
Genetika spinální muskulární atrofie
Genetics of spinal muscular atrophy
Spinal muscular atrophy (SMA) is a severe genetic disorder. The most frequent form of SMA is inherited in an autosomal recessive mode, caused by a homozygous deletion of a part of the SMN1 gene on the long arm of chromosome 5. Having frequency of carriers in Caucasian population about 1 : 40, it is the second most common autosomal recessive disease of childhood. The causal SMN1 gene was found in 1995. Since then, investigation of the number of copies of this gene has been performed by different techniques, the gold standard now being a multiplex ligation-dependent probe amplification. To predict the development of the disease, it is crucial to determine the number of SMN2 gene (so called pseudogene) copies, too. The patient’s phenotype is also impacted by other modification factors. Clinical genetics offers counselling to the families and, in relevant cases, recommends carrier testing. With the arrival of treatment possibilities in early stages of the disease, the prompt and early diagnostics has become essential.
Keywords:
spinal muscular atrophy – SMN1 gene – SMN2 gene – copy numbers – homozygous deletion – multiplex ligation-dependent probe amplification – carriers
Autoři:
P. Hedvičáková
Působiště autorů:
Ústav biologie a lékařské genetiky 2. LF UK a FN Motol
Vyšlo v časopise:
Cesk Slov Neurol N 2020; 83/116(Supplementum 2): 17-20
doi:
https://doi.org/10.48095/cccsnn20202S17
Souhrn
Spinální muskulární atrofie (SMA) je závažné genetické onemocnění. Nejčastější forma SMA je autozomálně recesivně děděná, způsobená homozygotní delecí části genu SMN1 na dlouhém raménku pátého chromozomu. S frekvencí přenašečů v kavkazské populaci přibližně 1 : 40 je to druhá nejčastější autozomálně recesivní dětská choroba. Kauzální gen SMN1 byl popsán v roce 1995. Vyšetřování počtu kopií tohoto genu je od té doby možné provádět různými technikami, zlatým standardem je dnes metoda multiplexní na ligaci závislé amplifikace sond. Pro predikci vývoje onemocnění je zásadní také počet kopií genu SMN2, tzv. pseudogenu. Fenotyp pacientů ovlivňují i další modifikátory. Genetické ambulance poskytují rodinám poradenství a genetickou konzultaci, v relevantních případech doporučují vyšetření přenašečství. S příchodem možností léčby při časném nasazení je rychlost a včasnost diagnostiky zásadní.
Klíčová slova:
spinální muskulární atrofie – gen SMN1 – gen SMN2 – počet kopií – homozygotní delece – multiplexní na ligaci závislá amplifikace sond – přenašeči
Spinální muskulární atrofie (SMA) je onemocnění heterogenní nejen klinicky, ale i geneticky. Do skupiny SMA patří nejčastější proximální forma SMA, distální forma SMA, progresivní bulbární obrna dětského věku, skapulo-peroneální forma SMA a facio-skapulo-humerální forma SMA.
S příchodem technik sekvenování nové generace se zrychlilo objevování nových genů, jen mezi lety 2011–2014 bylo popsáno 13 nových genů SMA. Patofyziologické jevy související se SMA zahrnují poruchy metabolizmu a sestřihu RNA, axonálního transportu, vývoje motoneuronů a konektivity [1].
Ve většině případů (cca 95 %) se jedná o SMA autozomálně recesivní (AR), způsobenou mutacemi v genu SMN1 (survival of motor neurons). Je to druhá nejčastější letální AR dětská choroba po cystické fibróze, s incidencí 1 : 6 000–10 000 živě narozených a frekvencí heterozygotů 1 : 40 až 1 : 60.
Historie genetiky SMA se začala psát v roce 1995, kdy Suzie Lefebvre et al v Cell [2] popsali metodami vazebné analýzy a kombinací genetického a fyzikálního mapování 2 kandidátní geny na dlouhém raménku pátého chromozomu v oblasti 5q13. Přesněji řečeno popsali 500 kilobází (kb) dlouhou invertovanou duplikaci a zpřesnili kritickou oblast na 140 kb v její telomerické části. Dvacet kb dlouhý gen v této oblasti chyběl nebo byl porušený u velké většiny jejich pacientů. V duplikované centromerické části detekovali přítomnost vysoce homologního genu, dnes označovaného jako pseudogen nebo SMN2 gen.
Sekvenačními technikami zjistili, že gen SMN má osm exonů. O rok později Burglen et al [3] podrobnější charakterizací genu ukázali, že jde ve skutečnosti o exonů 9, a ve snaze zabránit nedorozuměním rozdělili exon 2 na 2a a 2b. Většina prací proto dnes označuje jako zásadní pro vznik onemocnění přítomnost nebo nepřítomnost exonu 7 z původního značení (LRG_676t1 naopak označuje jako kauzální změnu v exonu 8, při celkovém počtu devíti exonů). V tradičním značení není exon 8 překládán, stop kodon se nachází v sedmém exonu.
Duplikovaná 500kb oblast vznikla v pozdějším evolučním období u primátů, ale dva rozdílné SMN geny má pouze člověk [4] (obr. 1).
Fig. 1. Inverted duplication region with SMN1 and SMN2 genes on 5q chromosome [29].
![Oblast invertované duplikace s geny SMN1 a SMN2 na 5q chromozomu [29].<br>
Fig. 1. Inverted duplication region with SMN1 and SMN2 genes on 5q chromosome [29].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/b464a5382db7bb6597b22d907bef6a23.png)
Telomerický a centromerický SMN gen se liší v pěti nukleotidech. Pro vznik onemocnění je podstatná translačně tichá záměna v nukleotidu 873, v šestém nukleotidu exonu 7, v kodonu 280 (C > T). Analýza RNA odhalila, že v centromerické kopii genu dochází k odlišnému sestřihu exonu 7 (obr. 2).
Fig. 2. Different splicing of SMN1 and SMN2 [29].
![Rozdílný sestřih SMN1 a SMN2 [29].<br>
Fig. 2. Different splicing of SMN1 and SMN2 [29].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/aede16fbc9e4fff3df4ce6f330cdb402.png)
Až 90 % pre-mRNA genu SMN1 je sestřiženo na SMN protein s úplnou délkou, zatímco 80 % pre-mRNA genu SMN2 je sestřiženo na SMN protein bez exonu 7. SMN bez exonu 7 je nestabilní a je rychle degradován.
Modifikátory fenotypu [5] ovlivňují expresi a stabilitu RNA nebo proteinu, mohou to být faktory působící v cis nebo trans, epigenetické faktory, proteiny téže dráhy, jiné proteiny a negenetické faktory nebo faktory okolního prostředí (nedostatečná výživa, hypoxie). Jejich výzkum vede k vývoji cílené terapie [6].
Hlavním modifikátorem závažnosti SMA je SMN2 gen, a proto je také primárním cílem terapie. Mutace v SMN2 mohou mít vliv na cis regulační prvky a zvyšovat nebo snižovat hladiny transkriptů o plné délce. Jako protektivní modifikátor SMA působí plastin 3 [7].
Závažnost fenotypu u pacientů s homozygotní delecí závisí na počtu kopií pseudogenu, na tom, zda je příčinou postižení skutečná delece, nebo konverze telomerické verze v centromerickou (obr. 3).
Fig. 3. SMA types based on the way of
disabling of functional SMN1 gene [29].
![Typy SMA podle způsobu vyřazení
funkčního genu SMN1 [29].<br>
Fig. 3. SMA types based on the way of
disabling of functional SMN1 gene [29].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/c1c8e82779abe8c063980e7cf6a90aee.png)
SMA – spinal muscular atrophy
Korelace počtu kopií pseudogenu s mírou postižení však není absolutní, roli hrají další výše zmíněné faktory, jak genetické, tak faktory prostředí. Fenotyp závažnější, než by se očekávalo u pacientů se 3 kopiemi SMN2, je možné vysvětlit hypermetylací DNA, v jejímž důsledku je částečně inaktivována jedna nebo více kopií genu SMN2 [8]. Predikce vývoje na základě počtu kopií SMN2 je tak obtížná zejména u pacientů se třemi kopiemi, které se vyskytují u všech typů závažnosti onemocnění. Snaha o nalezení biologických markerů je proto stále velkou výzvou. Ukazuje se, že přítomnost varianty c.859G>C v SMN2 vede k lepší prognóze u pacientů se dvěma kopiemi SMN2, který v tomto případě produkuje více transkriptů o plné délce. Nevysvětluje však všechny případy [9].
Diskrepance mohou vysvětlovat také další modifikující geny (jak pozitivně, tak negativně). V neposlední řadě výsledky takových průzkumů ovlivňují vyšetřovací metody, kvalita vyšetřovaných vzorků DNA, přítomnost jednonukleotidových polymorfismů apod. Navíc současné techniky umožňují velmi přesné stanovení počtu kopií 1–3, ale při 4 a více kopiích je rozlišení výrazně méně přesné. Calucho et al [10] provedli studii na velkém počtu pacientů různých etnik, kterou připojili k datům z databází (celkem 3 459 pacientů); na základě výsledků se domnívají, že predikovat vývoj onemocnění podle počtu kopií SMN2 je možné, přihlédne-li se k důkladnému klinickému vyšetření a stanovení fenotypu. U všech jedinců se 4 a více kopiemi se předpokládá, že budou typu II–IV, tedy chodící. Většina případů s jednou a dvěma kopiemi rozvine fenotyp SMA I, tedy nejzávažnější formu postižení. Více než polovina pacientů se třemi kopiemi pak bude mít SMA typu II.
Část pacientů má deletovaný pouze exon 7, exon 8 je přítomen. Je to způsobeno výše zmíněnou konverzí mezi geny SMN1 a SMN2, vznikem chimérického genu s exonem 7 SMN2 genu a exonem 8 SMN1 genu [11]. V obecné populaci je vyšší počet kopií SMN1 spojen s nižším počtem kopií SMN2, dochází tedy také ke konverzi SMN2 na SMN1 [12]. Přítomnost dvou kopií SMN1 na jednom chromozomu je možné v jiném scénáři vysvětlit jako důsledek nerovnoměrného crossing-overu v oblasti, kde se vyskytují repetice, a je tedy náchylná k rekombinacím.
Ke stanovení počtu kopií SMN1 se používá zjištěný počet kopií exonu 7. V případě chimérického genu s chybějícím exonem 8 SMN1 stále dochází k tvorbě funkčního proteinu. Na světě je popsáno několik málo případů, kdy se za důvod SMA považuje izolovaná homozygotní delece exonu 8 SMN1 genu, původně spojovaná s mírnější formou onemocnění [13]. Později byl publikován i případ SMA typu I s bilaterální atrofií optiku, způsobený izolovanou homozygotní delecí exonu 8 [14].
Delece pseudogenu SMN2 se vyskytuje i u zdravé populace, v literatuře je však popsán i jeden případ, kde je považována za příčinu SMA [15]. Jednalo se o SMA se slabostí spíše distálních svalů; obecně se o deleci SMN2 mluví zejména v možné souvislosti se syndromem distálního motorického neuronu [16]. Delece SMN2 genu je popisována u 5–9 % zdravé populace.
Počet kopií SMN2 nehraje žádnou roli, resp. nepřináší žádnou informaci relevantní pro stanovení přenašečství [17].
Homozygotní delece SMN1 i SMN2 je považována za embryonálně letální [18].
Někteří autoři také došli k názoru, že počty kopií nejen SMN2, ale také sousedního genu NAIP souvisejí s věkem nástupu nemoci [19].
Asi v 5 % se u pacientů vyskytuje kombinace delece na jedné alele a bodové mutace na alele druhé, pacienti jsou složenými heterozygoty se stejným způsobem AR dědičnosti [20]. Za 2 % případů odpovídá vznik mutace de novo, kdy jeden z rodičů není přenašečem [21].
Některé mutace se vyskytují opakovaně, např. duplikace 11 nukleotidů c.770_780 [22].
Molekulárně genetická diagnostika – metody
Na základě rozdílů v sekvenci SMN1 a SMN2 je možné je rozlišit. Toho se využívá v diagnostice SMA.
V současnosti je zlatým standardem vyšetření delece exonů 7 a 8 genu SMN1 metodou MLPA (Multiplex Ligation-dependent Probe Amplification) (obr. 4). Senzitivita metody je 100 %, specificita > 99 % (nepokrývá bodové mutace). Vyšetření se provádí z DNA pacienta izolované z leukocytů periferní krve, případně u prenatálního vyšetření z buněk nativní plodové vody, nativních choriových klků, kultivovaných choriových klků nebo kultivovaných buněk plodové vody.
Fig. 4. MLPA analysis using Coff alyser.net software; a patient with a homozygous deletion of exons 7 and 8 of the SMN1 gene and three copies of the SMN2 gen; MLPA kit
P060.
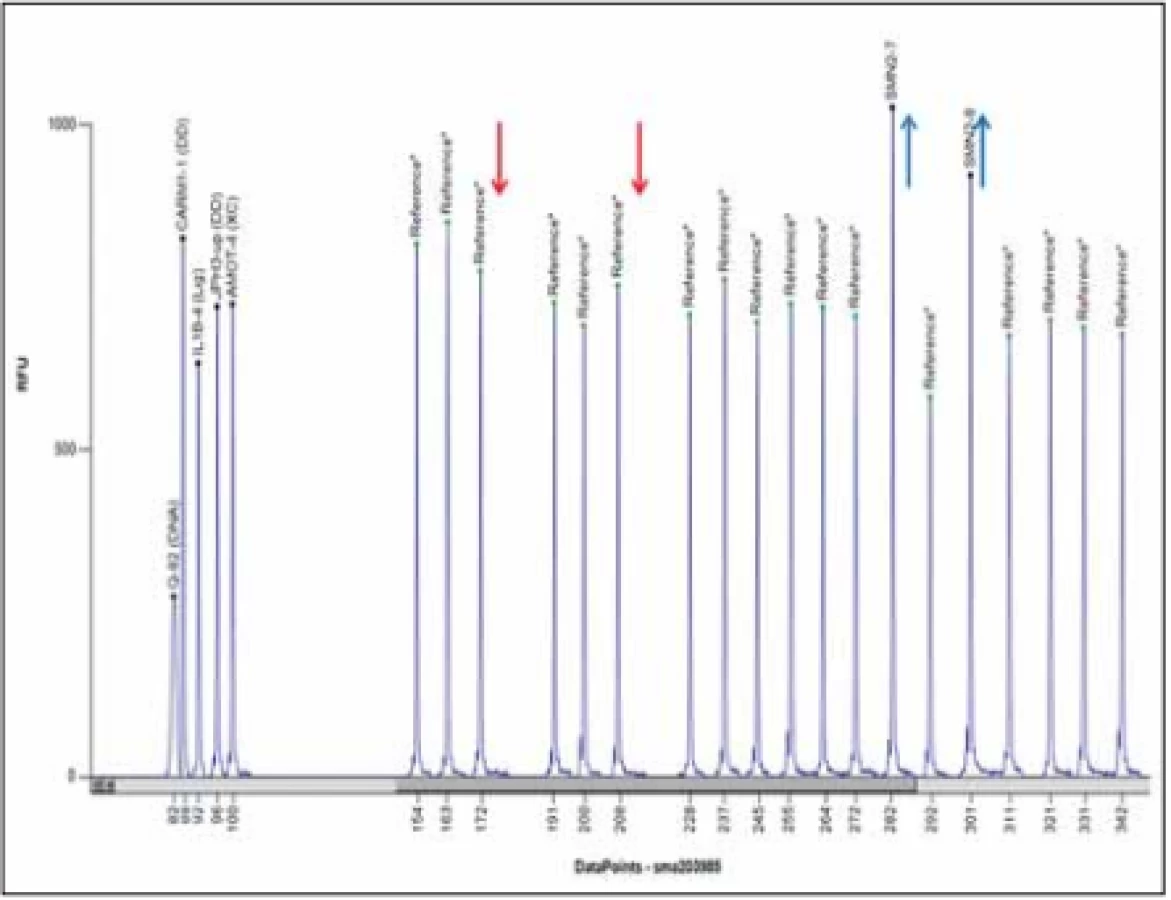
Rodinám pacientů, u nichž se potvrdí homozygotní delece exonů 7 a 8 nebo jenom exonu 7, je vhodné doporučit genetickou konzultaci. Klinický genetik poskytne genetické poradenství, doporučí vyšetření relevantních příbuzných na přenašečství.
Přenašeči heterozygotní delece exonů 7 a 8 mají 50% riziko předání této delece potomkům. Dva partneři-přenašeči tedy mají 25% riziko, že se jim narodí dítě postižené SMA. Potvrzeným přenašečům proto genetik doporučí vyšetření partnera. Pokud je jeden z partnerů přenašeč delece a u druhého se zjistí přítomnost dvou kopií SMN1, mají riziko postižení dítěte 1 : 2 035, vč. započítání de novo mutační frekvence [23].
Zjistí-li se (po vyloučení možnosti nonpaternity) u některého z rodičů pacienta s homozygotní delecí SMN1, že má 2 kopie SMN1, jedná se s nejvyšší pravděpodobností o případ maskovaného přenašečství, kdy obě kopie SMN1 jsou na jednom chromozomu, zatímco na druhém je delece. Je pak velmi vhodné vyšetřit celou rodinu. Většinou se dohledá příbuzný se 3 kopiemi (některý z prarodičů nebo sourozenců postiženého dítěte). Poradenství v takových rodinách pak musí upozornit na možnost, že děti jedinců se třemi kopiemi budou vystaveny riziku, že se u nich kvantitativními metodami případné přenašečství nerozezná [24].
V testování přenašečství je třeba mít na paměti, že celá 2 % případů SMA vznikají v důsledku de novo mutací; je to kvůli již zmíněné přítomnosti repetic, a tedy častých nerovnoměrných crossing-overů a rekombinačních událostí v oblasti. K de novo mutacím dochází zejména během paternální meiózy. V populaci není výskyt 3 kopií SMN1 u jedince vzácný – jde asi o 5 % normální populace. Vyšetření přenašečství SMA se stále častěji stává součástí prekoncepční péče. V rodinách, kde se SMA vyskytlo, je klinickým genetikem nabízeno rutinně. SMA zcela splňuje kritéria pro populační skríning přenašečství, neboť je onemocněním klinicky závažným, s vysokou frekvencí přenašečů, s dostupným testem o vysoké citlivosti i specifičnosti, s možností prenatální diagnostiky a s možností genetického poradenství [25]. Pro přenašeče je dostupné další genetické sledování a mají k dispozici prenatální a preimplantační diagnostiku. Ani negativní skríning však z výše uvedených důvodů nevyloučí přenašečství na 100 %. Důležité je také vědomí, že testování je dobrovolné a zakládá se na informovaném souhlasu a důkladném poučení o tom, co jeho výsledky mohou zjistit.
Četnost výskytu přenašečů je různá u různých etnik. Pro kavkazskou populaci v severní Americe se uvádí 2,7 %, pro afroamerickou 1,1 % a pro hispánskou dokonce 0,8 %. Naopak vysoká frekvence alel s více kopiemi SMN1 je v afroamerické populaci – 27 % ve srovnání s 3,3–8,1 % [26].
V poradenství se setkáváme nejčastěji s případem zdravého jedince bez rodinné anamnézy, jehož partner/ ka naopak SMA v rodinné anamnéze má. Takový jedinec má nejčastěji 2 kopie SMN1 genu. Podle výpočtu frekvence alel, který zahrnuje i jedince s heterozygotní bodovou mutací, je frekvence nosičů se dvěma kopiemi SMN1 genu přibližně 5,5 % [27].
Souhrnné údaje z roku 2017 [28] uvádějí pro kavkazskou populaci frekvenci přenašečů s jednou kopií SMN1 genu 2,2 %, přenašečů se dvěma kopiemi SMN1 (na jednom chromozomu) je v kavkazské populaci 0,13 %, přičemž frekvence alel se dvěma kopiemi je 3,5 %. Incidence se odhaduje na 1 z 7 829.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
RNDr. Petra Hedvičáková
Ústav biologie a lékařské genetiky
2. LF UK a FN Motol
V Úvalu 84/1
150 06 Praha
e-mail. petra.hedvicakova@fnmotol.cz
Zdroje
1. Farrar MA, Kiernan MC. The genetics of spinal muscular atrophy: progress and challenges. Neurotherapeutics 2015; 12(2): 290–302. doi: 10.1007/ s13311-014-0314-x.
2. Lefebvre S, Bürglen L, Reboullet S et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80(1): 155–165. doi: 10.1016/ 0092-8674(95)90460-3.
3. Bürglen L, Lefebvre S, Clermon O et al. Structure and organization of the human survival motor neurone (SMN) gene. Genomics 1996; 32(3): 479–482. doi: 10.1006/ geno.1996.0147.
4. Rochette CF, Gilbert N, Simard LR. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum Genet 2001; 108(3): 255–266. doi: 10.1007/ s004390100473.
5. Wirth B, Garbes L, Riessland M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr Opin Genet Dev 2013; 23(3): 330–338. doi: 10.1016/ j.gde.2013.03.003.
6. Vondráček P, Zapletalová E, Ošlejšková H et al. Ovlivnění exprese mRNA genu SMN2 inhibitory histonových deacetyláz a jejich vliv na fenotyp spinální svalové atrofie I. a II. typu. Ceskl Slov Neurol N 2007; 70/ 103(4): 413–418.
7. Oprea GE, Kröber S, McWhorter ML et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008; 320(5875): 524–527. doi: 10.1126/ science.1155085.
8. Hauke J, Riessland M, Lunka S et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet 2009; 18(2): 304–317. doi: 10.1093/ hmg/ ddn357.
9. Bernal S, Alías L, Barceló MJ et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J Med Genet 2010; 47(9): 640–642. doi: 10.1136/ jmg.2010.079004.
10. Calucho M, Bernal S, Alías L et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord 2018; 28(3): 208–215. doi: 10.1016/ j.nmd.2018.01.003.
11. Van Der Steege G, Grootscholten PM, Cobben JM et al. Apparent gene conversions involving the SMN gene in the region of the spinal muscular atrophy locus on chromosome 5. Am J Hum Genet 1996; 59(4): 834–838.
12. Ogino S, Gao S, Leonard DG et al. Inverse correlation between SMN1 and SMN2 copy numbers: evidence for gene conversion from SMN2 to SMN1. Eur J Hum Genet 2003; 11(3): 275–277. doi: 10.1038/ sj.ejhg.5200957.
13. Gambardella A, Mazzei R, Toscano A et al. Spinal muscular atrophy due to an isolated deletion of exon 8 of the telomeric survival motor neuron gene. Ann Neurol 1998; 44(5): 836–839. doi: 10.1002/ ana.410440522.
14. Maiti D, Bhattacharya M, Yadav S. Isolated exon 8 deletion in type 1 spinal muscular atrophy with bilateral optic atrophy: unusual genetic mutation leading to unusual manifestation? J Postgrad Med 2012; 58(4): 294–295. doi: 10.4103/ 0022-3859.105451.
15. Srivastava S, Mukherjee M, Panigrahi I et al. SMN2-deletion in childhood-onset spinal muscular atrophy. Am J Med Genet 2001; 101(3): 198–202. doi: 10.1002/ ajmg.1386.
16. Moulard B, Salachas F, Chassande B et al. Association between centromeric deletions of the SMN gene and sporadic adult-onset lower motor neuron disease. Ann Neurol 1998; 43(5): 640–644. doi: 10.1002/ ana.410430513.
17. Prior TW. Strategy for the molecular testing of spinal muscular atrophy. In: Prior TW. Spinal Muscular Atrophy. Cambridge, MA, USA: Academic Press 2017: 63–71.
18. Rudnik-Schöneborn S, Eggermann T, Kress W et al. Clinical utility gene card for: proximal spinal muscular atrophy (SMA) – update 2015. Eur J Hum Genet 2015; 23(11). doi: 10.1038/ ejhg.2015.90.
19. Fang P, Li L, Zeng J et al. Molecular characterization and copy number of SMN1, SMN2 and NAIP in Chinese patients with spinal muscular atrophy and unrelated healthy controls. BMC Musculoskelet Disord 2015; 16(1): 11. doi: 10.1186/ s12891-015-0457-x.
20. Cooper DS, Darki L, Beydoun SR. Spinal muscular atrophy – two case reports of compound heterozygosity. US Neurol 2019; 15(2): 97. doi: 10.17925/ usn.2019.15.2.97.
21. Wirth B, Schmidt T, Hahnen E et al. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet 1997; 61(5): 1102–1111. doi: 10.1086/ 301608.
22. Gonçalves-Rocha M, Oliveira J, Rodrigues L et al. New approaches in molecular diagnosis and population carrier screening for spinal muscular atrophy. Genet Test Mol Biomarkers 2011; 15(5): 319–326. doi: 10.1089/ gtmb.2010.0164.
23. Smith M, Calabro V, Chong B et al. Population screening and cascade testing for carriers of SMA. Eur J Hum Genet 2007; 15(7): 759–766. doi: 10.1038/ sj.ejhg.5201821.
24. Scheffer H, Cobben JM, Matthijs G et al. Best practice guidelines for molecular analysis in spinal muscular atrophy. Eur J Hum Genet 2001; 9(7): 484–491. doi: 10.1038/ sj.ejhg.5200667.
25. Prior TW. Carrier screening for spinal muscular atrophy. Genet Med 2008; 11(11): 840–842. doi: 10.1097/ GIM.0b013e318188d069.
26. Hendrickson BC, Donohoe C, Akmaev VR et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet 2009; 46(9): 641–644. doi: 10.1136/ jmg.2009.066969.
27. Ogino S, Leonard DG, Rennert H et al. Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet 2002; 110(4): 301–307. doi: 10.1002/ ajmg.10425.
28. Verhaart IE, Robertson A, Wilson IJ et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis 2017; 12(1): 124. doi: 10.1186/ s13023-017-0671-8.
29. Hereditary Motor Syndromes, Spinal Muscular Atrophy. [online]. Available from URL: https:/ / neuromuscular.wustl.edu/ synmot.html#sma5q.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2020 Číslo Supplementum 2
Nejčtenější v tomto čísle
- Klinický obraz spinální muskulární atrofie u dospělých pacientů
- Genetika spinální muskulární atrofie
- Léčba spinální svalové atrofie
- Rehabilitace u spinální muskulární atrofie