazonu (až na 8 –16 mg/den). V tuto chvíli lze takovou strategii ospravedlnit vzhledem k ireverzibilitě deteriorace a vzhledem k nutnosti agresivního potlačení neurologických symptomů. I přesto je důležité, aby navýšení dávky se dělo postupně v čase, se sledováním reakcí pacienta. Jestliže pacient nereaguje na zvýšení dávky dexametazonu během 48 hod, nelze již ovlivnit zafixovaný neurologický deficit a je vhodné snížit dávku zpět na poslední úroveň. Reagují-li symptomy na zvýšení dávky, je vhodné pokračovat po dobu cca čtyř dnů. Poté opět dávku pomalu snižovat až na nejnižší dávku, která má benefit. Po celou dobu je nutné pacienta sledovat. Snižování dávky kortikoterapie je ospravedlnitelné i v případě existence netolerovatelných vedlejších efektů, vymizení benefitu a při ztrátě kontroly deteriorace. Po celou dobu je vhodné diskutovat s pacientem a jeho okolím o důvodech úprav medikace, jejich přínosů a zatížení.
- V konečné fázi terminálního stadia dochází k postupné deterioraci neurologického stavu, ztrácí se schopnost polykat, a tím schopnost přijímat medikamenty perorální cestou. Jde o etické dilema, zdali pokračovat v subkutánní nebo parenterální kortikoterapii až do konce života. Jedna retrospektivní studie zaměřená na paliativní péči i u pacientů s nádory mozku ukázala, že jen 2 % pacientů přešla v terminálním stadiu z dlouhodobé perorální kortikoterapie na parenterální [105].
Autoři deklarují, že v souvislosti s předmětemstudie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Ondřej Kalita, Ph.D., MBA
Neurochirurgická klinika
LF UP a FN Olomouc
I. P. Pavlova 6
779 00 Olomouc
e-mail: ondrej.kalita@fnol.cz
Přijato k recenzi: 12. 1. 2016
Přijato do tisku: 2. 4. 2016
Sources
1. Talar-Williams C, Sneller MC. Complications of corticosteroid therapy. Eur Arch Otorhinolaryngol 1994; 251 (3): 131–6.
2. Johnson AB, O’Malley BW. Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM) -based cancer therapy. Mol Cell Endocrinol 2012; 348 (2): 430–9. doi: 10.1016/j.mce.2011.04.021.
3. Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther 2012; 134 (1): 54–67. doi: 10.1016/j.pharmthera.2011.12.004.
4. Sandercock PA, Soane T. Corticosteroids for acute ischaemic stroke. Cochrane Database Syst Rev 2011; 9: CD000064. doi: 10.1002/14651858.CD000064.pub2.
5. Andersen C, Astrup J, Gyldensted C. Quantitation of peritumoural oedema and the effect of steroids using NMR relaxation time imaging and blood-brain barrier analysis. Acta Neurochir Suppl 1994; 60: 413–5.
6. Armitage PA, Schwindack C, Bastin ME, et al. Quantitative assessment of intracranial tumor response to dexamethasone using diffusion, perfusion and permeability magnetic resonance imaging. Magn Reson Imaging 2007; 25 (3): 303–10.
7. Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: how to „open“ the blood brain barrier. Curr Neuropharmacol 2008; 6 (3): 179–92. doi: 10.2174/157015908785777210.
8. Papadopoulos MC, Saadoun S, Woodrow CJ, et al. Occludin expression in microvessels of neoplastic and non-neoplastic human brain. Neuropathol Appl Neurobiol 2001; 27 (5): 384–95.
9. Liebner S, Fischmann A, Rascher G, et al. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol 2000; 100 (3): 323–31.
10. Saadoun S, Papadopoulos MC, Davies DC, et al. Aquaporin-4 expression is increased in oedematous human brain tumours. J Neurol Neurosurg Psychiatry 2002; 72 (2): 262–5.
11. Machein MR, Plate KH. VEGF in brain tumors. J Neurooncol 2000; 50 (1–2): 109–20.
12. Machein MR, Kullmer J, Fiebich BL, et al. Vascular endothelial growth factor expression, vascular volume, and, capillary permeability in human brain tumors. Neurosurgery 1999; 44 (4): 732–40.
13. Peak SJ, Levin VA. Role of bevacizumab therapy in the management of glioblastoma. Cancer Manag Res 2010; 2: 97–104.
14. Black KL, Hoff JT, McGillicuddy JE, et al. Increased leukotriene C4 and vasogenic edema surrounding brain tumors in humans. Ann Neurol 1986; 19 (6): 592–5.
15. Nathoo N, Barnett GH, Golubic M. The eicosanoid cascade: possible role in gliomas and meningiomas. J Clin Pathol 2004; 57 (1): 6–13.
16. Piette C, Munaut C, Foidart JM, et al. Treating gliomas with glucocorticoids: from bedside to bench. Acta Neuropathol 2006; 112 (6): 651–64.
17. Osawa T, Tosaka M, Nagaishi M, et al. Factors affecting peritumoral brain edema in meningioma: special histological subtypes with prominently extensive edema. J Neurooncol 2013; 111 (1): 49–57. doi: 10.1007/s11060-012-0989-y.
18. Kim H, Lee JM, Park JS, et al. Dexamethasone coordinately regulates angiopoietin-1 and VEGF: a mechanism of glucocorticoid-induced stabilization of bloodbrain barrier. Biochem Biophys Res Commun 2008; 372 (1): 243–8. doi: 10.1016/j.bbrc.2008.05.025.
19. Kreisl TN, Kim L, Moore K, et al. Phase II trial of single- agent bevacizumab followed by bevacizumab plus iri- notecan at tumor progression in recurrent glioblastoma. J Clin Oncol 2009; 27 (5): 740–5. doi: 10.1200/JCO.2008.16.3055.
20. Badie B, Schartner JM, Paul J, et al. Dexamethasone-induced abolition of the inflammatory response in an experimental glioma model: a flow cytometry study. J Neurosurg 2000; 93 (4): 634–9.
21. Salvador E, Shityakov S, Forster C. Glucocorticoids and endothelial cell barrier function. Cell Tissue Res 2014; 355 (3): 597–605. doi: 10.1007/s00441-013-1762-z.
22. Bebawy JF. Perioperative steroids for peritumoral intracranial edema: a review of mechanisms, efficacy, and side effects. J Neurosurg Anesthesiol 2012; 24 (3): 173–7. doi: 10.1097/ANA.0b013e3182578bb5.
23. Macdonald DR, Cascino TL, Schold SC jr, et al. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 1990; 8 (7): 1277–80.
24. Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 2010; 28: 1963–72. doi: 10.1200/JCO.2009.26.3541.
25. Sionov RV, Spokoini R, Kfir-Erenfeld S, et al. Mechanisms regulating the susceptibility of hematopoietic malignancies to glucocorticoidinduced apoptosis. Adv Cancer Res 2008; 101: 127–248. doi: 10.1016/S0065-230X (08) 00406-5.
26. Kullmann MK, Grubbauer C, Goetsch K, et al. The p27-Skp2 axis mediates glucocorticoid-induced cell cycle arrest in T-lymphoma cells. Cell Cycle 2013; 12 (16): 2625–35. doi: 10.4161/cc.25622.
27. Roth P, Stupp R, Eisele G, et al. Treatment of primary CNS lymphoma. Curr Treat Options Neurol 2014; 16 (1): 277. doi: 10.1007/s11940-013-0277-y.
28. Langeveld CH, van Waas MP, Stoof JC, et al. Implication of glucocorticoid receptors in the stimulation of human glioma cell proliferation by dexamethasone. J Neurosci Res 1992; 31 (3): 524–31.
29. Zibera C, Gibelli N, Butti G, et al. Proliferative effect of dexamethasone on a human glioblastoma cell line (HU 197) is mediated by glucocorticoid receptors. Anticancer Res 1992; 12 (5): 1571–4.
30. Weller M, Schmidt C, Roth W, et al. Chemotherapy of human malignant glioma: prevention of efficacy by dexamethasone? Neurology 1997; 48 (6): 1704–9.
31. Das A, Banik NL, Ray SK. Modulatory effects of acetazolomide and dexamethasone on temozolomide-mediated apoptosis in human glioblastoma T98G and U87MG cells. Cancer Invest 2008; 26 (4): 352–8. doi: 10.1080/07357900701788080.
32. Friese MA, Platten M, Lutz SZ, et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res 2003; 63 (24): 8996–9006.
33. Grunberg SM. Antiemetic activity of corticosteroids in patients receiving cancer chemotherapy: dosing, efficacy, and tolerability analysis. Ann Oncol 2007; 18 (2): 233–40.
34. Mantovani G, Maccio A, Esu S, et al. Evidence that cisplatin induces serotonin release from human peripheral blood mononuclear cells and that methylprednisolone inhibits this effect. Eur J Cancer 1996; 32A (11): 1983–5.
35. Suzuki T, Sugimoto M, Koyama H, et al. Inhibitory effect of glucocorticoids on human-cloned 5-hydroxytryptamine3A receptor expressed in xenopus oocytes. Anesthesiology 2004; 101 (3): 660–5.
36. Ho CM, Ho ST, Wang JJ, et al. Dexamethasone has a central antiemetic mechanism in decerebrated cats. Anesth Analg 2004; 99 (3): 734–9.
37. Kršek M. Systémová léčba glukokortikoidy: praktický pohled. Vnitř Lék 2015; 61 (10): 905–12.
38. Clore JN, Estep H, Ross-Clunis H, et al. Adrenocorticotropin and cortisol-induced changes in urinary sodium and potassium excretion in man: effects of spironolactone and RU 486. J Clin Endocrinol Metab 1988; 67 (4): 824–31.
39. Freiberg JM, Kinsella J, Sacktor B. Glucocorticoids increase the Na+ H+ exchange and decrease the Na+ gradient-dependent phosphate-uptake systems in renal brush border membrane vesicles. Proc Natl Acad Sci USA 1982; 79 (16): 4932–6.
40. Pilkey J, Daeninck PJ. A retrospective analysis of dexamethasone use on a Canadian palliative care unit. Prog Palliat Care 2008; 16: 63–8.
41. Hardy JR, Rees E, Ling J, et al. A prospective survey of the use of dexamethasone on a palliative care unit. Palliat Med 2001; 15 (1): 3–8.
42. Weissman DE, Dufer D, Vogel V, et al. Corticosteroid toxicity in neuro-oncology patients. J Neurooncol 1987; 5 (2): 125–8.
43. Sturdza A, Millar BA, Bana N, et al. The use and toxicity of steroids in the management of patients with brain metastases. Support Care Cancer 2008; 16 (9): 1041–8. doi: 10.1007/s00520-007-0395-8.
44. Huerta C, Johansson S, Wallander MA, et al. Risk factors and short-term mortality of venous thromboembolism diagnosed in the primary care setting in the United Kingdom. Arch Intern Med 2007; 167 (9): 935–43.
45. Johannesdottir SA, Horvath-Puho E, Dekkers OM, et al. Use of glucocorticoids and risk of venous thromboembolism: a nationwide population-based case-control study. JAMA Intern Med 2013; 173 (9): 743–52. doi: 10.1001/jamainternmed.2013.122.
46. Grossman E, Messerli FH. Drug-induced hypertension: an unappreciated cause of secondary hypertension. Am J Med 2012; 125 (1): 14–22. doi: 10.1016/j.amjmed.2011.05.024.
47. Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78 (11): 748–56. doi: 10.3949/ccjm.78a.10180.
48. McGirt MJ, Chaichana KL, Gathinji M, et al. Persistent outpatient hyperglycemia is independently associated with decreased survival after primary resection of malignant brain astrocytomas. Neurosurgery 2008; 63 (2): 286–91. doi: 10.1227/01.NEU.0000315282.61035.48.
49. Henson JW, Jalaj JK, Walker RW, et al. Pneumocystis carinii pneumonia in patients with primary brain tumors. Arch Neurol 1991; 48 (4): 406–9.
50. Enomoto T, Azuma A, Matsumoto A, et al. Preventive effect of sulfamethoxasole-trimethoprim on Pneumocystis jiroveci pneumonia in patients with interstitial pneumonia. Intern Med 2008; 47 (1): 15–20.
51. Pereira RM, Freire de Carvalho J. Glucocorticoid-induced myopathy. Joint Bone Spine 2011; 78 (1): 41–4. doi: 10.1016/j.jbspin.2010.02.025.
52. Williams TJ, O’Hehir RE, Czarny D, et al. Acute myopathy in severe acute asthma treated with intravenously administered corticosteroids. Am Rev Respir Dis 1988; 137 (2): 460–3.
53. Amaya-Villar R, Garnacho-Montero J, Garcia-Garmendia JL, et al. Steroid-induced myopathy in patients intubated due to exacerbation of chronic obstructive pulmonary disease. Intensive Care Med 2005; 31 (1): 157–61.
54. Levin OS, Polunina AG, Demyanova MA, et al. Steroid myopathy in patients with chronic respiratory diseases. J Neurol Sci 2014; 338 (1–2): 96–101. doi: 10.1016/j.jns.2013.12.023.
55. Qian T, Guo X, Levi AD, et al. High-dose methylprednisolone may cause myopathy in acute spinal cord injury patients. Spinal Cord 2005; 43 (4): 199–203.
56. Steinberg KP, Hudson LD, Goodman RB, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med 2006; 354 (16): 1671–84.
57. Dekhuijzen PN, Decramer M. Steroid-induced myopathy and its significance to respiratory disease: a known disease rediscovered. Eur Respir J 1992; 5 (8): 997–1003.
58. Frieze DA. Musculoskeletal pain associated with corticosteroid therapy in cancer. Curr Pain Headache Rep 2010; 14 (4): 256–60. doi: 10.1007/s11916-010-0120-z.
59. Lewis DA, Smith RE. Steroid-induced psychiatric syndromes. A report of 14 cases and a review of the literature. J Affect Disord 1983; 5 (4): 319–32.
60. Bolanos SH, Khan DA, Hanczyc M, et al. Assessment of mood states in patients receiving long-term corticosteroid therapy and in controls with patient-rated and clinician-rated scales. Ann Allergy Asthma Immunol 2004; 92 (5): 500–5.
61. Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6 (2): 82–8. doi: 10.1038/nrrheum.2009.259.
62. Maricic M. Update on glucocorticoid-induced osteoporosis. Rheum Dis Clin North Am 2011; 37 (3): 415–31. doi: 10.1016/j.rdc.2011.07.003.
63. Steinbuch M, Youket TE, Cohen S. Oral glucocorticoid use is associated with an increased risk of fracture. Osteoporos Int 2004; 15 (4): 323–8.
64. Kanis JA, Johansson H, Oden A, et al. A meta-analysis of prior corticosteroid use and fracture risk. J Bone Miner Res 2004; 19 (6): 893–9.
65. Farhat G, Yamout B, Mikati MA, et al. Effect of antiepileptic drugs on bone density in ambulatory patients. Neurology 2002; 58 (9): 1348–53.
66. Feldkamp J, Becker A, Witte OW, et al. Long-term anticonvulsant therapy leads to low bone mineral density evidence for direct drug effects of phenytoin and carbamazepine on human osteoblast-like cells. Exp Clin Endocrinol Diabetes 2000; 108 (1): 37–43.
67. Shen C, Chen F, Zhang Y, et al. Association between use of antiepileptic drugs and fracture risk: a systematic review and meta-analysis. Bone 2014; 64: 246–53. doi: 10.1016/j.bone.2014.04.018.
68. Mathew BS, Carson KA, Grossman SA. Initial response to glucocorticoids. Cancer 2006; 106 (2): 383–7.
69. Batchelor T, Loeffler JS. Primary CNS lymphoma. J Clin Oncol 2006; 24 (8): 1281–8.
70. Mancini T, Kola B, Mantero F, et al. High cardiovascular risk in patients with Cushing’s syndrome according to 1999 WHO/ISH guidelines. Clin Endocrinol 2004; 61 (6): 768–77.
71. van Raalte DH, Ouwens DM, Diamant M. Novel insights into glucocorticoid-mediated diabetogenic effects: towards expansion of therapeutic options? Eur J Clin Invest 2009; 39 (2): 81–93. doi: 10.1111/j.1365-2362.2008.02067.x.
72. Mazziotti G, Gazzaruso C, Giustina A. Diabetes in Cushing syndrome: basic and clinical aspects. Trends Endocrinol Metab 2011; 22 (12): 499–506. doi: 10.1016/j.tem.2011.09.001.
73. Munir A, Newell-Price J. Management of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology 2010; 92 (Suppl 1): S82–5.
74. Vlček J. Lékové interakce z pohledu klinického farmaceuta. Practicus 2009; 1: 10–5.
75. Stanbury RM, Graham EM. Systemic corticosteroid therapy-side effects and their management. Br J Ophthalmol 1998; 82: 704–8.
76. Manson SC, Brown RE, Cerulli A, et al. The cumulative burden of oral corticosteroid side effects and the economic implications of steroid use. Respir Med 2009; 103 (7): 975–94. doi: 10.1016/j.rmed.2009.01.003.
77. Weissman DE, Dufer D, Vogel V, et al. Corticosteroid toxicity in neuro-oncology patients. J Neurooncol 1987; 5 (2): 125–8.
78. The Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther 1972; 13 (5): 694–8.
79. Encio IJ, Detera-Wadleigh SD. The genomic structure of the human glucocorticoid receptor. J Biol Chem 1991; 266 (11): 7182–8.
80. Turner JD, Schote AB, Macedo JA, et al. Tissue specific glucocorticoid receptor expression, a role for alternative first exon usage? Biochem Pharmacol 2006; 72 (11): 1529–37.
81. Zhou J, Cidlowski JA. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids 2005; 70 (5–7): 407–17.
82. Schaaf MJ, Cidlowski JA. Molecular determinants of glucocorticoid receptor mobility in living cells: the importance of ligand affinity. Mol Cell Biol 2003; 23 (6): 1922–34.
83. Cadepond F, Schweizer-Groyer G, Segard-Maurel I, et al. Heat shock protein 90 as a critical factor in maintaining glucocorticosteroid receptor in a nonfunctional state. J Biol Chem 1991; 266 (9): 5834–41.
84. Johnson AB, O’Malley BW. Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM) -based cancer therapy. Mol Cell Endocrinol 2012; 348 (2): 430–9. doi: 10.1016/j.mce.2011.04.021.
85. Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther 2012; 134 (1): 54–67. doi: 10.1016/j.pharmthera.2011.12.004.
86. Xu J, Winkler J, Derendorf H. A pharmacokinetic/pharmacodynamic approach to predict total prednisolone concentrations in human plasma. J Pharmacokinet Pharmacodyn 2007; 34 (3): 355–72.
87. Hempen C, Weiss E, Hess CF. Dexamethasone treatment in patients with brain metastases and primary brain tumors: do the benefits outweigh the side-effects? Support Care Cancer 2002; 10 (4): 322–8.
88. Galicich JH, French LA. Use of dexamethasone in the treatment of cerebral edema resulting from brain tumors and brain surgery. Am Pract Dig Treat 1961; 12: 169–74.
89. Mitchell CD, Richards SM, Kinsey SE, et al. Research Council Childhood Leukaemia Working. Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 2005; 129 (6): 734–45.
90. Hanks GW, Trueman T, Twycross RG. Corticosteroids in terminal cancer – a prospective analysis of current practice. Postgrad Med J 1983; 59 (697): 702–6.
91. Aulakh R, Singh S. Strategies for minimizing corticosteroid toxicity: a review. Indian J Pediatr 2008; 75 (10): 1067–73. doi: 10.1007/s12098-008-0211-6.
92. Kirkham SR. The palliation of cerebral tumours with high dose dexamethasone: a review. Palliat Med 1988; 2: 27–33.
93. Vecht CJ, Hovestadt A, Verbiest HB, et al. Dose-effect relationship of dexamethasone on Karnofsky performance in metastatic brain tumors: a randomized study of doses of 4, 8, and 16 mg per day. Neurology 1994; 44 (4): 675–80.
94. Marantidou A, Levy C, Duquesne E, et al. Steroid requirements during radiotherapy for malignant gliomas. J Neurooncol 2010; 100 (1): 89–94. doi: 10.1007/s11060-010-0142-8.
95. Weissman DE, Janjan NA, Erickson B, et al. Twice-daily tapering dexamethasone treatment during cranial radiation for newly diagnosed brain metastases. J Neurooncol 1991; 11 (3): 235–9.
96. Ryken TC, McDermott M, Robinson PD, et al. The role of steroids in the management of brain metastases: a systematic review and evidence-based clinical practice guideline. J Neurooncol 2010; 96 (1): 103–14. doi: 10.1007/s11060-009-0057-4.
97. Kostaras X, Cusano F, Kline GA, et al. Use of dexamethasone in patients with high-grade glioma: a clinical practice guideline. Curr Oncol 2014; 21 (3): e493–503. doi: 10.3747/co.21.1769.
98. Debono M, Mallappa A, Gounden V, et al. Hormonal circadian rhythms in patients with congenital adrenal hyperplasia: identifying optimal monitoring times and novel disease biomarkers. Eur J Endocrinol 2015; 173 (6): 727–37. doi: 10.1530/EJE-15-0064.
99. Kala M. Neurologické diagnózy v diferencované hospicové péči – dvě kazuistiky. Cesk Slov Neurol N 2013; 76/109 (6): 756–8.
100. Vredenburgh J, Cloughesy TF, Samant M, et al. Corticosteroid use in patients with glioblastoma at first or second relapse treated with bevacizumab in the brain study. Oncologist 2010; 15 (12): 1329–34. doi: 10.1634/theoncologist.2010-0105.
101. Cohen MH, Shen YL, Keegan P, et al. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009; 14 (11): 1131–8. doi: 10.1634/theoncologist.2009-0121.
102. Agar M, Koh ES, Gibbs E, et al. Validating self-report and proxy reports of the Dexamethasone Symptom Questionnaire Chronic for the evaluation of longer-term corticosteroid toxicity. Support Care Cancer 2016; 24 (3): 1209–18. doi: 10.1007/s00520-015-2897-0.
103. Roth P, Happold C, Weller M. Corticosteroid use in neuro-oncology: an update. Neurooncol Pract 2015; 2 (1): 6–12.
104. Schiff D, Lee EQ, Nayak L, et al. Medical management of brain tumors and the sequelae of treatment. Neuro Oncol 2015; 17 (4): 488–504. doi: 10.1093/neuonc/nou304.
105. Gannon C, McNamara P. A retrospective observation of corticosteroid use at the end of life in a hospice. J Pain Symptom Manage 2002; 24 (3): 328–35.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2016 Issue 5
Most read in this issue
- Current Corticosteroid Treatment in Brain Tumours
- Rasmussen’s Encephalitis
- Traumatic Brachial Plexus Injuries Represents Serious Peripheral Nerve Palsies
- Detection of Spirochetal DNA from Patients with Neuroborreliosis