
Získaná neuromyotonie s nevelkými centrálními příznaky s průkazem protilátek proti napěťově řízeným kaliovým kanálům – kazuistika
Acquired Neuromyotonia with Minor Central Symptoms and Antibodies against Voltage- Gated Potassium Channels – a Case Report
A 52- ye ar- old still he althy man developed, over several weeks, a myalgic syndrome with fatigue and muscle we akness, tingling and fasciculati on, predominantly in the lower limbs. Such disorders ca used inability to walk independently. Upon admissi on, the clinical findings included muscle we akness and muscle stiffness with tenderness, a utonomic symptoms with swe ating, intermittent tachycardi a, constipati on, and also personality and behavi oral changes with insomni a and late- night confusi on. The electromyography showed a persisting activity with discharges, which were provoked by voluntary activity, and in particular by the stimulati on of motor fibres. The pati ent was tre ated with carbamazepine and methylprednisolone. Hypertoni a, myalgi a and central symptoms subsided. At that stage, the results came back with highly elevated antibodi es against voltage- gated potassi um channels in serum. However, the pati ent suddenly di ed of malignant arrhythmi a.
Key words:
neuromyotonia – Morvan’s disease – voltage-gated potassium channels – arrhythmia – myokymia – fasciculation
Authors:
J. Latta 1; E. Ehler 1; J. Zámečník 2
Authors‘ workplace:
Neurologická klinika, Pardubická krajská nemocnice, a. s., 2Ústav patologie a molekulární medicíny, UK 2. LF a FN Motol, Praha
1
Published in:
Cesk Slov Neurol N 2009; 72/105(4): 373-377
Category:
Case Report
Overview
U 52letého, dosud zdravého muže se v průběhu několika týdnů rozvinul myalgický syndrom s únavností, slabostí, brněním a záškuby ve svalech, a to zejména na dolních končetinách (DK). Pro tyto potíže pak už nebyl schopen samostatné chůze. V klinickém nálezu při přijetí byla slabost a tuhost svalů s bolestivo u palpací, vegetativní příznaky s pocením, intermitentní tachykardi í, zácpo u, dále osobnostní a behavi orální změny s nespavostí a noční zmateností. V elektromyografii (EMG) byla patrna klidová trvalá aktivita s výboji, které byly provokovány volní aktivito u a zejména stimulací motorických vláken. Nemocného jsme léčili karbamazepinem a metylprednizolonem. Došlo k ústupu hypertoni e, bolestí svalů i centrálních příznaků. V té fázi došel silně pozitivní nález a utoprotilátek proti kali ovým kanálům. Paci ent však náhle zemřel na maligní arytmii.
Klíčová slova:
neuromyotonie – Morvanův syndrom – centrální příznaky Morvanova syndromu – protilátky proti kaliovým kanálům – maligní arytmie – moykymie – fascikulace
Úvod
„Stiff-muscles“ (tuhé, neohebné svaly) jsou podmíněny trvalou svalovou aktivitou. Nejedná se o přímé onemocnění svalu a tento stav je nutno odlišit od rigidity či spasticity. Stiff-muscles se nejčastěji vyskytují u syndromu kontinuální svalové aktivity (neuromyotonie), encefalomyelitidy se svalovou hypertonií, stiff-person syndromu a stiff-leg syndromu [1]. Neuromyotonie se vyznačuje pseudomyotonickou poruchou svalové relaxace v kombinaci s myokymiemi a fascikulacemi. V klinickém nálezu se nachází tuhost svalů a krampy [2].
V roce 1890 Augustin Marie Morvan popsal pacienta s myokymií, bolestmi svalstva, nadměrným pocením a poruchou spánku [3]. Morvanův syndrom je vzácné autoimunitní onemocnění charakterizované nepravidelnými kontrakcemi svalstva, krampy, slabostí, hyperhidrózou, nespavostí a zmateností [4]. V anglicky psané literatuře bylo doposud popsáno jen asi 14 případů Morvanova syndromu a jen několik případů s kompletním spektrem centrálních příznaků [5]. Předkládáme případ dospělého muže, u kterého se vyvinuly typické projevy získané neuromyotonie s centrálními příznaky Morvanova syndromu.
Kazuistika
52letý muž byl přijat pro tři týdny progredující bolesti a brnění chodidel a lýtek s postupným šířením proximálně až do bederní oblasti a třísel. Bolesti byly doprovázeny výraznými fascikulacemi a slabostí při chůzi. Byl schopen jen několika krůčků s pomocí dvou podpažních berlí. Pracoval jako řidič, byl sledován pro syndrom karpálního tunelu profesionálního původu a před čtyřmi lety podstoupil operaci vlevo s výrazným efektem. Pět let se léčil pro hypertenzi (isradipin, perindopril), jinak byl vždy zdráv a křeče či záškuby ve svalech nemíval.
V klinickém nálezu při přijetí dominovala slabost a tuhost svalů, ve svalech byly spontánní záškuby charakteru myokymií i rychlé izolované nerytmické záškuby. Při poklepu na svaly – více na dolních končetinách, ale i horních končetinách a jazyku – se akcentovaly myokymie i náhlé záškuby (nejspíše fascikulace). Idiomuskulární dráždivost byla také zvýšena a vibrační čití bylo zkráceno na 5/8. Pohyby byly pomalé, mimika ztuhlá, až maskovitá, patrno bylo i „myotonické“ zpoždění víček při pohledu dolů a mydriáza s omezeným rozsahem fotoreakce. Dále hyperhidróza na končetinách, trupu i obličeji a zácpa. Během hospitalizace se postupně vyvinuly i psychické změny. V popředí dominovala noční agitovanost nasedající na krátké stavy zmatenosti při častém buzení ze spánku, naopak během dne stěží udržel pozornost a pospával. Byly přítomny i alterované reakce na některé nepříjemné vyšetřovací a léčebné metody (per rectum vyšetření, zavádění čípků, klyzma).
V laboratorních odběrech jsme zjistili zvýšenou hodnotu kreatinkinázy (CK): 12,82 ukat/l (0,58–3,87) a její izoenzym CK-MB: 0,42 ukat/l (0,00–0,10). Volný myoglobin v séru byl výrazně vyšší: 196,4 ug/l (25–72). Jaterní enzymy byly zvýšeny jen lehce. Z dalších vyšetření byla vyšší hladina imunoglobulinů třídy G: 22,09 g/l (6,81–16,4), gamaglobulinů v séru: 0,236 (0,10–0,19) a C‑reaktivní protein: 33,8 mg/l (0–10). Nádorové markery (PSA, AFP, CEA, C19.9, C12.5, C15.3) v séru byly negativní.
Následně byla provedena lumbální punkce s normálním cytologickým nálezem s proteiny 0,59 g/l (0,20–0,40). Při vyšetření proteinových frakcí byl prokázán zvýšený orosomukoid 17,8 mg/l (1,5–4,5) a při izoelektrické fokuzaci nebyly nalezeny žádné oligoklonální pásy.
Při motorické neurografii byla nalezena jen delší distální latence pro n. medianus vpravo (8,70 ms) a repetitivní následné výboje po stimulaci motorických vláken. Pro tyto následné a bezprostředně nastupující výboje nebylo možno vyšetřit F-vlny (obr. 1) a ani H-reflex (obr. 2). Senzitivní nervové akční potenciály byly nižší amplitudy, rychlost vedení senzitivním nervem byla lehce snížena. Při vyšetření svalů koncentrickou jehlovou elektrodou byly v popředí spontánní a často repetitivní až rytmické výboje charakteru dupletů, tripletů i multipletů a někdy i vysokofrekvenčního krátce trvajícího výboje, fascikulace a myokymie (obr. 3, 4). Patologická aktivita (multiplety, výboje) se provokovala jak pohybem jehly, poklepem na sval, tak v menší míře i volní kontrakcí. Při analýze potenciálů motorických jednotek byl jen zvýšený podíl polyfázických potenciálů, jinak trvání, amplituda i frekvence pálení motoneuronů byly v mezích normy.

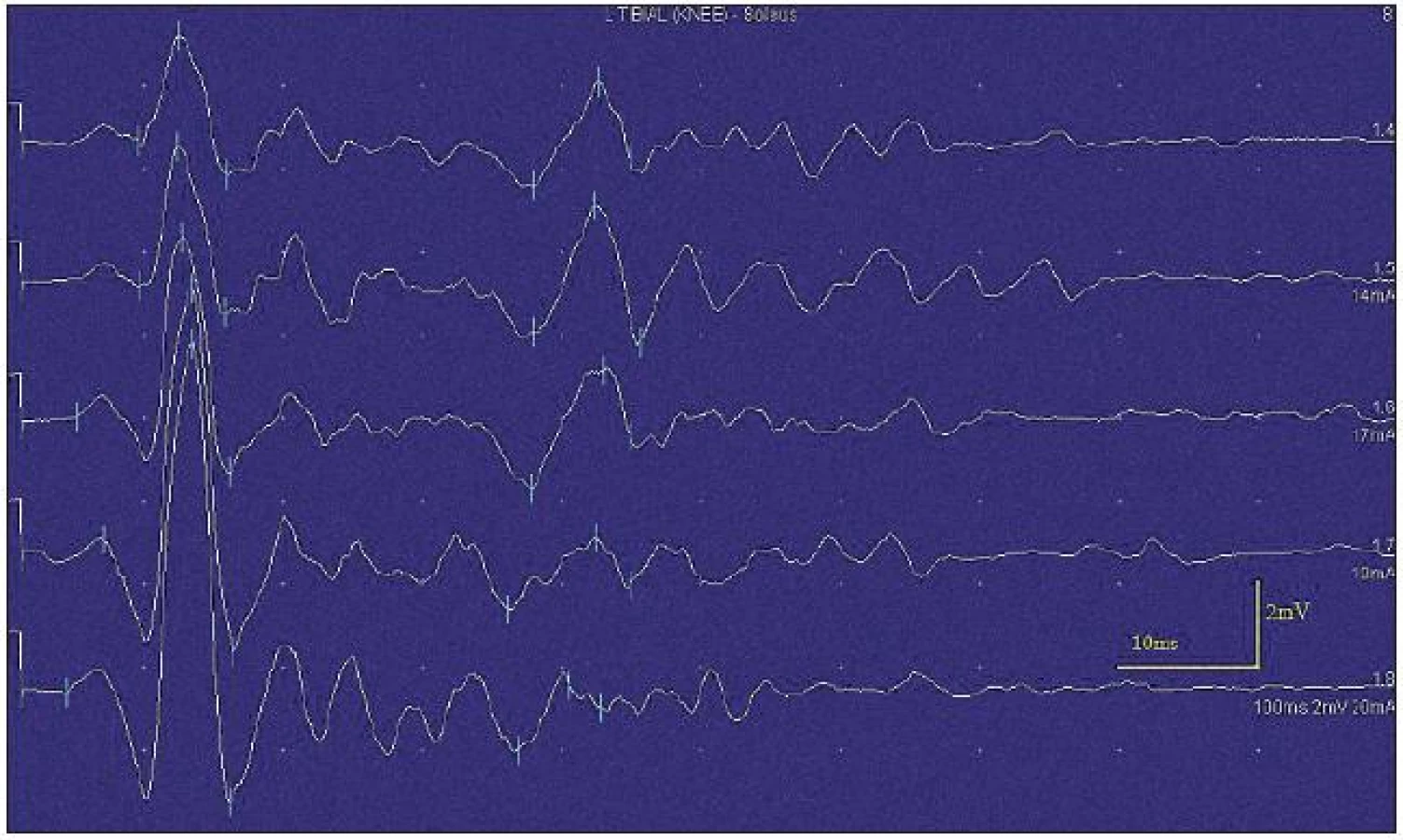
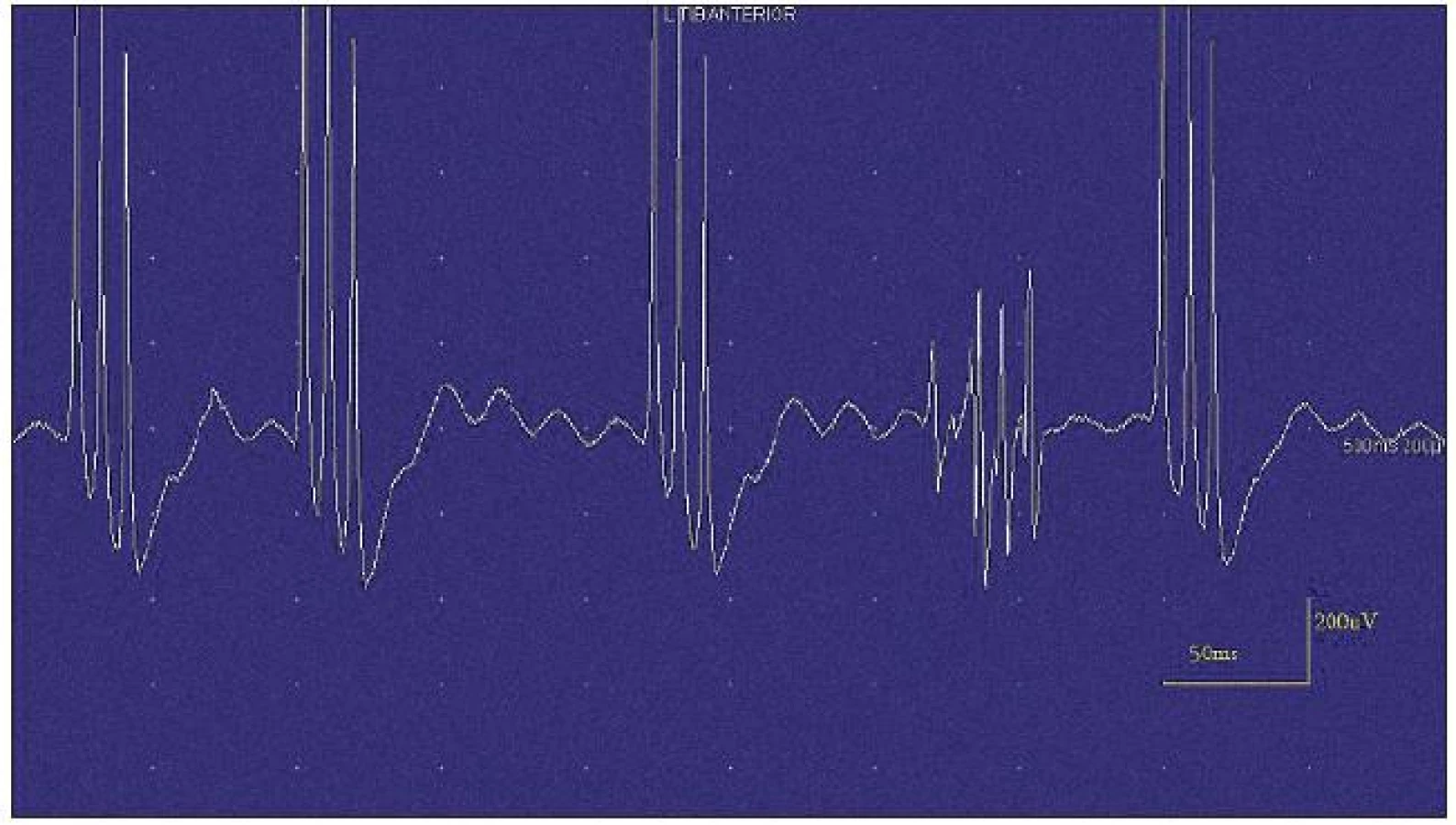
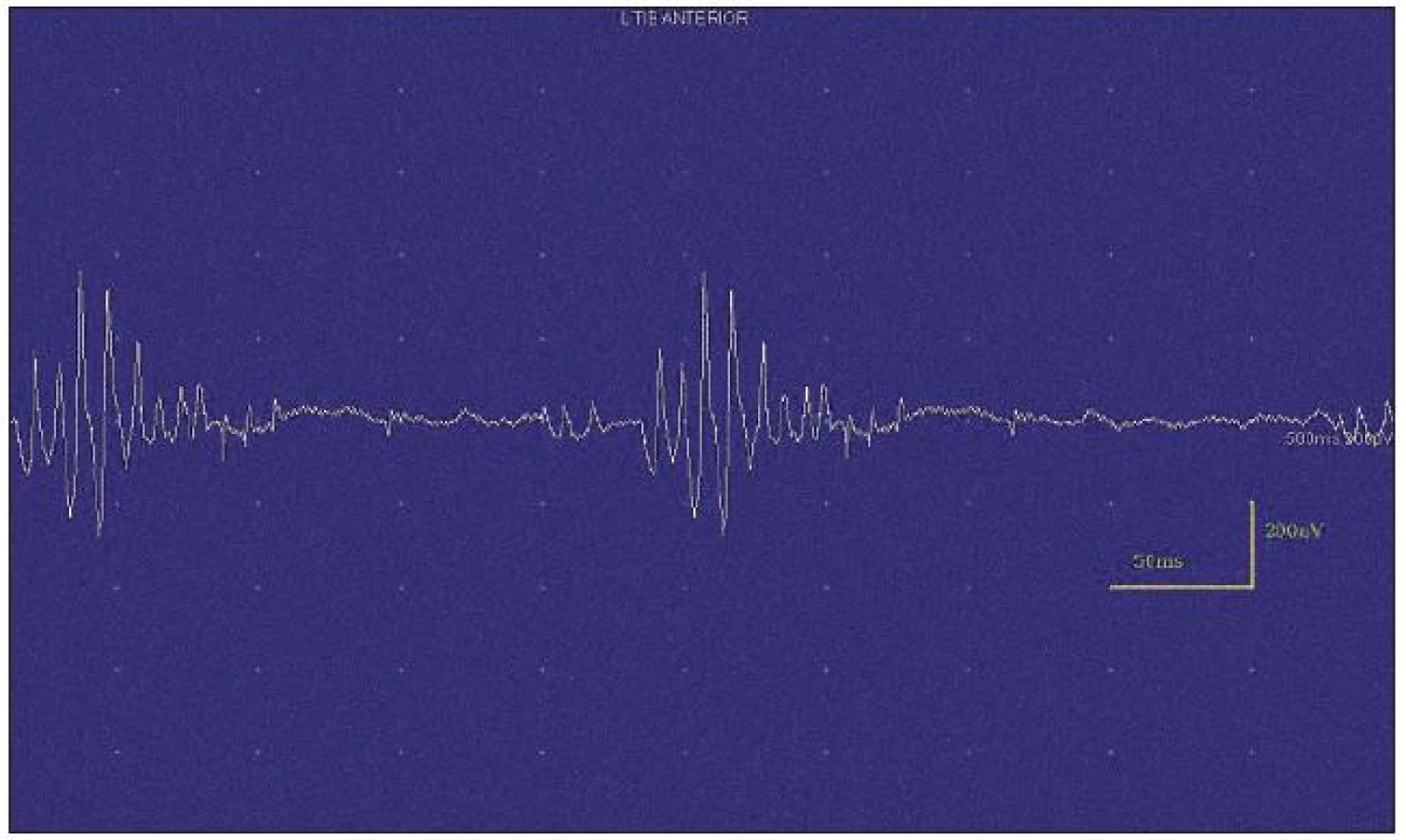
EEG neprokázala patologickou aktivitu a brain mapping zobrazil dominanci alfa aktivity 10 Hz nad zadními kvadranty. EKG ukázalo klidovou sinusovou tachykardii, QTc (Bazettův index) byl v normě. Bez průkazu změn QT úseku (do 400 ms).
Pro vyloučení paraneoplastické etiologie jsme provedli vyšetření paraneoplastických protilátek (anti‑Hu, anti‑Ri, anti‑Yo, anti‑myelin), UZ břicha, CT hrudníku, biopsii uzliny v levé nadklíčkové jamce a kožní biopsii s negativním nálezem.
Biopsie levého m. vastus medialis neprokázala žádné myopatické ani neurogenní změny. Standardní enzymově histochemické vyšetření i imunohistologie s průkazem sarkolemálních proteinů byly v normě. Patologické změny nebyly pozorovány ani při elektronmikroskopickém vyšetření.
Vzhledem k možnému autoimunitnímu charakteru postižení jsme nechali vyšetřit protilátky proti gangliosidům a anti‑GAD (protilátky proti glutamátdekarboxyláze), které byly rovněž s negativním výsledkem.
EMG nález svědčil pro neuromyotonii a byl v souladu s klinickým obrazem i laboratorním nálezem. Nemocného jsme začali léčit karbamazepinem (až 2 × 400 mg) v kombinaci se slabými opioidy (tramadol a pak dihydrokodein 2 × 90 mg) a zahájili jsme imunosupresivní léčbu metylprednizolonem i. v. (250 mg denně). Následně jsme obdrželi výsledek vyšetření protilátek proti napěťově řízením kaliovým kanálům (anti‑VGKC), které bylo silně pozitivní: 1 439 pM/l (Oxford Radcliff Hospital – radio immunoassay, norma 0–100). Nemocný se postupně zlepšoval a byl schopen již samostatné chůze, bylo menší pocení, zácpa i tuhost svalstva. Přechodně ustoupily i fascikulace a myokymie. Z centrálních příznaků přetrvávala již jen porucha usínání.
Přes zlepšení klinického nálezu i subjektivních potíží však pacient po 30 dnech hospitalizace náhle umírá. Kromě známek akutního srdečního selhání při maligní arytmii nebyly pitevním vyšetřením prokázány žádné další změny na vnitřních orgánech, včetně mozku a míchy.
Diskuze
Existuje celá řada poruch periferního neurogenního původu, která může produkovat kontinuální svalovou aktivitu a je pak podkladem svalové tuhosti a křečí. (tab. 1). Tato porucha byla popsána jako kontinuální aktivita svalového vlákna či Isaacsův syndrom (Isaacs 1961). Pro svalovou hypertonii s poruchou relaxace připomínající myotonii ji Mertens a Zschocke v roce 1965 pojmenovali neuromyotonií. Další názvy vždy obsahovaly popis jak hypertonie, tak i záškubů svalových snopců – myokymie s poruchou svalové relaxace či pseudomyotonie s myokymií (tab. 2) [1,3].
![Charakteristiky neuromyotonie [1,9].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/332e33df6aae6bab91ccc73ab8ba50f9.jpg)
![Klasifikace neuromyotonie [16].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/b3da5a0b9df87cb6589bde587a63c019.jpg)
Nemoc se vyvíjí postupně a je klinicky charakterizována nárůstem svalové tuhosti v klidu, trvalými záškuby ve svalech – twitching (fascikulace) a kontrakcemi svalů šířícími se ve vlnách – rippling (myokymie), často sval připomíná pohybující se červy („sack of worms“) [6]. Křeče, mnohdy bolestivé, navazují na volní svalovou kontrakci a jsou důsledkem opožděné relaxace svalu (pseudomyotonie). Spontánní bolesti jsou vzácné, ale téměř pravidelně jsou přítomny bolesti svalů při křečích a je palpační bolestivost svalů. Nejprve bývají postiženy distální svaly dolních končetin (DK) a postupně se nemoc šíří proximálně, na horní končetiny (HK), trup i bulbární svaly. Touto distribucí se neuromyotonie odlišuje od stiff-person syndromu (nejprve bývá postiženo svalstvo trupu a proximálních segmentů končetin) a naopak se blíží až obrazu tetanických křečí (karpo-pedální) [1,7]. U našeho nemocného se objevila tuhost svalů a křeče zpočátku na akrech DK a postupně se šířily na HK i proximálně na trup. Ihned od počátku byly provázeny akroparesteziemi. Fascikulace i myokymie se objevily asi s týdenním zpožděním po ztuhlosti svalů.
Sérové hladiny svalových proteinů (volný myoglobin, CK, aldoláza, laktát dehydrogenáza) mohou být u neuromyotonie zvýšeny. Je to pravděpodobně známka svalového poškození při nadměrné svalové zátěži [8].
Výboje při myokymiích jsou krátké pršky jednotlivých motorických jednotek, které pálí frekvencí 5–150 Hz. Tyto výboje se projevují ve formě dupletů, tripletů či multipletů a jsou následovány krátkou periodou elektrického ticha. Mohou být pravidelné i nepravidelné a jejich frekvence často závisí na délce myokymických výbojů – delší pršky se objevují méně často. Generátor myokymických výbojů může být umístěn na periferním motoneuronu velmi proximálně (např. u roztroušené sklerózy či gliomu pontu) i velmi distálně (např. u akutní polyradikuloneuritidy) [1,7]. Výboje se nejčastěji objevují spontánně. Méně často jsou indukovány volní aktivitou, kdy volně spuštěný akční potenciál prochází přes generátor – tedy oblast axonu, ve které je iniciováno repetitivní pálení potenciálu motorické jednotky. Dysfunkce VGKC v axonální membráně vede k výraznému zvýšení supernormality a ke spontánním repetitivním výbojům na podkladě hyperexcitabilní axolemy. Role efaptické transmise je v místě generátoru nejistá [9].
Neuromyotonické výboje mají stejnou patofyziologii i lokalizaci generátoru, avšak mají vyšší frekvence (150–300 Hz). Pršky výbojů jsou prodloužené, mají náhlý začátek i náhlý konec a dochází ke snižování amplitudy jednotlivých potenciálů v průběhu výboje. Výboje bývají spontánní i spouštěné volní aktivitou, pohyby jehly, hypoxií či poklepem na sval. Od myokymií se liší pouze delším trváním a klesající amplitudou [9,10].
Neuromyotonické i myokymické výboje přetrvávají i po proximálních blokádách nervu lokálními anestetiky a mizí u blokády nervosvalové ploténky (kurare, botulotoxin). Jsou přítomny při celkové anestezii i ve spánku [9,11]. U autoimunitně podmíněných neuromytonických syndromů se obvykle nachází generátor výbojů distálně od terminálního větvení axonu, a přitom však nepostihuje presynaptickou část nervosvalové ploténky (rozdíly mezi spontánními a volně spouštěnými motorickými akčními potenciály při metodě stanovení velikosti motorických jednotek) [12]. My jsme klinicky i v EMG nacházeli myokymie i neuromyotonické fenomény, také však fascikulace, ojediněle i rytmické fibrilace. Tyto fenomény byly přítomny spontánně, jejich četnost se mírně zvýšila při volní aktivitě (s delší latencí), výrazně se zvýšila po poklepu na sval (s rychlým nástupem) i při mechanické iritaci EMG jehlou. Rovněž při motorické neurografii a zejména při vyšetření F-vln vždy bezprostředně následovala série následných výbojů. Na rozdíl od literárních údajů jsme latenci repetitivních výbojů neprokázali [2,11]. Docházelo však ke snižování amplitudy motorických odpovědí.
Neuromyotonické syndromy jsou poruchy periferních nervů. Jsou to léze lokalizované – fokální či spíše multifokální, porucha je v axonální membráně, a to na podkladě dysfunkce (upregulace) pomalých kaliových kanálů v nodální oblasti [1,9,13]. Se získanou neuromyotonií a Morvanovým syndromem jsou vázané „Shaker‑type“ kaliové kanály (Kv1) [14]. Nejsou však postiženy pouze motorické axony, ale také vegetativní nervy, někdy i převodní systém v srdci, rovněž i senzitivní vlákna. Kromě motorických projevů jsme prokázali i výrazné poruchy autonomních nervů – mydriázu s omezeným rozsahem fotoreakce, výrazné pocení, slzení, tachykardii a zácpu. Již od počátku se objevilo brnění na akrech HK i DK, což bylo podkladem pro úvahu o akutní polyradikuloneuritidě. Po zahájení léčby karbamazepinem se zmírnila nejen tuhost svalstva a omezil se i výskyt fascikulací a myokymií, ale ustoupilo i pocení, mydriáza a zčásti tachykardie i úporná zácpa. Ustoupila nespavost a noční zmatenost.
Potřeba spánku je u Morvanova syndromu výrazně redukována až na 2–4 hodiny za den. Klinické projevy spojené s insomnií jsou denní ospalost, narušená struktura REM spánku a porucha usínání spojená s halucinatorními projevy [5]. Obdobné příznaky jsme pozorovali i u našeho nemocného.
V roce 1890 popsal Morvan myokymie s bolestmi svalů, excesivním pocením a poruchou spánku. Tento stav nazval „fibrilární choreou“. Jeho nemocný zemřel pět týdnů po vzniku potíží. Jako Morvanův syndrom se označuje neuromyotonie jak s periferními projevy, tak i projevy centrálními – nespavost, deprese, psychické změny, halucinace. U tohoto syndromu bývají obzvláště výrazné změny autonomních funkcí a zejména srdeční arytmie. Popisují se frekventní supraventrikulární extrasystoly až „syndrom pomalého Q-T“. Mozkové a kardiální změny jsou v souvislosti s poruchou kaliových kanálů, a tedy s výskytem protilátek proti VGKC [3,15]. Mírní se s podáním imunosupresivní léčby či zejména po plazmaferéze [15]. Náš nemocný se vyznačoval výraznými vegetativními příznaky i výraznými psychickými problémy (nespavost, fixace k jednotlivým příznakům – i vegetativním, noční agitace, behaviorální a osobností změny). Po karbamazepinu a po zahájení imunosupresivní léčby se zmírnila hypertonie svalů, psychické projevy i vegetativní příznaky (pocení, zácpa, méně arytmií). Pátrali jsme po převodní poruše i po změnách v repolarizační fázi komor (ekg, Bazettův index), avšak žádnou z těchto kardiálních poruch jsme neprokázali. Nemocný však zemřel náhle, na maligní arytmii. Bylo to sedm týdnů od začátku onemocnění. Klinický obraz a průběh nemoci našeho pacienta do značné míry odpovídal Morvanovu syndromu.
Závěr
Neuromyotonické syndromy se vyznačují zvýšeným svalovým napětím, pseudomyotonickými fenomény, přítomností myokymií a fascikulací ve svalech, počátečním postižením akrálních svalů a postupným šířením na trup a HK, akroparesteziemi, poruchami autonomního systému, charakteristickým neurofyziologickým nálezem a dysfunkcí axonální membrány periferních nervů s poruchou kaliových kanálů. I když se může projevit dosti různorodým obrazem, přesto jsou základní neurofyziologické charakteristiky jasně přítomny a nemoc má autoimunitní charakter, s průkazem protilátek a je dobře ovlivnitelná léčbou.
MUDr. Jan Latta
Neurologická klinika
Pardubická krajská nemocnice, a.s.
Kyjevská 44
532 03 Pardubice
e-mail: janci82@gmail.com
Sources
1. Fahn S, Jankovic J. Principles and practice of movement disorders. Philadelphi a: Churchill Livingstone Elsevi er 2007.
2. Kadaňka Z, Bednařík J. Ne uromyotoni e – nová kanalopati e. Cesk Slov Ne urol N 2000; 63/ 96(3): 128– 133.
3. Liguori R, Vincent A, Clover L, Avoni P, Plazzi G, Cortelli P et al. Morvan’s syndrome: peripheral and central nervo us system and cardi ac involvement with antibodi es to voltage- gated potassi um channels. Brain 2001; 124(12): 2417– 2426.
4. Lee EK, Maselli RA, Ellis WG, Agi us MA. Morvan’s fibrillary chore a: a parane oplastic manifestati on of thymoma. J Ne urol Ne urosurg Psychi atry 1998; 65(6): 857– 862.
5. Bajaj BK, Shrestha S. An interesting case report of Morvan’s syndrome from the Indi an subcontinent. Ne urol Indi a 2007; 55(1): 67– 69.
6. Oh SJ. Principles of clinical electromyography. Case studi es. Baltimore: Willi ams & Wilkins 1993.
7. Valls- Solé J, Montero J. Role of EMG evalu ati on in muscle hyperactivity syndromes. J Ne urol 2004; 251(3): 251– 260.
8. Han IK, Newsom- Davis J. Ne uromyotoni a (Isaacs‘ Syndrome). In: Lane RJM (ed). Handbo ok of Muscle Dise ase. 1st ed. New York: Informa He althcare 1996: 355– 363.
9. Gutmann L, Gutmann L. Myokymi a and ne uromyotoni a. J Ne urol 2004; 251(2): 138– 142.
10. Maddison P. Ne uromyotoni a. Clin Ne urophysi ol 2006; 117(10): 2118– 2127.
11. Bednarík J, Kadanka Z. Voliti onal and stimulati on induced ne uromyotonic discharges: unusu al electrophysi ological pattern in acquired ne uromyotoni a. J Ne urol Ne urosurg Psychi atry 2001; 70(3): 406– 407.
12. Arimura K, Arimura Y, Ng A, Uehara A, Nakae M, Osame M et al. The origin of spontane o us discharges in acquired ne uromyotoni a. A Macro EMG study. Clin Ne urophysi ol 2005; 116(8): 1835– 1839.
13. Nodera H, Kaji R. Nerve excitability testing and its clinical applicati on to ne uromuscular dise ases. Clin Ne urophysi ol 2006; 117(9): 1902– 1916.
14. Kle opa KA, Elman LB, Lang B, Vincent A, Scherer SS. Ne uromyotoni a and limbic encephalitis sera target mature Shaker‑type K+ channels: subunit specificity correlates with clinical manifestati ons. Brain 2006; 129(6): 1570– 1584.
15. Irani S, Lang B. Auto antibody- medi ated disorders of the central nervo us system. Auto immunity 2008; 41(1): 55– 65.
16. Hart I, Vincent A, Willison H. Ne uromyotoni a. In: Engel A. Myastheni a Gravis and Myasthenic Disorders. Oxford: Oxford University Press US 1999: 230.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2009 Issue 4
Most read in this issue
- Nádory tretej mozgovej komory
- Získaná neuromyotonie s nevelkými centrálními příznaky s průkazem protilátek proti napěťově řízeným kaliovým kanálům – kazuistika
- Botulotoxin v léčbě spasticity
- Paroxysmální kinezigenní dyskineze: případ mladé ženy s alternující hemidystoni í – kazuistika