
Paroxysmal Kinesigenic Dystonia as a Primomanifestation of Multiple Sclerosis – a Case Report
Paroxyzmálna kinezigénna dystónia ako primomanifestácia roztrúsenej sklerózy – kazuistika
Paroxyzmálne dyskinézy sú pomerne zriedkavé ochorenia charakterizované rôznymi typmi mimovoľných pohybov, ktoré vznikajú v súvislosti s identifikovateľným spúšťačom, ako je únava, emocionálny stres, kofeín, náhly pohyb a podobne. Okrem primárnych, geneticky determinovaných ochorení, existuje skupina sekundárnych paroxyzmálnych dyskinéz, ktorých najčastejšou príčinou býva roztrúsená skleróza. V článku opisujeme prípad mladého pacienta s náhlym vznikom kinezigénne navodených atakov paroxyzmálnej dystónie bez iných ďalších prejavov. Diagnostikovaný bol klinický izolovaný syndróm v rámci roztrúsenej sklerózy, pričom za príčinu dyskinéz považujeme demyelinizačný plak v oblasti cervikálnej miechy. Detailne popisujeme diagnostické kroky, liečbu a ďalší priebeh u tohto pacienta v kontexte s dostupnými literárnymi údajmi o tejto klinickej jednotke.
Kľúčové slová:
paroxyzmálne dyskinézy – paroxyzmálna kinezigénna dystónia – roztrúsená skleróza – klinicky izolovaný syndróm
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Authors:
J. Necpál
Authors‘ workplace:
Department of Neurology, Zvolen, Hospital, Zvolen
Published in:
Cesk Slov Neurol N 2016; 79/112(5): 601-603
Category:
Case Report
doi:
https://doi.org/10.14735/amcsnn2016601
Overview
Paroxysmal dyskinesias are rare disorders characterized by various involuntary movements induced by triggers such as fatigue, emotional stress, caffeine, sudden movement and others. Except for primary, genetically determined diseases, there are secondary paroxysmal dyskinesias caused most commonly by multiple sclerosis. In this article, we describe a case of a young man with sudden onset attacks of kinesigenic paroxysmal dystonia without any other symptoms. The diagnosis of clinically isolated syndrome in multiple sclerosis was made and demyelinating plaque within the cervical spine was suggested as the cause of dyskinesia. We discuss in detail diagnostic steps, treatment and outcome of this patient in context of literature available about this condition.
Key words:
paroxysmal dyskinesias – paroxysmal kinesigenic dystonia – multiple sclerosis – clinically isolated syndrome
Introduction
Paroxysmal dyskinesias (PD) form a hetero- genous group of rare disorders with recurrent, mostly short-lasting episodes of choreatic, dystonic, athetoid or combined involuntary movements. In 1938, Pitha de-scribed a case of a student of medicine with 10 to 30 s lasting PD at his six years, provoked by movement or startle after a period of rest [1]. The same dyskinesias were labelled in 1967 „kinesigenic“ [2]. Two years after the Pitha’s case, Mount and Reback described a hereditary disease with paroxysmal choreoathetosis triggered by some foods, fatigue or stress [3]. The third form, described in 1977, was characterized by attacks induced by prolonged exercise [4]. Based on the trigger of involuntary movements, three forms of PD are recognized today: paroxysmal kinesigenic dyskinesias (PKD), paroxysmal non-kinesige- nic dyskinesias (PKND) and paroxysmal exercise-induced dyskinesias (PED) [5]. According to the cause, each of these may be primary (autosomal dominant hereditary forms or less often sporadic forms) or secondary (imaging or laboratory findings detect an evident cause of dyskinesias) [6]. Primary PKD is characterized by choreatic, athetoid or dystonic attacks provoked by sudden movement or startle after a period of rest. The attacks usually last several seconds. Their frequency varies from sporadic occurrence to multiple daily attacks. An attack itself, ranging from slight finger movements to generalized movements, is often preceded by aura, perhaps with paresthesias of the limbs or nonspecific epigastric sensations. After an attack, there often is a refractory period lasting several seconds, during which there no paroxysms. The disorder is familial in 70% with an during childhood or adolescence and symptoms fading out with time. Therapeutically, carbamazepine, phenytoin or acetalozamide can be helpful. Attacks of primary PKND last longer (from 15 min to one hour) and are provoked by sleep deprivation, fatigue, emotional stress, alcohol, coffee, chocolate or tea. They are also preceded by aura sensations but their frequency is much lower than in PKD (no more than once daily). Except for avoidance of triggers, treatment with benzodiazepines may be beneficial. Primary PED is most often characterized by episodes of dystonia of lower limbs triggered by repeated physical exercise (e. g. by walking or running) and lasting five to 30 min. The disease manifests in childhood or early adulthood. Frequency of attacks is diverse and sensoric aura is not present. Benzodiazepines as well as ketogenic diet in case of GLUT-1 deficiency syndrome- -associated PED may be effective [7]. Previously reported paroxysmal hypnogenic dyskinesias are now considered to be frontal epileptic seizures. Mutations in PRRT2 gene and myofibrillogenesis gene MR-1 (or KCNMA1 in minority of cases) are causes of primary PKD and PKND, respectively. Patients with mutations in SLC2A1 gene with GLUT-1 deficiency syndrome may manifest along with other signs by PED fenotype. It has been shown that such fenotype-genotype correlation is not strictly seen and that carriers of mutations mentioned above may manifest with various types of PD [6]. One of the most common secondary causes of PD is multiple sclerosis (MS) [8].
Case report
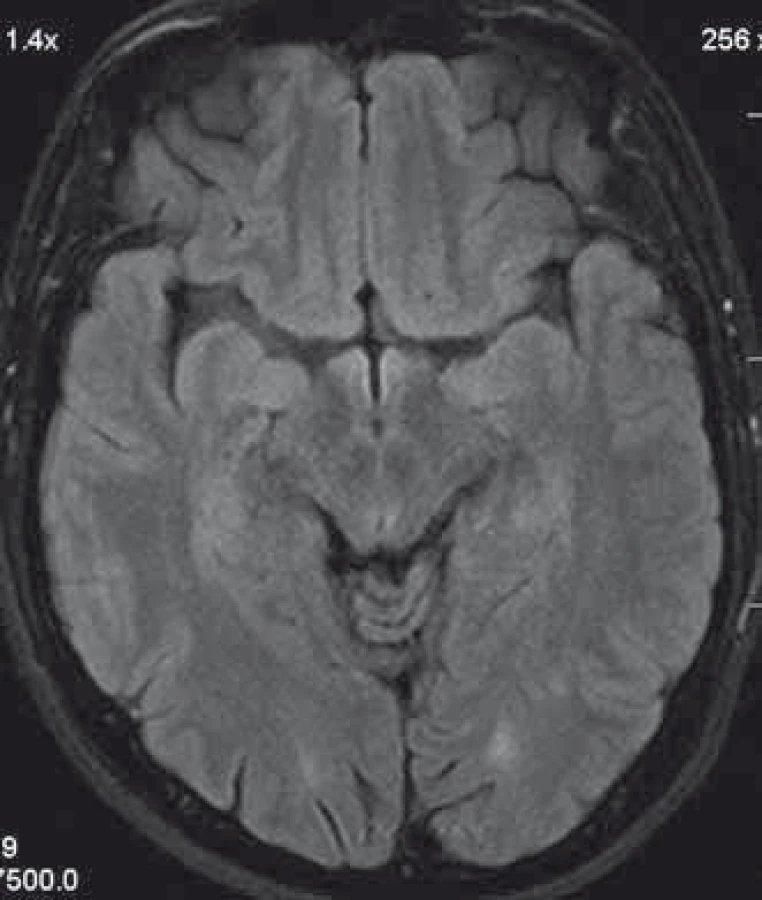
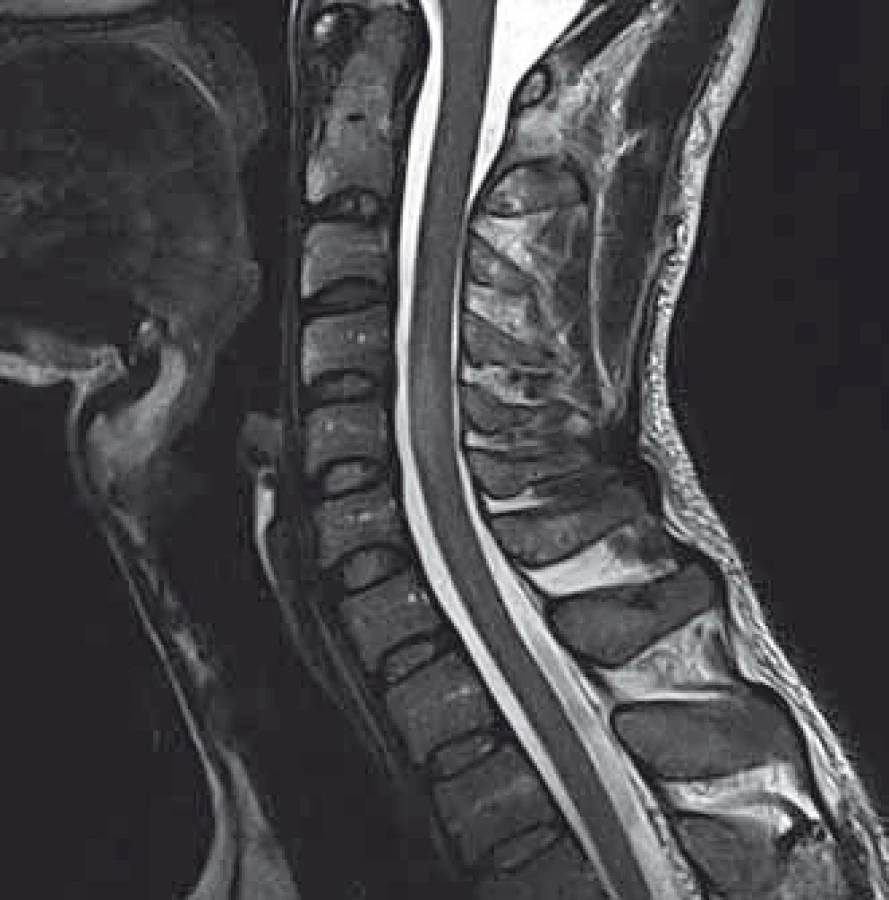
We present a case of a 37-year-old otherwise healthy man. He was admitted to our department because of four-day history of uncontrollable repetitive jerky movements with tonic component and onset on the left lower extremity. He noticed these for the first time after he had rested following a night shift. He reported a nonspecific abdominal discomfort signaling an onset of involuntary movements described as a „heating“ sensation in the abdomen. He had a „sensation of anticipation of some kind of a problem“. Subsequently, he experienced the involuntary movements described above. Left extremities including the trunk were affected, much less right limbs, facial region was always spared. The attacks of involuntary movements took less then 10 s and appeared several times a day. The patient had no other complaints. Family history of epilepsy or other involuntary movements as well as history of antipsychotic medication was negative. Interictal neurologic examination revealed symmetric hyperreflexia of lower limbs and positive Babinski sign on the left. Dystonic hyperkinetic attacks were evidently movement-induced they appeared on standing up, placing the leg on the floor or walking. Patient was able to anticipate them, therefore preferred a supine motionless position. Stress, caffeine, alcohol, sleep deprivation or other stimuli did not induce the attacks. 20 min native EEG repeatedly did not identify any epileptic activity. Features of paroxysmal hypnogenic dyskinesias during recording were not present. T2-weighted MRI scan of the brain revealed three little hyperintensive periventricular and paraventricular lesions (the largest was laterally from the occipital horn of the left lateral ventricule) (Fig. 1). MRI of the cervical spine found a solitary, slightly expansive enhancing lesion at the C4–C5 level (Fig. 2). Thoracal spine region was unaffected. Lumbar puncture revealed a mild protein-cytologic association with protein content of 578 mg/l and 17 elements, mainly mononuclears. Both the disruption of the blood-brain barrier and intrathecal oligoclonal synthesis of IgG were present (by method of isoelectric focusing at alkaline pH: 10 oligoclonal bands in liquor, 3 in serum, RIgG 19.3 mg/l, Reiber index 0.85). Visual evoked potentials showed asymmetry and prolonged latencies of both P 100 waves (110 ms on the left, 118 ms on the right). Basic biochemical parametres, CBC, rheumatoid factor, TSH, routine coagulation parametres, lupus anticoagulant and serologic testing for Lyme disease were normal. Tests for autoimmune antibodies (Ro-52, SS-A, SS-B, RNP, Sm, Scl 70, Jo-1), anti-neuronal antibodies (Hu, Yo, Ri, Ma2, CV2, SOX1, titin, recoverin), anti-cardiolipin, anti-gliadin, anti-transglutaminase as well as anti-aquaporin-4 antibodies were negative. The patient bore heterozygous mutation of factor V Leiden. All these results suggest that the paroxysmal kinesigenic dystonia in this patient represented clinically isolated syndrome in MS. A total dose of 1 g of methylprednisolone in intravenous pulses was administered over five days, followed by oral prednisone and carbamazepine, gradually titrated to 800 mg daily. Involuntary movements completely resolved after about 10 days of this therapy. Immunomodulation therapy with interferon beta 1b was started. Follow up brain MRI after one year displayed six small T2 hyperintensive lesions located periventricularly and in the centrum semi- ovale bilaterally (in addition to the periventricular lesion in the left occipital area described above). Two years after the PKD, cervical spine MRI showed regression of the intramedullar lesion that did not enhancem and had edematous character. Two years from the diagnosis, the patient did not experience any further attacks of PKD. The patient takes 800 mg of carbamazepine daily and injects interferon-1-beta-1b. He is free of any clinical features and complaints associated with the activity of MS.
Discussion
Secondary PD has a known cause of involuntary movements (mainly neurologic and systemic diseases). They were described in cerebrovascular diseases, cerebral trauma, perinatal hypoxic encephalopathy, hypocalcemia, hypoparathyreosis, thyreotoxicosis, hypoglycemia, diabetes mellitus, various infections, AIDS etc. The most common secondary PD are PD in MS (PKD and PKND type) [7,9]. The majority of published data about this association are case reports. Clinical presentation mainly involves unilateral painful tonic spasms that do not correspond with the duration or severity of MS. They may represent the first sign of the disease. They are triggered by various movements, less frequently by hyperventilation or they occur spontaneously. Paroxysms of dyskinesias are of short duration and may appear quite frequently – sometimes up to 50-times daily. They usually cease after several weeks. Anticonvulsants are the mainstay of treatment [7]. Formation of a demyelinated plaque has been suggested as a cause of involuntary movements. Damage to various regions of the brain may result in PD, including thalamus, mesencpehalon pedunculus [8], internal capsule [10], basal ganglia [10] and even cervical spine [7]. Rare case reports of PD in MS with causal pathology lying in the cervical spine have been described that in many aspects resemble our own case [11–14]. PD resulting from a cervical spine affection may be caused by other disorders such as neuromyelitis optica [15] or even compressive cervical cord discopathy [16]. PDs, such as spinal segmental and propriospinal myoclonus, belong to a group of spinally generated movement disorders. Their pathophysiological background in MS may involve ephaptic transmission between partially demyelinated axones [17]. Morgan et al. published an interesting case report of duplicity of two secondary PD-paroxysmal hemidystonias in MS with episodes lasting up to two minutes and longer-lasting psychogenic PD that occured one year later and in association with missing β-interferon injections [18]. Our patient suffered from involuntary movements that could be described as tonic spasms or dystonic dyskinesias. Their paroxysmal character, induction by kinesigenic stimuli and presence of sensoric aura suggest paroxysmal kinesigenic dystonia. Results from neuroimaging and laboratory tests were consistent with MS as a cause of secondary PKD. Sporadic incidence of epileptic seizures in MS, paroxysmal nature as well as presence of abdominal aura could suggest epilepsy but the clear kinesigenity and absence of typical epileptic graphoelements on EEG are in conflict with this diagnosis. Although paroxysmal dystonic features in MS are more often unilateral [7], all four limbs were involved in our patient, with dominance on the left side. This may be due to the demyelinating lesion being located in the area of cervical spine; this can also explain sparing of the the facial region. The absence of multi-level involvement of the cervical cord and aquaporin-4 antibodies negativity exclude neuromyelitis optica (Devic’s disease). In our patient, PKD represented a sole manifestation of MS and thus this can be considered the clinically isolated syndrome. This paper illustrates another case of association between MS and PD in the literature and it also shows PD in the light of broader differential diagnosis of MS.
The authors declare they have no potential confl icts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Jan Necpál, MD
Department of Neurology
Zvolen Hospital
Kuzmányho nabrežie 28
960 01 Zvolen
e-mail: necpal.neuro@gmail.com
Accepted for review: 13. 12. 2015
Accepted for print: 29. 4. 2016
Sources
1. Pitha V. Reflex epilepsy. Rev Neurol 1938; 70: 178–81.
2. Kertesz A. Paroxysmal kinesigenic choreoathetosis: an entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology 1967; 17 (7): 680–90.
3. Mount L, Reback S. Familial paroxysmal choreoathetosis. Arch Neurol 1940; 44: 841–7.
4. Lance JW. Familial paroxysmal dystonic choreoathetosis and its differentiation from related syndromes. Ann Neurol 1977; 2 (4): 285–93.
5. Demirkiran M, Jankovic J. Paroxysmal dyskinesias: clinical features and classification. Ann Neurol 1995; 38 (4): 571–9.
6. Erro R, Sheerin UM, Bhatia KP. Paroxysmal dyskinesias revisited: a review of 500 genetically proven cases and a new classification. Mov Disord 2014; 29 (9): 1108–16. doi: 10.1002/mds.25933.
7. Donaldson IM, Marsden CD, Schneider SA, eds. Marsden’s book of movement disorders. New York: Oxford University Press Inc 2012: 1552.
8. Zittel S, Bester M, Gerloff C, et al. Symptomatic paroxysmal kinesigenic choreoathetosis as primary menifestation of multiple sclerosis. J Neurol 2012; 259 (3): 557–8. doi: 10.1007/s00415-011-6188-5.
9. Hao SS, Feng YH, Zhang GB, et al. Neuropathophysiology of paroxysmal, systemic and other related movement disorders. Eur Rev Med Pharmacol Sci 2015; 19 (13): 2452–60.
10. Uca AU, Altas M. Paroxysmal dystonia as the first manifestation of multiples sclerosis with internal capsular plaque. Arch Neuropsychiatr 2014; 51: 295–6.
11. Previdi P, Buzzi P. Paroxysmal dystonia due to a lesion of the cervical cord: case report. Ital J Neurol Sci 1992; 13 (6): 521–3.
12. Fragoso YD, Araujo MG, Branco NL. Kinesigenic paroxysmal hemidyskinesia as the initial presentation of multiple sclerosis. MedGenMed 2006; 8 (4): 3.
13. Rozza L, Bortolotti P, Sica A, et al. Kinesigenic dystonia as the first manifestation of multiple sclerosis with cervical and brainstem lesions. Eur Neurol 1993; 33 (4): 331–2.
14. Cosentino C, Torres L, Flores M, et al. Paroxysmal kinesigenic dystonia and spinal cord lesion. Mov Disord 1996; 11 (4): 453–5.
15. Schmidt FR, Costa FH, Silva FM, et al. Paroxysmal dystonia and neuromyelitis optica. Arq Neuropsiquiatr 2012; 70 (4): 271–3.
16. Yulug B, Bakar M, Özer H, et al. Paroxysmal kinesigenic dyskinesia and cervical disc prolapse with cord cord compression: more than a coincidence? J Neuropsychiatry Clin Neurosci 2008; 20 (2): 237–9. doi: 10.1176/appi.neuropsych.20.2.237.
17. Termasarasab P, Thammongkolchai T, Fruch SJ. Spinal-generated movement disorders: a clinical review. J Clin Mov Disord 2015; 2: 18. doi: 10.1186/s40734-015-0028-1.
18. Morgan JC, Hughes M, Figueroa RA, et al. Psychogenic paroxysmal dyskinesia following paroxysmal hemidystonia in multiple sclerosis. Neurology 2005; 65 (6): E12.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2016 Issue 5
Most read in this issue
- Current Corticosteroid Treatment in Brain Tumours
- Rasmussen’s Encephalitis
- Traumatic Brachial Plexus Injuries Represents Serious Peripheral Nerve Palsies
- Detection of Spirochetal DNA from Patients with Neuroborreliosis