
Klinický obraz spinální svalové atrofie v dětském věku
Clinical features of spinal muscular atrophy in children
Spinal muscular atrophy (SMA) is a neurodegenerative disease characterized by a loss of motor neurons in the anterior horn of the spinal cord and by resultant progressive, mainly proximal, weakness. A phenotypic spectrum includes pulmonary, gastrointestinal, nutritional complications, scoliosis and contractures. The most common form of SMA, accounting for 95% of cases, is autosomal recessive proximal SMA associated with mutations in the survival of motor neurons (SMN1) gene. It is a rare disorder with the incidence 1 : 6,000–1 : 10,000. The clinical spectrum ranges from early infant death to normal adult life with only mild weakness. SMA is classified into four types depending on the age of onset and highest level of motor function achieved. Actual treatment and multidisciplinary care change the course of the disease and improve life expectancy and quality of life.
Keywords:
spinal muscular atrophy – classification of spinal muscular atrophy – phenotype spinal muscular atrophy
Autoři:
J. Staněk
Působiště autorů:
Oddělení dětské neurologie, FN Ostrava
Vyšlo v časopise:
Cesk Slov Neurol N 2020; 83/116(Supplementum 2): 8-12
doi:
https://doi.org/10.48095/cccsnn20202S8
Souhrn
Spinální svalová atrofie (SMA) je neurodegenerativním onemocněním se ztrátou motoneuronů v předních rozích míšních. Hlavním klinickým příznakem je především proximální progredující svalová slabost. Klinický obraz zahrnuje dále respirační, gastrointestinální a nutriční komplikace, skoliózy, kontraktury. Nejčastější formou tvořící 95 % případů je autozomálně recesivní SMA spojená s mutací survival motor neuron (SMN1) genu. Jedná se o vzácné onemocnění s incidencí 1 : 6 000 až 1 : 10 000 živých novorozenců. Klinický obraz zahrnuje široké spektrum postižení od imobilních novorozenců s časným úmrtím po dospělé pacienty s jen mírnou slabostí. Klasifikace rozlišuje základní čtyři typy podle věku, ve kterém se objevily počáteční příznaky, a podle dosaženého motorického maxima. Současné možnosti terapie a multioborová péče významným způsobem mění průběh onemocnění, zlepšují předpokládané dožití a kvalitu života pacientů se SMA.
Klíčová slova:
spinální svalová atrofie – klasifikace spinální svalové atrofie – fenotyp spinální svalové atrofie
Úvod
Spinální svalové atrofie (SMA) jsou skupinou genetických nemocí s degenerací motoneuronů předních rohů míšních a motorických jader hlavových nervů. Nemoc je nejčastěji způsobena mutací survival motor neuron genu (SMN1) na 5q13 chromozomu.
Jedná se o vzácné onemocnění s incidencí 1 : 6 000 až 1 : 10 000 živých novorozenců s frekvencí nosičů 1 z 40. V ČR se každoročně předpokládá narození 10 dětí se SMA. Tato incidence činí SMA druhým nejčastějším vrozeným nervosvalovým onemocněním po Duchenneově svalové dystrofii. Nemoc bývala nejčastější příčinou úmrtí na vrozené onemocnění v kojeneckém věku [1,2].
Spinální svalová atrofie byla nejdříve popsána u případů dvou bratrů Guidem Werdnigem v roce 1891 a u dalších sedmi případů Johannem Hoffmannem mezi lety 1893 a 1900. Ačkoliv se eponymum Werdnig-Hoffmannova nemoc stala označením pro těžkou infantilní formu SMA, jejich případy byly intermediální. První popis těžké infantilní formy byl uskutečněn Sylvestrem v roce 1899 a Beevorem v roce 1903. Lehčí formy SMA, u kterých zůstala schopnost stoje a chůze, byly popsány před rokem 1950 Wohl-fartem, Fezem a Eliassonem a poté podrobněji Kugelbergem a Welanderovou. V roce 1964 Dubowitz popsal fenotyp intermediálního typu SMA II u 12 pacientů a rozlišil tři základní typy SMA.
Během dalších let probíhala kontroverze, zda infantilní, juvenilní a adultní formy SMA reprezentují jednu nebo více nemocí.
V roce 1995 bylo zjištěno, že 95 % případů SMA nezávisle na typu je způsobeno homozygotní delecí SMN1 genu na chromozomu 5q13 [3]. Patogeneze je předmětem další části supplementa.
Klasifikace
V roce 1991 byla přijata Mezinárodním konsorciem SMA klasifikace SMA, která stanovila tři typy SMA založené na věku pacienta s počátečními příznaky onemocnění a na nejvyšší úrovni motorických funkcí (tab. 1). Pozdější modifikace přičlenila typ IV pro dospělé případy a typ 0 pro pacienty s prenatálním počátkem a úmrtím během týdnů až měsíců po narození. Typ SMA I byl subklasifikován na typ Ia s novorozeneckým nebo antenatálním počátkem, typ Ib – typický m. Werdnig-Hoffmann s počátkem po novorozeneckém údobí – a Ic s pozdějším počátkem, se schopností sedu s oporou, lehkými bulbárními a respiračními příznaky během prvních 6 měsíců života. Klasifikace rozlišuje typ IIIa s počátkem do 3 let věku a typ IIIb s počátkem po 3. roce věku [3,4].
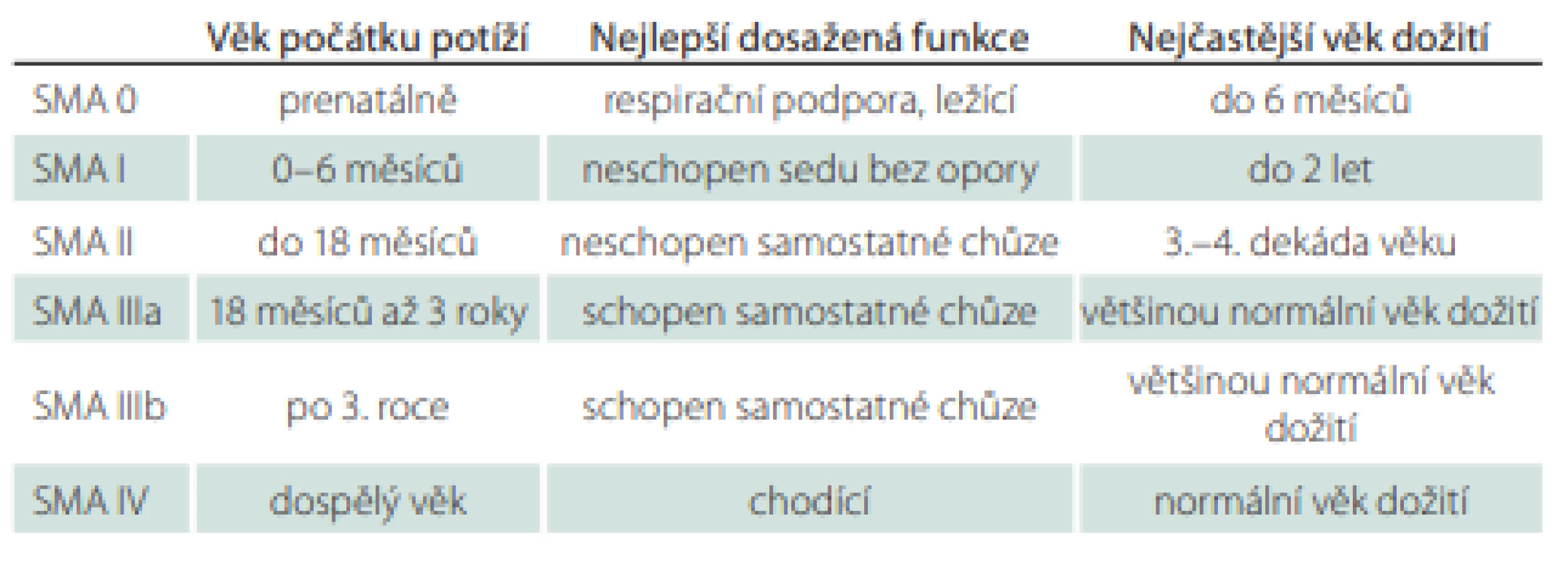
Třebaže téměř 25 % pacientů nelze přesně klasifikovat, tato klasifikace zůstává relevantní v genetické éře a poskytuje užitečné klinické a prognostické informace [3].
Klinická specifika typů SMA
SMA typ I – m. Werdnig-Hoffmann, infantilní SMA, je těžkou a nejčastější klinickou formou. Vyskytuje se přibližně u poloviny pacientů se SMA. Obtíže jsou patrné již při narození nebo se vyvíjejí do 6 měsíců věku.
V klinickém nálezu dominuje relativně rychle progredující hypotonie se svalovou slabostí, která vede k opoždění motorického vývoje a poruše držení hlavy při slabosti šíjového svalstva. Svalová slabost je symetrická, více vyjádřená na dolních končetinách a na proximálních svalech končetin. Děti nikdy nejsou schopny samostatného sedu. Šlachosvalové reflexy jsou nevýbavné. Postižené svaly podléhají atrofii, která bývá maskována vrstvou podkožního tuku. Děti mají slabý křik a kašel. Fenotyp dítěte se označuje jako floppy infant a je projevem časné generalizované hypotonie s typickou hyperabdukcí v kyčlích (obr. 1, 2). Na jazyku pozorujeme atrofii a fascikulace. Průběh je progresivní s prohlubováním motorického deficitu. Většinou zůstává částečně zachovalá motorika aker horních končetin. Díky tomuto je pacient často schopen ovládat elektrický vozík a PC. U dětí je nápadná diskrepance mezi chabou motorikou a dobrým sociálním kontaktem [1,2,5].
Fig. 1. Severe hypotonia in an infant with SMA I
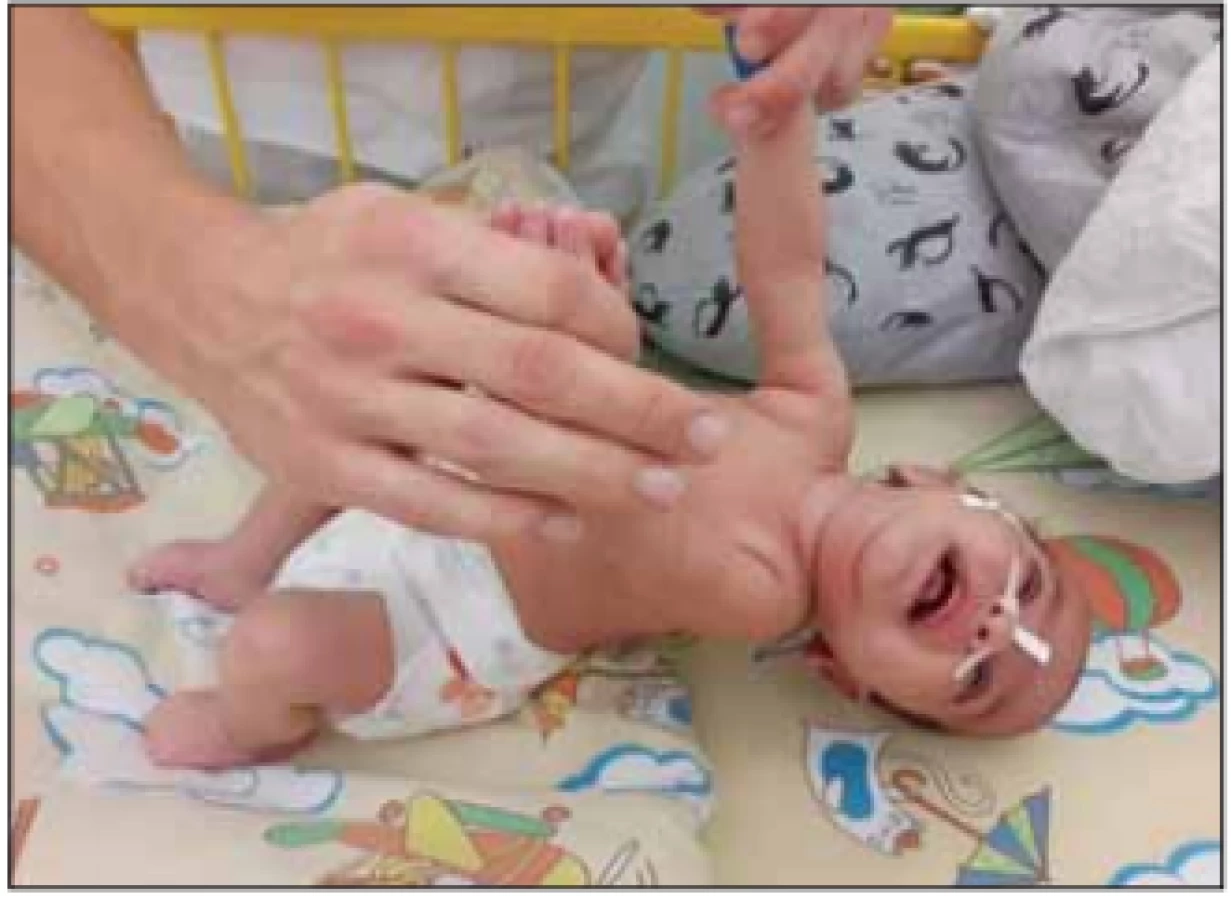
Fig. 2. A floppy infant – SMA I.
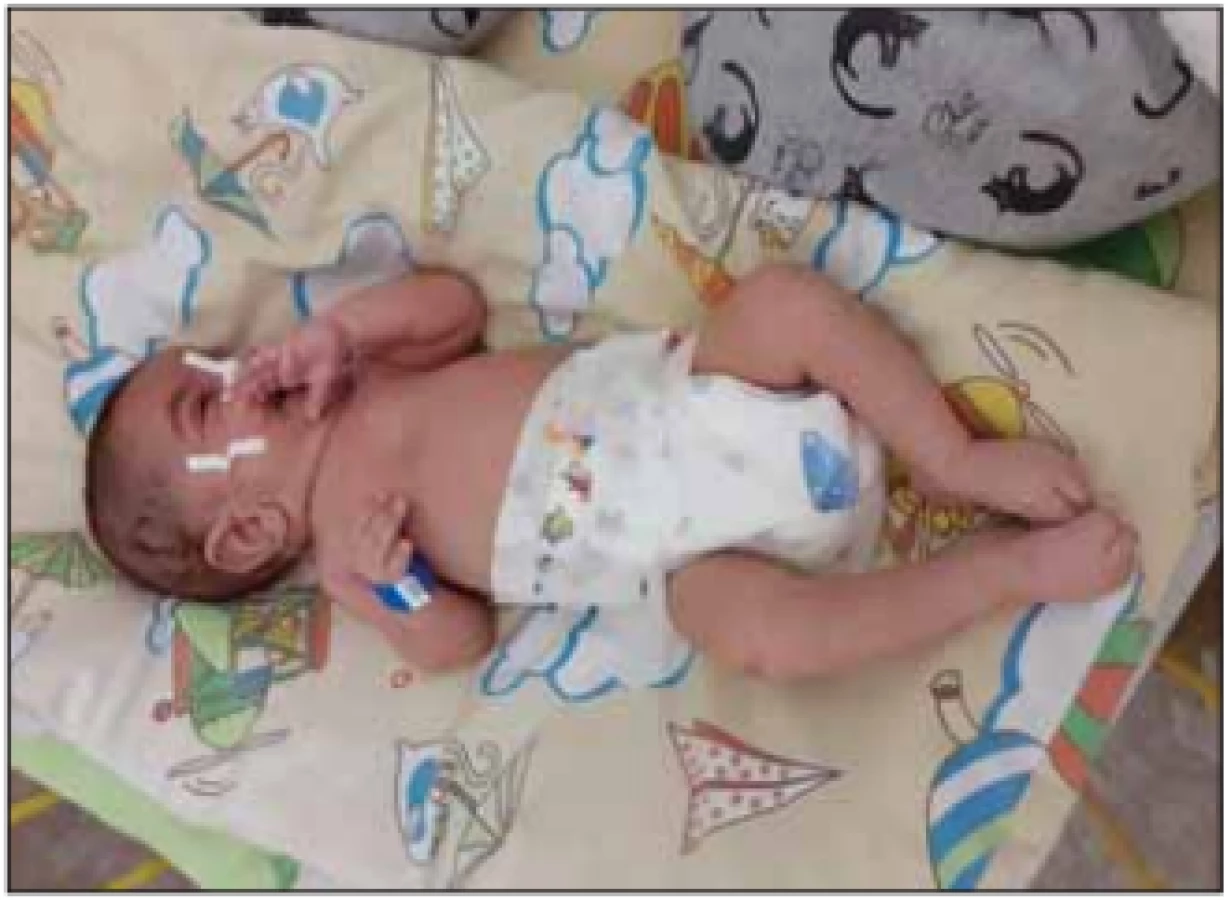
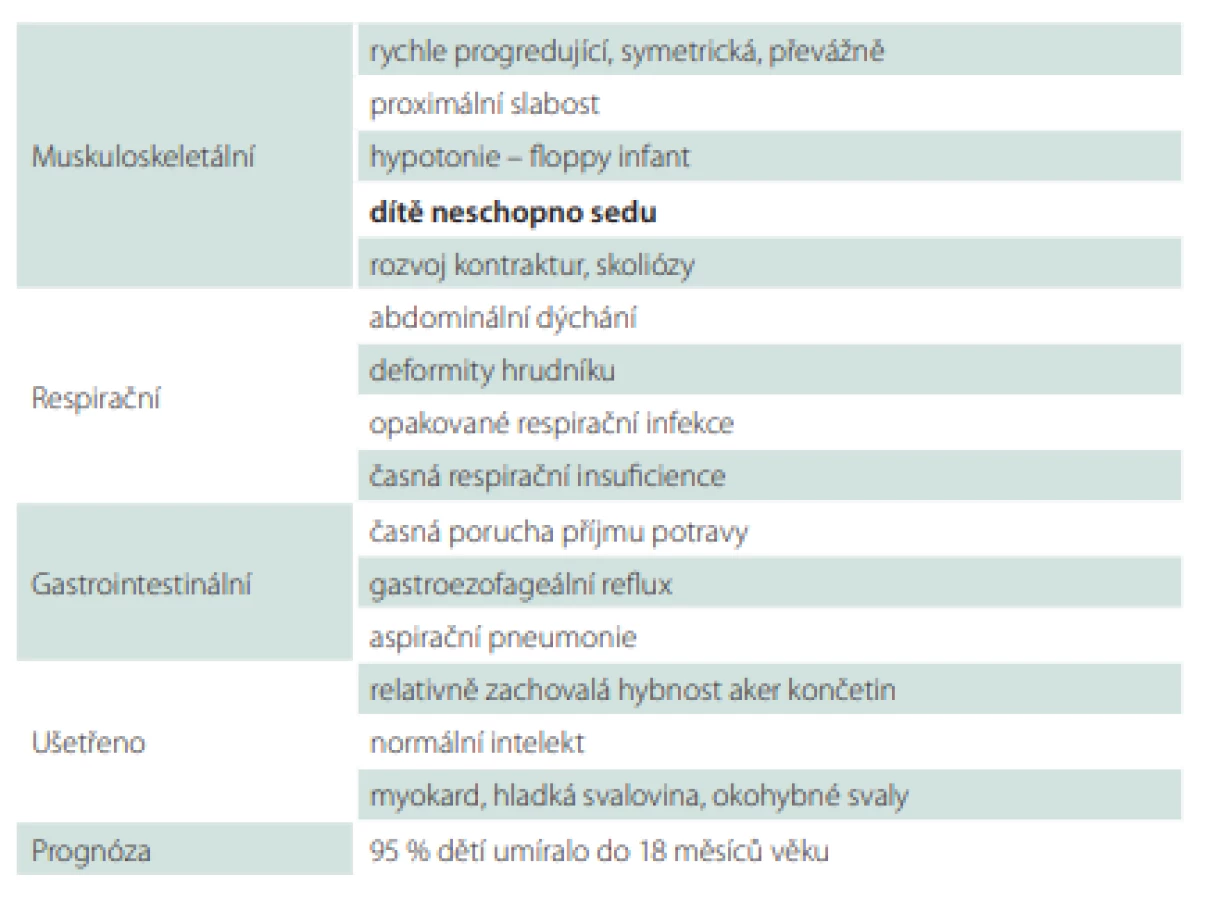
Slabost interkostálních svalů při relativním ušetření bránice vede k typickému bráničnímu paradoxnímu „abdominálnímu“ dýchání s břišní protruzí a k deformitám hrudníku. Efektivitu ventilace výrazně ovlivňuje také skolióza, porucha polykání a gastroezofageální reflux (GER). Slabost inspiratorních i exspiratorních svalů vede k hypoventilaci nejdříve ve spánku, slabému kašli s nedostatečnou expektorací a k poruše vývoje stěny hrudníku a plic s nízkou kapacitou plic. Noční hypoventilace s hyposaturací a hyperkapnií progreduje do denní respirační insuficience, kterou dále zhoršují plicní infekce (obr. 3). Opakované respirační infekce zhoršují povšechnou svalovou slabost.
Fig. 3. A 5-year old girl (SMA I), ventilation necessary from 3 years
of age.

Děti představují zvýšené riziko postanestetických komplikací, které vedou k prodloužené intubaci, nozokomiálním infekcím, atelektázám, nutnosti tracheostomie a případně smrti [4].
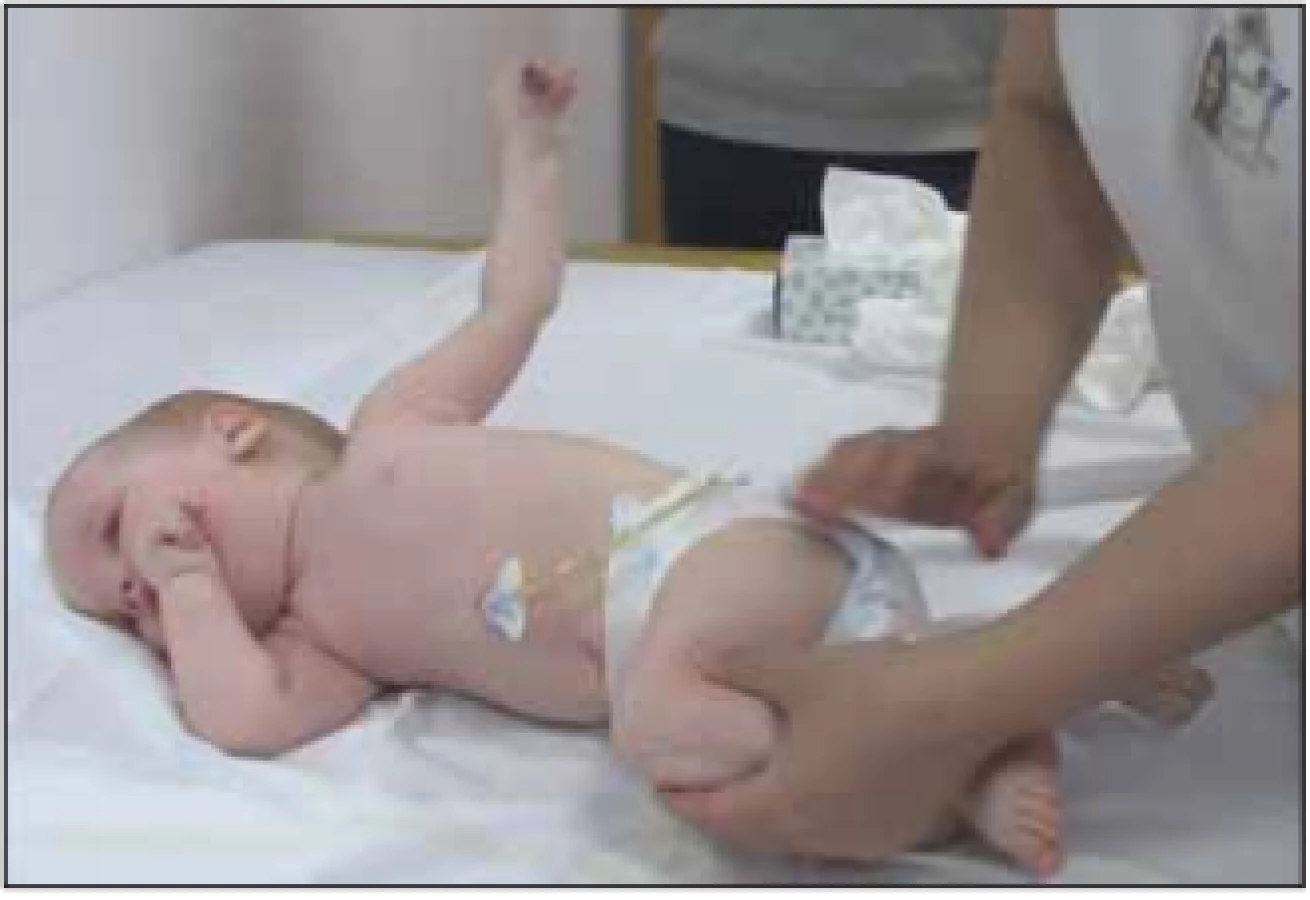
Ke konci prvního roku života se u většiny dětí objevují potíže s příjmem potravy. Je nutné sondování, zavedení perkutánní gastrostomie (obr. 4). Bulbární dysfunkce u pacientů s těžkou slabostí vede k poruše kousání, žvýkání, polykání a aspiračním pneumoniím. Snížený rozsah pohybů mandibuly omezuje otevírání úst a vede k prodloužení doby jídla. Slabá stabilita hlavy je rovněž faktorem ovlivňujícím příjem stravy. Gastrointestinální problémy zahrnují také obstipaci, opožděné vyprazdňování žaludku a GER. Bulbární potíže krmení výrazně prodlužují, zvýrazňují únavu a vedou k dušení a kašli během krmení. Opakované pneumonie jsou možným indikátorem aspirací.
Fig. 4. A 13-month old child (SMA I), therapy with nusinersen
from 3 months of age, PEG from 1 year of age.
Gastroezofageální reflux je významným faktorem mortality a morbidity dětí se SMA. Může být spojen s aspirací a život ohrožujícími událostmi. Některé děti mohou odmítat jídlo pro obtížné polykání s následnou poruchou výživy. Zpomalená peristaltika vede k obstipaci a vzedmutí břicha. Pacienti mají sklon k hypoglykémii a katabolizmu. Častá je porucha růstu [4].
Myokard a hladká svalovina jsou obvykle intaktní. Ušetřeny jsou okohybné svaly. Není přítomna mentální retardace a sfinkterová porucha. Postižení senzitivních vláken nervů a porucha čití nejsou přítomny [4,5].
Častá je indikace operačního řešení progredující skoliózy nejdříve v předškolním věku, výjimečně v batolecím věku. Progredují kloubní kontraktury. Většina dětí má zachovalou schopnost řeči. Pacienti mají normální intelekt, mnozí jsou schopni studia a alespoň částečného pracovního zařazení a ekonomické soběstačnosti. V ČR je to spíše výjimečné na rozdíl od pacientů v USA a západní Evropě [1,2]. U malé části pacientů především se SMA typu 0 a I se mohou vyskytnout postižení dalších orgánů jako mozku, srdce, cév a pankreatu s poruchou glukózového metabolizmu [5]. Hladina kreatinkinázy (CK) je většinou normální, ale může být i lehce zvýšena [5].
Celkem 95 % dětí bez ventilační podpory umíralo do 18 měsíců věku. Respirační insuficience a infekce jsou nejčastější příčinou úmrtí [2]. Tab. 2 shrnuje hlavní znaky SMA I.
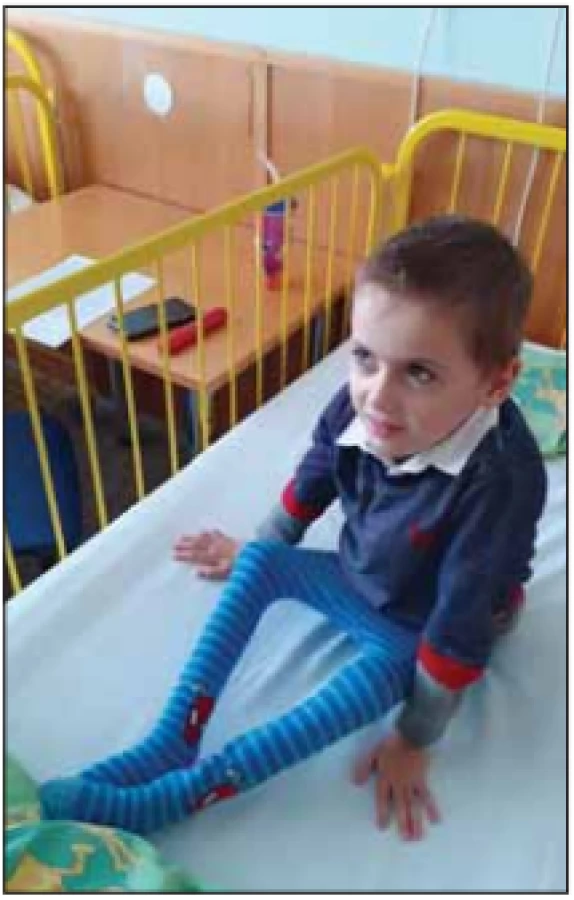
SMA typ II – intermediální a druhá nejčastější forma SMA. Děti se během prvních 6 měsíců života vyvíjejí většinou normálně. Rozvoj onemocnění nastává do 18. měsíce věku se zástavou a následnou regresí motorického vývoje. Svalová slabost je symetrická a více vyjádřená na proximálních svalech dolních končetin. Děti jsou hypotonické s hyporeflexií až areflexií šlachosvalových reflexů. Jsou schopny sedu bez opory, nikdy nejsou schopny samostatné chůze (obr. 5). Některé děti jsou schopny stoje ve stojanu nebo s ortézami. Je přítomné vysoké riziko rozvoje kloubních kontraktur a kyfoskoliózy (obr. 6). Hypertrofie lýtkových svalů není obvyklá. Typický je jemný třes aker horních končetin. Sérová CK může být lehce zvýšená [5]. Potíže s příjmem potravy jsou málo časté [3], ale někdy vedou k neprospívání dětí (obr. 7). U jiných dětí je obezita při nízkém energetickém výdeji [4]. Někteří pacienti mají rovněž brániční typ dýchání. Pacienti mají sníženou efektivitu kašle a riziko zahlenění dýchacích cest [4]. S postupujícím věkem vzrůstá riziko respirační insuficience s nutností podpory ventilace. Délka života je zkrácená. Celkem 70 % pacientů přežívá 25 let [4]. Pacienti se většinou dožívají 4. dekády [7]. Tab. 3 shrnuje hlavní znaky SMA II.
Fig. 5. A child with SMA II

Fig. 6. A 17-year old girl with SMA II. Severe dextroscoliosis of
Th-L spine with vertebral rotation and smaller sinistroscoliosis
of the cranial part of the thoracic spine with vertebral rotation.
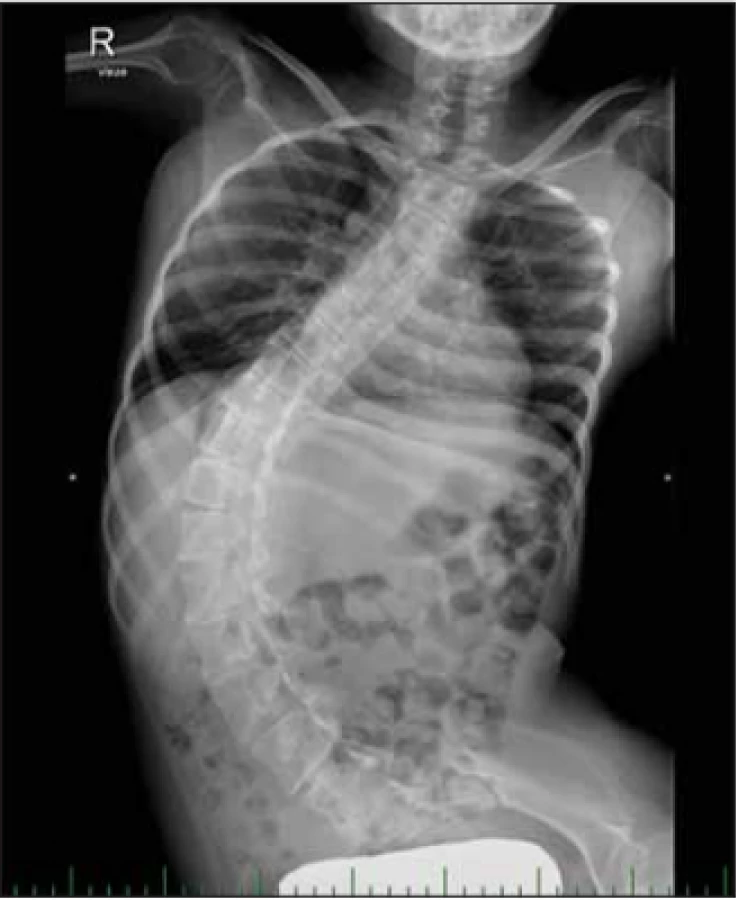
Fig. 7. A boy with SMA II and malnutrition.

SMA typ III – m. Kugelberg-Welander, juvenilní mírná forma s přežitím do dospělého věku. Z dětských forem SMA je nejméně častá. Klinicky se první potíže objevují po 18. měsíci věku. Tíže fenotypu je velice variabilní. Čím dříve se nemoc projeví, tím těžší je fenotyp. Děti jsou schopny samostatného stoje a chůze (obr. 8). Vedoucím počátečním příznakem je porucha chůze při symetrickém oslabení proximálních svalů dolních končetin. Často je přítomna pseudohypertrofie lýtkových svalů. Fenotyp onemocnění připomíná myopatii. U některých pacientů může být lehká elevace CK [5]. Častý je třes aker horních končetin. Progrese onemocnění je pomalá. V pozdějších stadiích jsou přítomny kontraktury zejména lýtek a skolióza. U některých pacientů se slabost generalizuje se ztrátou schopnosti samostatné chůze již během dětství. Respirační insuficience s noční hypoventilací a bulbární symptomatologie jsou vzácné. Obezita je častá při nízkém energetickém výdeji [4]. Časté jsou bolesti svalů a kloubní hypermobilita. Délka života většinou není zkrácena [3,7].
Fig. 8. Sisters (twins) with SMA III.

Tab. 4 shrnuje hlavní znaky SMA III.

SMA typ 0 – tzv. fetální forma, v novější klasifikaci odlišena od typu I, je nejtěžší formou onemocnění s počátkem slabosti již prenatálně se slabými pohyby plodu nebo jejich absencí. Při porodu je patrna těžká generalizovaná hypotonie, velmi chudá spontánní hybnost a mnohočetné kontraktury. Brzy po porodu nastává respirační insuficience. Bez umělé plicní ventilace většina dětí umírá v prvních týdnech až měsících života [2,3].
SMA typ IV má začátek v dospělosti a je předmětem další části supplementa.
Popsaný přirozený klinický obraz a vývoj pacientů se SMA se významně mění s rozvojem multioborové péče a inovativní terapie. Více než 100 let od prvního popisu nemoci terapie zahrnovala hlavně podpůrnou a paliativní péči. Během posledních dekád se v souvislosti s proaktivní klinickou péčí a zejména neinvazivní ventilací výrazně zlepšily možnosti řešení respiračních, nutričních, ortopedických a dalších problémů. Tento multioborový přístup, možnosti symptomatické péče, zavedení terapie ovlivňující splicing SMN2 genu a genová terapie významným způsobem mění přirozený průběh onemocnění, zlepšují předpokládané dožití a kvalitu života dětských pacientů se SMA [1,8,9].
Non-5q varianty SMA
Mimo uvedené typy SMA s vazbou na mutace SMN genu existují tzv. varianty SMA (non-5q SMA). Jedná se o klinicky a geneticky heterogenní skupinu nemocí s různými typy dědičnosti. Mimo časnou denervaci, svalovou slabost a atrofie jsou přítomny i další abnormity jako artrogrypóza, růstová retardace, parézy bránice s časným respiračním selháním, pontocerebelární degenerace a další. Slabost bývá častěji distální [1,2,10]. Popis této skupiny přesahuje rámec sdělení.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
MUDr. Jan Staněk
Oddělení dětské neurologie
FN Ostrava
17. listopadu 1790
708 52 Ostrava – Poruba
e-mail: jan.stanek@fno.cz
Zdroje
1. Oskoui M, Levy G, Garland CJ et al. The changing natural history of spinal muscular atrophy type 1. Neurology 2007; 69(20): 1931–1936. doi: 10.1212/ 01.wnl.0000290830.40544.b9.
2. Haberlová J, Slabá A, Hedvičakova P et al. Spinální svalové atrofie – diagnostika, léčba, výzkum. Neurol praxi 2016; 17(6): 349–353.
3. Kolb SJ, Kissel JT. Spinal muscular atrophy a timely review. Arch Neurol 2011; 68(8): 979–984. doi: 10.1001/ archneurol.2011.74.
4. Wang CH, Finkel RS, Bertini ES et al. Consensus Stateman for standard of care in spinal muscular atrophy. J Child Neurol 2007; 22(8): 1027–1049. doi: 10.1177/ 08830 73807305788.
5. Sarnat BH, Menkes JH. Onemocnění motorické jednotky. In: Menkes JH, Sarnat HB, Maria BL. Dětská neurologie. 7th ed. Praha: Triton 2011: 1545–1553.
6. Finkel RS, Mercuri E, Meyer OH et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscular Disorders 2018; 28(3): 197–207. doi: 10.1016/ j.nmd.2017.11.004.
7. Ferrar MA, Vucic S, Johnston HM et al. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr 2013; 162(1): 155–159. doi: 10.1016/ j.jpeds.2012.05.067.
8. Markowitz JA, Singh PP, Darras BT. Spinal muscular atrophy: a clinical and research update. Pediatr Neurol 2012; 46(1): 1–12. doi: 10.1016/ j.pediatrneurol.2011.09.001.
9. Darras BT. Spinal muscular atrophies. Pediatr Clin North Am 2015; 62(3): 743–766. doi: 1016/ j.pcl.2015.03.010.
10. Tomek A, Maťoška V, Goetz P et al. Molekulární etiopatogeneze spinální muskulární atrofie. Cesk Slov Neurol N 2002; 65(5): 313–320.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2020 Číslo Supplementum 2
Nejčtenější v tomto čísle
- Klinický obraz spinální muskulární atrofie u dospělých pacientů
- Genetika spinální muskulární atrofie
- Léčba spinální svalové atrofie
- Rehabilitace u spinální muskulární atrofie