
Paroxysmální kinezigenní dyskineze: případ mladé ženy s alternující hemidystoni í – kazuistika
Paroxysmal Kinesigenic Dyskinesi a – a Case Report of a Yo ung Woman with Alternating Hemidystoni a
Paroxysmal kinesigenic dyskinesi a is a ne urological disorder characterized by bri ef episodes of dystoni a and chore athetosis triggered by sudden voluntary movement. The attacks typically last less than one minute and may occur even several times a day. Besides famili ar idi opathic forms with a utosomal dominant inheritance, sporadic cases may occur, or symptomatic forms may be associ ated with some CNS dise ases and may manifest with the same type of paroxysms. At the beginning we revi ew current knowledge on this disorder, including the historical development of the concept of paroxysmal dyskinesi a. Then we present the case of a yo ung woman who has suffered from sporadic idi opathic paroxysmal kinesigenic dyskinesi a since she was 13 ye ars old. Di agnosis was proved during vide o- EEG monitoring and her paroxysms can be characterised as alternating hemidystoni a. She has been successfully tre ated with 300 mg of lamotrigine per day.
Key words:
paroxysmal dyskinesia – alternating hemidystonia
Authors:
H. Krijtová; P. Marusič
Authors‘ workplace:
Neurologická klinika UK 2. LF a FN Motol, Praha
Published in:
Cesk Slov Neurol N 2009; 72/105(4): 368-372
Category:
Case Report
Overview
Paroxysmální kinezigenní dyskineze je porucha projevující se krátkými záchvaty mimovolních pohybů provokovaných náhlým pohybem. Záchvaty typicky trvají méně než jednu minutu a moho u se vyskytovat i několikrát denně. Kromě formy idi opatické a utozomálně dominantně dědičné nebo sporadické existují i formy symptomatické, kdy se stejnými záchvaty moho u manifestovat některá onemocnění postihující CNS. V úvodu podáváme so uhrn so učasných představ o této poruše i historický vývoj těchto představ v kontextu skupiny paroxysmálních dyskinezí. Na závěr prezentujeme případ mladé ženy se sporadicko u idi opaticko u paroxysmální kinezigenní dyskinezí od 13 let věku, jejíž di agnóza byla stanovena při vide o- EEG monitorování a záchvaty měly charakter alternující hemidystoni e. Obtíže usto upily při léčbě lamotriginem v dávce 300 mg denně.
Klíčová slova:
paroxysmální dyskineze – alternující hemidystonie
Úvod
Historie
Poprvé tento nejčastěji se vyskytující typparoxysmálních dyskinezí popsal již v roce 1892 japonský psychiatr Shuzo Kure. Ve svém článku zaznamenal případ mladého muže, u něhož se začaly ve věku 10 let objevovat záchvaty podivných neúčelných pohybů, které byly provokovány náhlým pohybem. Začínaly na dolních končetinách, odkud se šířily na celé tělo, někdy s pravostrannou převahou. Dyskinezi předcházel zvláštní pocit, senzorická aura v postižených končetinách, a pacient se naučil pohyby tlumit nebo potlačit pohybováním nohama nebo představou následujícího pohybu ještě vsedě předtím, než se postavil a vykročil. Záchvaty měly velmi krátké trvání, postupně se objevovaly stále častěji, až jejich frekvence byla téměř každodenní. Přestože si Kure uvědomil, že mezi záchvaty je neurologický nález normální, domníval se chybně, že jeho pacient trpí Thomsenovou kongenitální myotonií, a proto bylo prvenství popisu této poruchy delší dobu připisováno až krátké zmínce v knize Gowera z roku 1901 [1]. V průběhu 20. století byly shromažďovány údaje o existenci celé skupiny tzv. paroxysmálních dyskinezí, jejichž uspořádání podle vyvolávajícího momentu provedli v roce 1995 ve své klasifikaci Demirkiran a Jankovic [2]. Rozlišovali dyskineze: 1. kinezigenní provokované náhlým pohybem, 2. nekinezigenní provokované různými faktory, jako je káva, čaj, alkohol a únava, a mající delší trvání a 3. dyskineze provokované cvičením nebo námahou (nejčastěji charakteru delší chůze nebo běhu) a 4. dyskineze provokované spánkem. Osud této klasifikace ilustruje svízelnost, s níž se vyvíjejí naše znalosti o povaze paroxysmálních dyskinez. Nejprve se z ní vydělily dyskineze provokované spánkem, neboť se ukázalo, že u tzv. paroxysmální noční choreoatetózy se vlastně jedná o formu frontální epilepsie s autozomálně dominantní dědičností (ADNFLE) [3,4]. Rovněž původní představy o možném společném patofyziologickém mechanizmu pro celou skupinu paroxysmálních dyskinez se nepotvrdily [5]. Guerrini se například domníval, že všechny tyto jednotky jsou podmíněny poruchou iontových kanálů, ale jak se později ukázalo, paroxysmální nekinezigenní dyskinezi vyvolávají mutace MR 1 – genu kódujícího regulátor myofibrilogeneze 1 [6] a paroxysmální dyskinezi provokovanou cvičením (námahou) způsobují mutace SLC2A1 – genu pro GLUT1, což je transportér glukózy přes hematoencefalickou bariéru [7]. Porucha na úrovni iontových kanálů zůstává však stále jedním z potenciálních faktorů v patogenezi paroxysmální kinezigenní dyskineze (PKD) [8].
Klinický obraz
Paroxysmální kinezigenní dyskineze je porucha charakterizovaná krátkými záchvaty mimovolních pohybů, které jsou provokovány náhlým pohybem. Diagnóza je stanovena na základě klinického obrazu, neboť v současné době neexistuje vyšetření, které by bylo diagnosticky specifické. U typických případů jsou všechna pomocná vyšetření – jako je magnetická rezonance mozku a laboratorní vyšetření – bez jednoznačných patologických odchylek.
Charakter abnormálních pohybů může být variabilní – pohyby mohou mít ráz choreoatetózy, mohou být balistické, často se jedná o dystonii nebo hemidystonii, nebo se různé typy pohybů kombinují. Většinou začínají fokálně z dolních končetin, odkud se šíří ipsi- i kontralaterálně a mohou se až generalizovat. Rozmanitosti klinických projevů odpovídá i pestrost v užívané terminologii, u různých autorů se tak setkáme s odlišnými názvy – paroxysmální kinezigenní choreoatetóza, paroxysmální kinezigenní dystonie či paroxysmální kinezigenní dyskineze nebo epizodická kinezigenní dyskineze (EKD). Trvání atak je krátké, většinou několik desítek sekund – jako horní hranice se v současné době uvádí jedna minuta. Výskyt záchvatů je zpravidla každodenní, mohou se jich objevit až desítky denně. Opakovaně bylo zaznamenáno, že muži jsou postiženi častěji než ženy. Záchvaty začínají nejčastěji ve školním věku (5–15 let), ale při stanovování diagnózy idiopatické PKD se jako možné věkové rozmezí začátku záchvatů připouští období 1–20 let [9].
Formy onemocnění
Idiopatická familiární forma se dědí autozomálně dominantně s neúplnou penetrancí. Pátrání po kauzálním genu je velmi intenzivní, ale doposud objeven nebyl. Dva geny byly mapovány do pericentromerické oblasti 16. chromozomu – EKD1 16p12.1–q13a EKD2 16q13–q22.1 [10–13]. Předpokládáse ještě existence dalšího genu (EKD3) mimo 16. chromozom [14].
Do stejné oblasti na 16. chromozomu byly mapovány i benigní familiární infantilní konvulze, které se mohou vyskytnout v rodinách pacientů s PKD, a dokonce se obě jednotky mohou objevit u téhož jedince v různých obdobích jeho života. Hovoří se pak o syndromu infantilních konvulzí s choreoatetózou a je možné, že se jedná o různý způsob exprese téže genetické poruchy [15], která se manifestuje variabilně buď v oblasti bazálních gangliích, či v oblasti senzorimotorického kortexu v průběhu dozrávání CNS [1].
Kromě nejčastější formy familiární jsou hojné též případy sporadické, méně časté jsou formy symptomatické. Stejným typem záchvatů se totiž mohou manifestovat některá onemocnění CNS – nejčastěji je PKD popisována u pacientů s roztroušenou sklerózou nebo s vaskulárním postižením CNS. Ojediněle byly PKD popsány např. u lymfomu či Arnold-Chiariho malformace nebo syndromu centrální pontinní myelinolýzy [16–20]. Mohou se tak projevit i poruchy metabolizmu vápníku – hypoparatyreoidizmus a pseudohypoparatyreoidizmus [21].
Patofyziologie
Přestože klinické rysy PKD jsou dobře popsány, její patofyziologie zůstává nevyjasněna. Má některé společné rysy s epilepsií, a proto byla historicky považována za zvláštní formu reflexní epilepsie [5]. V současné době se ale předpokládá, že se jedná o dvě odlišné poruchy s různými patofyziologickými mechanizmy [1].
Ve snaze objasnit patofyziologické děje u PKD byly prováděny studie sledující změny perfuze mozku metodou SPECT. Interiktálně byla pozorována hypoperfuze posteriorních částí ncl. caudatus oboustranně [22] nebo byly popisovány změny v perfuzi thalamu [23]. Iktálně většinou docházelo k hyperperfuzi bazálních ganglií oboustranně nebo kontralaterálně k fokálnímu začátku dyskinez [24]. To podporuje úvahu o důležité roli extrapyramidového systému při vzniku PKD, nicméně zatím není jasné, zda jsou tyto změny primární nebo zda vznikají až sekundárně v průběhu záchvatů.
Elektrofyziologické studie zaznamenaly u pacientů s PKD funkční odchylky v rámci inhibičních systémů – intrakortikální, transkalosální a reciproční inhibici, z nichž některé se upravovaly po podávání karbamazepinu. Na základě těchto nálezů se předpokládá, že příčina vzniku PKD by mohla spočívat v poruše kortikospinálních inhibičních mechanizmů [25]. Společné rysy s epilepsií, včetně promptního účinku antiepileptik, jako je karbamazepin, vedly opakovaně k úvahám, že se pravděpodobně jedná o poruchu funkce sodíkových kanálů, ať již geneticky podmíněnou, či navozenou jiným onemocněním CNS [5,10,17,26].
Paroxysmální dyskineze a jiné záchvaty
Paroxysmální dyskineze mají řadu společných rysů s epilepsií, ale dosud nebyla mezi těmito dvěma poruchami vytvořena jednoznačná hranice, ani nejsou definovány případné společné nebo odlišné patofyziologické děje.
Již výše byl uveden syndrom infantilních konvulzí s choreoatetózou, který se vyznačuje současným výskytem benigních infantilních konvulzí a PKD v rámci postižených rodin, nebo dokonce u jednoho jedince. Kromě toho byly popsány rodiny, v nichž se vyskytly současně paroxysmální dyskineze a různé syndromy epileptické. Například v rodině s paroxysmální dyskinezí provokovanou cvičením (námahou) se může vyskytnout generalizovaná (i fokální) epilepsie, nebo byly zaznamenány současně písařská křeč a benigní rolandická epilepsie. Uváděny jsou i případy jiných forem epilepsie, migrény, ataxie, myoklonu či koincidence záchvatů kinezigenních a nekinezigenních v rodinách s PKD [9,26].
Prognóza a léčba
Paroxysmální kinezigenní dyskineze je benigní onemocnění, které se většinou zmírňuje nebo mizí v průběhu života. Opakovaně bylo popsáno, že excelentně reaguje na podávání antiepileptik [9]. Z nich se v literatuře uvádí jako ověřený karbamazepin, fenytoin, oxkarbazepin, lamotrigin a topiramát [9,24,27–29].
Kazuistika
Pacientka – 23letá vysokoškolačka – byla odeslána na video-EEG monitorování k upřesnění klasifikace záchvatů. Její mladší bratr byl v předškolním věku přechodně léčen pro absence, matka trpěla těžkými záchvaty migrény s aurou. Naše pacientka neprodělala žádné vážné onemocnění, od 13 let se u ní začaly vyskytovat migrény s aurou podobného rázu jako u její matky – aura zahrnovala hemiparestezie, výpadek zorného pole a někdy fatickou poruchu. Frekvence migrén byla relativně nízká. Ve stejné době se také začaly objevovat záchvaty, které byly natolik zvláštní, že se s nimi pacientka dlouho nikomu nesvěřila – obávala se, že by jí nikdo nevěřil. Pacientka je uměla velmi dobře popsat. Důležitým rysem záchvatů byla jejich provokace náhlým pohybem, pak následovaly „podivné“ pohyby, které nedokázala ovládnout.
Dobře si vybavovala první příhodu – ve škole při tělocviku, když nacvičovali na běžecké dráze „starty“, se jí krátce po vyběhnutí zkroutila pravá noha a ona upadla. Záchvaty se začaly opakovat a jejich frekvence postupně narůstala, až se objevovaly i několikrát za den. Nejprve ucítila napětí či brnění v jedné dolní končetině až celé polovině těla, pak následovala křeč začínající většinou na akru jedné dolní končetiny, podle tíže záchvatu došlo současně či následně ke křečovitému zkroucení či prudšímu pohybu ipsilaterální horní končetiny, někdy doprovázené stažením koutku úst či jazyka v ústech, stažením celé tváře až s „vyvalením oka“ a stočením trupu do strany. Pohyby nebyly nikdy provázeny či následovány poruchou vědomí, nikdy se nerozvinul generalizovaný tonicko‑klonický záchvat. Někdy nemohla při postižení orofaciální oblasti během záchvatu mluvit. Mimovolní pohyby zpočátku postihovaly pravou polovinu těla, ale v dalším vývoji onemocnění se vyskytovaly stranově střídavě a v době video-EEG monitorování postihovaly stranu levou (obr. 1).
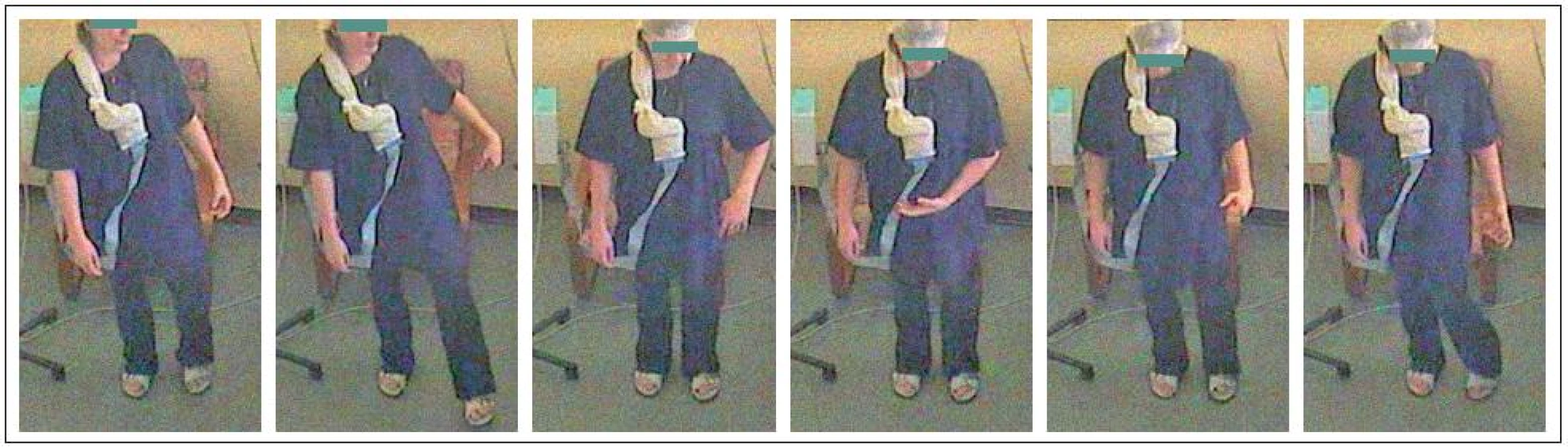
Na své okolí působila pacientka „líným“ dojmem. Vždy jí dlouho trvalo, než na zavolání přiběhla k mamince. Nejtypičtější situace spouštějící záchvaty nastávaly v dopravních prostředcích při vystupování nebo ve škole, když byla vyvolána ke zkoušení u tabule. Tedy při volním pohybu, který však nebylo možné předem přesně naplánovat. Frekvence se sice ustálila na několika záchvatech za den, ale zvyšovala se stresem, takže například ve zkouškovém období jich byly denně až desítky. Záchvaty trvaly většinou několik sekund až půl minuty – délka trvání i jejich intenzita narůstaly, pokud pacientka pokračovala v pohybu, který je vyvolal. V některých situacích se naučila jim bránit – pokud věděla, že bude muset vstát, tak se vsedě snažila rychlým pohybem nohy záchvat vyvolat. Ten proběhl ještě vsedě a ona pak mohla vstát a například vystoupit z tramvaje, protože následovala „refrakterní“ fáze.
Při vyšetření jsme neshledali žádné odchylky v neurologickém nálezu, laboratorní hodnoty včetně vyšetření metabolizmu kalcia byly v mezích normy. MR mozku byla bez patologických změn. V interiktálním EEG jsme pozorovali intermitentní theta vyšší amplitudy s maximem nad předními kvadranty, někdy až ostrého tvaru. Místy bylo patrné amplitudové maximum fronto-temporálně se střídavou stranovou převahou.
Během video-EEG monitorování jsme zpočátku žádné záchvaty nezaznamenali. Proto jsme se pokusili záchvaty vyprovokovat provedením několika dřepů, což se opakovaně podařilo. Záchvaty mimovolních pohybů trvaly většinou 10–20 sekund, měly charakter levostranné dystonie se složkou choreoatetózy. Postihovaly v počátku levou dolní končetinu a rychle se též šířily na levou horní končetinu. Pacientka subjektivně pocítila i stažení levého koutku. Iktální EEG bylo částečně překryto artefakty a jinak nevykazovalo žádnou abnormitu. Pacientce bylo doporučeno užívat lamotrigin – při dávce 300 mg denně záchvaty vymizely, pouze v období zvýšeného stresu se ještě ojediněle objeví.
Diskuze
Stanovení správné diagnózy u pacientů s PKD trvá průměrně 5–6 let od doby, kdy vyhledají lékařskou pomoc, a nejčastěji jsou podezříváni z neepileptických záchvatů psychogenních pro bizarnost okolností, při nichž se objevují, i pro podivný charakter motorických projevů [9]. Před video-EEG monitorováním jsme již v průběhu rozhovoru s nemocnou možnost neepileptických psychogenních záchvatů téměř vyloučili. Pak jsme se soustředili na specifický rys záchvatů – provokaci náhlým pohybem – a rozhodovali se mezi reflexními parciálními epileptickými záchvaty a PKD. Pohybem mohou být provokovány epileptické záchvaty vycházející z oblasti centrální (ze senzorimotorického kortexu) nebo ze suplementární senzorimotorické arey [30]. Při provokaci pohybem se na spuštění většinou podílejí somatosenzorické či proprioceptivní podněty. Záchvaty začínají často somatosenzorickou aurou v drážděné oblasti a následují buď myoklonické záškuby, či tonický záchvat, který se může šířit ipsilaterálně i bilaterálně, nebo může přejít v záchvat klonický i generalizovaný tonicko‑klonický. Pokud záchvaty začínají ze suplementární senzorimotorické arey, tak mají rychle charakter bilaterálního asymetrického tonického záchvatu [30].
Somatosenzorické aury se mohou vyskytnout u záchvatů epileptických i u PKD, ale extrapyramidový charakter mimovolních pohybů u naší pacientky spolu s alternujícím stranovým postižením, typický charakter provokujícího náhlého pohybu, krátké trvání záchvatů, zachované vědomí a typický věk při začátku onemocnění nás vedly k závěru, že se jedná o PKD. Alternující hemidystonie je v literatuře uváděna jako jeden z možných projevů PKD [9,31]. Bruno et al ji zaznamenali dokonce u 12 případů ve skupině 95 sledovaných pacientů s idiopatickou PKD [9].
Změny v interiktálním EEG mohou být u naší pacientky považovány až za výboje epileptiformní – mohou ale dobře korelovat s diagnózou migrény nebo s pozitivní rodinnou anamnézou epilepsie u bratra, neboť u příbuzných pacientů zvláště s idiopatickými epilepsiemi se někdy v EEG vyskytuje i specifická epileptiformní abnormita bez klinicky manifestních epileptických záchvatů. Epileptiformní výboje v EEG jsou naopak uváděny u některých pacientů s PKD i v literatuře [32].
Důkladným vyšetřením jsme vyloučili jiná onemocnění, o nichž je známo, že mohou vyvolávat symptomatické formy PKD, včetně poruchy metabolizmu vápníku.
V rodině pacientky se vykytují další poruchy záchvatového rázu – u bratra blíže neklasifikovaná benigní forma epilepsie nejspíše generalizované s absencemi, u matky a současně i u pacientky pak migréna s aurou. Současný výskyt epilepsie i migrény v rodinách nemocných s paroxysmální dyskinezí je uváděn, a to i v koincidenci s PKD [26]. Nicméně migréna sama je onemocnění natolik časté, že její výskyt v rodině naší pacientky nelze patrně považovat za více než náhodný.
Závěr
Paroxysmální kinezigenní dyskineze představuje vzácnou neurologickou poruchu mající některé společné rysy s epileptickými záchvaty spouštěnými pohybem. Pokud na PKD myslíme, je ve většině případů možné určit správnou diagnózu již na základě anamnestických údajů. Kromě typického náhlého (předem přesně neplánovaného) volního pohybu provokujícího záchvaty se vyznačuje plně zachovaným vědomím v průběhu záchvatů. Alternující stranové postižení je pro diagnózu PKD zřejmě rovněž typické. Při léčbě obou typů poruch – PKD i epilepsie – se používají antiepileptika, prognóza je u PKD ve většině případů příznivá.
MUDr. Hana Krijtová
Neurologická klinika UK 2. LF a FN Motol, Praha
V Úvalu 84
150 06 Praha 5-Motol
e-mail: hana.krijtova@fnmotol.cz
Sources
1. Kato N, Sadamatsu M, Kikuchi T, Niikawa N, Fukuyama Y. Paroxysmal kinesigenic chore o athetosis: from first discovery in 1892 to genetic linkage with benign famili al infantile convulsi ons. Epilepsy Res 2006; 70 (Suppl 1): S174– S184.
2. Demirkiran M, Jankovic J. Paroxysmal dyskinesi as: clinical fe atures and classificati on. Ann Ne urol 1995; 38(4): 571– 579.
3. Scheffer IE, Bhati a KP, Lopes- Cendes I, Fish DR, Marsden CD, Andermann F et al. Autosomal dominant frontal epilepsy misdi agnosed as sleep disorder. Lancet 1994; 343(8896): 515– 517.
4. Bhati a KP. Famili al (idi opathic) paroxysmal dyskinesi as: an update. Semin Ne urol 2001; 21(1): 69– 74.
5. Guerrini R. Idi opathic epilepsy and paroxysmal dyskinesi a. Epilepsi a 2001; 42 (Suppl 3): 36– 41.
6. Raini er S, Thomas D, Tokarz D, Ming L, Bui M, Plein Eet al. Myofibrillogenesis regulator 1 gene mutati ons ca use paroxysmal dystonic chore o athetosis. Arch Ne urol 2004; 61(7): 1025– 1029.
7. Suls A, Dedeken P, Goffin K, Van Esch H, Dupont P, Cassiman D et al. Paroxysmal exercise‑induced dyskinesi a and epilepsy is due to mutati ons in SLC2AI, encoding the glucose transporter GLUTI. Brain 2008; 131(7): 1831– 1844.
8. Rochette J, Roll P, Szepetowski P. Genetics of infantile seizures with paroxysmal dyskinesi a: the infantile convulsi ons and chore o athetosis (ISSA) and ICCA‑related syndromes. J Med Genet 2008; 45(12): 773– 779.
9. Bruno MK, Hallett M, Gwin‑Hardy K, Sorensen B, Considine E, Tucker S et al. Clinical evalu ati on of idi opathic paroxysmal kinesigenic dyskinesi a: new di agnostic criteri a. Ne urology 2004; 63(12): 2280– 2287.
10. Tomita H, Nagamitsu S, Wakui K, Fukushima Y,Yamada K, Sadamatsu M et al. Paroxysmal kinesigenic chore o athetosis locus maps to chromosome 16p11.2- q12.1. Am J Hum Genet 1999; 65(6): 1688– 1697.
11. Du T, Feng B, Wang X, Mao W, Zhu X, Li L et al. Localizati on and mutati on detecti on for paroxysmal kinesigenic chore o athetosis. J Mol Ne urosci 2008; 34(2): 101– 107.
12. Kikuchi T, Nomura M, Tomita H, Harada N, Kanai K,Konishi T et al. Paroxysmal kinesigenic chore o athetosis (PKC): confirmati on of linkage to 16p11- q21, but unsuccessful detecti on of mutati ons among 157 genes at the PKC- critical regi on in seven PKC famili es. J Hum Genet 2007; 52(4): 334– 341.
13. Valente EM, Spacey SD, Wali GM, Bhati a KP, Dixon PH, Wo od NW et al. A second paroxysmal kinesigenic chore o athetosis locus (EKD2) mapping on 16q13- q22.1 indicates a family of genes which give rise to paroxysmal disorders on human chromosome 16. Brain 2000; 123(10): 2040– 2045.
14. Spacey SD, Valente EM, Wali GM, Warner TT, Jarman PR, Schapira AH et al. Genetic and clinical heterogeneity in paroxysmal kinesigenic chore o athetosis: evidence for a third EKD gene. Mov Disord 2002; 17(4): 717– 725.
15. Caraballo R, Pavek S, Lemainque A, Gastaldi M, Echenne B, Motte J et al. Linkage of benign famili al infantile convulsi ons to chromosome 16p12- q12 suggests allelism to the infantile convulsi ons and chore o athetosis syndrome. Am J Hum Genet 2001; 68(3): 788- 794.
16. Blakeley J, Jankovic J. Secondary ca uses of paroxysmal dyskinesi a. Adv Ne urol 2002; 89: 401– 420.
17. Bonev VI, Gledhill RF. Paroxysmal kinesigenic chore o athetosis beca use of cryptogenic myelitis. Remissi on with carbamazepine and the pathogenetic role of altered sodi um channels. Eur J Ne urol 2002; 9(5): 517– 520.
18. Rollnik JD, Winkler T, Ganser A. A case of symptomatic paroxysmal kinesigenic dyskinesi a with primary central nervo us system lymphoma. Nervenarzt 2003; 74(4): 362– 365.
19. Zorzi G, Conti C, Erba A, Granata T, Angelini L, Nardocci N. Paroxysmal dyskinesi as in childho od. Pedi atr Ne urol 2003; 28(3): 168– 172.
20. Baba Y, Wszolek ZK, Normand MM. Paroxysmal kinesigenic chore o athetosis associ ated with central pontine myelinolysis. Parkinsonism Relat Disord 2003; 10(2): 113.
21. Mahmud FH, Linglart A, Bastepe M, Jüppner H, Lteif AN. Molecular di agnosis of pse udohypoparathyre o idism type 1b in a family with presumed paroxysmal dyskinesi a. Pedi atrics 2005; 115(2): 242– 244.
22. Jo o EY, Hong SB, Tae WS, Kim JH, Han SJ, Se o DW et al. Perfusi on abnormality of the ca udate nucle us in pati ents with paroxysmal kinesigenic chore o athetosis. Eur J Nucl Med Mol Imaging 2005; 32(10): 1205– 1209.
23. Shirane S, Sasaki M, Kogure D, Matsuda H, Hashimoto T. Incre ased ictal perfusi on of the thalamus in paroxysmal kinesigenic dyskinesi a. J Ne urol Ne urosurg Psychi atry 2001; 71(3): 408– 410.
24. Ko CH, Kong CK, Ngai WT, Ma KM. Ictal (99m)Tc ECD SPECT in paroxysmal kinesigenic chore o athetosis. Pedi atr Ne urol 2001; 24(3): 225– 227.
25. Mir P, Hu ang YZ, Gili o F, Edwards MJ, Berardelli A,Rothwell JC et al. Abnormal cortical and spinal inhibiti on in paroxysmal kinesigenic dyskinesi a. Brain 2005; 128(2): 291– 299.
26. Fo urcade G, Ro uberti e A, Do ummar D, Vidailhet M, Laba uge P. Paroxysmal kinesigenic dyskinesi a: a channelopathy? Study of 19 cases. Rev Ne urol (Paris) 2009; 165(2): 164– 169.
27. Gökçay A, Gökçay F. Oxcarbazepine therapy in paroxysmal kinesigenic chore o athetosis. Acta Ne urol Scand 2000; 101(5): 344– 345.
28. Uberall MA, Wenzel D. Effectiveness of lamotrigine in children with paroxysmal kinesigenic chore o athetosis. Dev Med Child Ne urol 2000; 42(10): 699– 700.
29. Hu ang YG, Chen YC, Du F, Li R, Xu GL, Ji ang Wet al. Topiramate therapy for paroxysmal kinesigenic chore o athetosis. Mov Disord 2005; 20(1): 75– 77.
30. Vignal JP, Biraben A, Cha uvel PY, Re utens DC. Reflex parti al seizures of sensorimotor cortex (including cortical reflex myoclonus and startle epilepsy). Adv Ne urol 1998; 75: 207– 226.
31. Ho user MK, Soland VL, Bhati a KP, Quinn NP, Marsden CD. Paroxysmal kinesigenic chore o athetosis: a report of 26 pati ents. J Ne urol 1999; 246(2): 120– 126.
32. Cuenca- Le on E, Cormand B, Thomson T, Macaya A.Paroxysmal kinesigenic dyskinesi a and generalised: clinical and genetic analysis in a Spanish pedigree. Ne uropedi atrics 2002; 33(6): 288– 293.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2009 Issue 4
Most read in this issue
- Nádory tretej mozgovej komory
- Získaná neuromyotonie s nevelkými centrálními příznaky s průkazem protilátek proti napěťově řízeným kaliovým kanálům – kazuistika
- Botulotoxin v léčbě spasticity
- Paroxysmální kinezigenní dyskineze: případ mladé ženy s alternující hemidystoni í – kazuistika