
Progresivní supranukleární obrna
Progressive supranuclear palsy
Progressive supranuclear palsy is a tauopathy belonging to atypical Parkinsonian syndromes. The main clinical symptoms include oculomotor dysfunctions, early postural instability, symmetrical hypokinetic-rigid syndrome with axial predominance and cognitive decline. The symptom variability and rate of progression depend on disease subtype. Diagnostics are based on clinical symptoms; MRI remains the most useful auxiliary method. The article is focused mainly on clinical perspectives and recent diagnostic approaches considering the latest recommendations. Symptomatic therapy remains of great importance as causal treatment is still lacking.
Keywords:
progressive supranuclear palsy – vertical gaze palsy – tauopathy – postural instability – parkinsonism – cognitive decline – Levodopa – amantadine
Autoři:
T. Bartošová 1; J. Klempíř- 1 3
Působiště autorů:
Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN v Praze
1; Anatomický ústav, 1. LF UK v Praze
2; Evropská referenční síť pro vzácná neurologická onemocnění
3
Vyšlo v časopise:
Cesk Slov Neurol N 2020; 83/116(6): 584-601
Kategorie:
Přehledný referát
doi:
https://doi.org/10.48095/cccsnn2020584
Souhrn
Progresivní supranukleární obrna je onemocnění z okruhu atypických parkinsonských syndromů řadící se mezi tauopatie. Mezi jeho hlavní příznaky patří poruchy okulomotoriky, časná posturální instabilita, symetrický hypokineticko-rigidní syndrom s axiální převahou a kognitivní deficit. Spektrum příznaků a rychlost progrese se liší v závislosti na konkrétní variantě onemocnění. Diagnostika se opírá především o klinický nález a z podpůrných metod zůstává na prvním místě MR. Text je zaměřen především na klinický obraz a současný pohled na diagnostiku, reflektující nově vznikající doporučení. Kauzální terapie neexistuje, proto je třeba maximálně využít možnosti symptomatické terapie.
Klíčová slova:
progresivní supranukleární obrna – paréza vertikálního pohledu – tauopatie – posturální instabilita – parkinsonský syndrom – kognitivní defi cit – Levodopa – amantadin
Epidemiologie
Prevalence progresivní supranukleární obrny (progressive supranuclear palsy; PSP) je v Evropě a v Severní Americe odhadována zhruba na 5–6/100 000 obyvatel [1–3]. V ČR nejsou přesná data známá. Roční incidence stoupá od 1,1 případů na 100 000 mezi 50.–59. rokem života až na 14,7/100 000 ve věku 80–89 let. Lze očekávat, že výskyt této nemoci je ve skutečnosti mnohem vyšší, neboť jsou popisovány stále nové fenotypy a vyvíjejí se i diagnostická kritéria [4]. První příznaky se zpravidla objevují v 6. dekádě. Patologicky prozatím nebyly potvrzeny případy mladší 40 let [4]. Rozdíly incidence s ohledem na pohlaví nebyly jednoznačně prokázány [1,5].
Z etiologického hlediska se v odborné literatuře diskutuje možná role pesticidů [6], těžkých kovů a alkaloidů ovlivňujících mitochondriální metabolizmus (např. neurotoxický alkaloid způsobující endemický atypický parkinsonismus známý z ostrova Guadeloupe) [7]. Život v zemědělských oblastech, kouření, nižší vzdělání a nižší příjem se jako rizikové faktory nepotvrdily [8]. Statisticky významným faktorem se zdá být dlouhodobá konzumace studnové vody [6].
Současný stav poznání ukazuje, že PSP je spíše syndromem než jedinou nemocí, který může vznikat na podkladě různých patofyziologických pochodů. Navíc v některých případech, které histopatologicky a/nebo klinicky splňovaly obraz PSP, byl opakovaně nalezen i současný výskyt neurodegenerativních onemocnění odlišné etiologie (např. Alzheimerovy nemoci [AN], multisystémové atrofie, frontotemporální lobární degenerace [FTLD], onemocnění motoneuronu). Rovněž existují příklady, které by neuropatologicky splňovaly kritéria PSP, avšak bez jakéhokoli klinického korelátu [9–11].
Patofyziologie a neuropatologie
Supranukleární obrna patří mezi tzv. tauopatie, tedy onemocnění spojená s poruchou funkce proteinu tau ze skupiny proteinů asociovaných s mikrotubuly (mitochondrial associated’s proteins; MAPs). Mezi tauopatie řadíme neurodegenerativní onemocnění s různou patofyziologií a klinickým obrazem jako např. kortikobazální degeneraci, FTLD, AN i nemoc s argyrofilními zrny. Fyziologicky dobře rozpustný tau protein se vyskytuje především v neuronech, v menší míře pak v astrocytech, oligodendrocytech a dokonce i extracelulárně. Jednou z jeho hlavních funkcí je účast na stabilizaci mikrotubulů s umožněním jejich polymerace, čímž napomáhá zajistit správnou funkci cytoskeletu, tedy i axonálního transportu, mezibuněčné signalizace, synaptické plasticity či stability genomu [12]. Za patologických okolností dochází k hyperfosforylaci tohoto proteinu, což vede k jeho chybné konformaci a tvorbě nerozpustných filament. Neurony se tak stávají vulnerabilnější vůči nejrůznějším insultům zahrnujícím oxidativní poškození, kalciovou dysregulaci nebo excitotoxicitu. Patologický protein navíc sám o sobě získává neurotoxické vlastnosti [13].
Zároveň dochází k omezení degradace takto změněného proteinu pomocí ubquitin-proteázového komplexu, který patří k hlavním mechanizmům jeho eliminace. To vede k jeho akumulaci a následnému ukládání v podobě nerozpustných agregátů [2,14]. Agregovaný tau protein se vyskytuje v neuronech jako charakteristická tau pozitivní „neurofibrilární klubka“ (neurofibrilar tangles/pretangles) či v podobě „vláken neutropilu“ (neutropil threads). Postiženy mohou být i gliové buňky s nálezem „chomáčovitých astrocytů“ (tufted astrocytes) a „oligodendrocytárních stočených/vinutých těl“ (colied bodies). Existuje předpoklad, že právě tvorba zmíněných agregátů je fyziologickou odpovědí organizmu za účelem omezení neurotoxicity hyperfosforylovaných oligomerů tau proteinu [15]. Patologická forma tau proteinu může způsobit mimo jiné i mitochondriální dysfunkci s omezením intracelulárního transportu mitochondrií, jejich fúzování, dělení i funkcí respiračního řetězce. Tím dochází k selhávání oxidativního metabolizmu se snížením produkce adenosintrifosfátu (adenosine triphosphate; ATP), k poklesu detoxifikačních funkcí i nadprodukci volných kyslíkových radikálů [14,16]. V poslední době je diskutována vlastnost proteinu tau šířit se napříč neurony ve smyslu jakési prion-like transmise [2]. Toto tvrzení je podporováno přítomností nesbaleného tau proteinu v intersticiální tekutině, předpokladem jeho sekrece do mozkomíšního moku a aktivním šířením do dalších neuronů. Experimentálně přitom byla pozorována jeho transmisibilita s potenciálem měnit normální tau protein v patologický [17]. Gliální tau protein je navíc nejspíš schopen šíření podél bílé hmoty nezávisle na neuronech [18].
Neuropatologicky mohou být pozorovány neuronální úbytek, glióza a gliální inkluze (subkortikálně i kortikálně) [19]. Makroskopicky může být pozorovatelná atrofie v oblasti globus pallidus, ncl. subthalamicus, tegmentum mesencephali, pontu a ncl. dentatus [20,21]. Patrné bývá odbarvení substantia nigra a hilu ncl. dentatus [22]. Postiženy mohou být i jiné oblasti, např. gyrus precentralis, frontalis medius, a temporalis medius i parietální kortex [20].
Míra poškození v určitých oblastech koreluje s klinickým postižením, a tedy odpovídá i rozdílům jednotlivých typů PSP. Např. větší postižení kortikálních oblastí může podmiňovat subtyp s kortikobazálním syndromem (PSP with corticobasal syndrome presentation; PSP-CBS) či postižením řeči (speech/language variant of PSP; PSP-SL), viz níže v textu. K typickému obrazu motorického deficitu přispívá i degenerace pedunkulopontinního jádra (pedunculopontine nucleus; PPN). Patologie zmíněného jádra může vést k poruchám pozornosti a narušení stability s časnými pády [23]. V důsledku úbytku různých subpopulací neuronů dochází i k postupné dysregulaci neurotransmiterů vč. acetylcholinu, dopaminu a gama-aminomáselné kyseliny (gamma-aminobutyric acid; GABA) [24].
Hledání biomarkerů
Nadále pokračuje snaha o nalezení spolehlivého laboratorního biomarkeru, který by byl schopen stanovit ante mortem definitivní diagnózu.
Studovány byly především hladiny tau proteinu v mozkomíšním moku (cerebrospinal fluid; CSF) s nálezem snížené hladiny t i p tau proteinu oproti zdravým kontrolám a pacientům s AN. Nicméně výsledky byly podobné jako u Parkinsonovy nemoci (PN) [2,25,26]. V minulosti bylo taktéž patrné snížení sérového amyloidu (Ab 42) v porovnání s pacienty s PN [26].
Podobně jako u mnohých jiných neurodegenerací, i zde je v CSF přítomna zvýšená hladina lehkých řetězců neurofilament svědčící o neuronálním poškození. Samotné zvýšení tedy není dostatečně specifické pro diferenciální diagnostiku. Jejich hladiny by však mohly určit závažnost progrese, eventuálně predikovat dobu přežití [27,28].
K rozlišení oproti synukleinopatiím by v budoucnu mohla přispět i hladina sérového adiponektinu, přičemž u multisystémové atrofie (MSA) a PN byly nalezeny jeho hodnoty statisticky vyšší [29] (což svědčí pro odlišný lipidový mechanizmus v patofyziologii obou skupin onemocnění).
Magnetická rezonance
Obraz MR se v kombinaci s klinickým nálezem ukazuje jako pomocný diagnostický marker onemocnění. Pomocí sekvencí T1-, T2-vážených a FLAIR (fluid attenuated inversion recovery) může být v různé míře pozorována atrofie mezencefala, pontinního tegmenta, pedunculli cerebellares superiores, frontálního laloku a dále rozšíření III. a IV. komory mozkové. V T2-vážených sekvenci mohu být nalezeny změny signálu periaqueduktální zóny, globus pallidus a pedunculi cerebellares superiores [21,30].
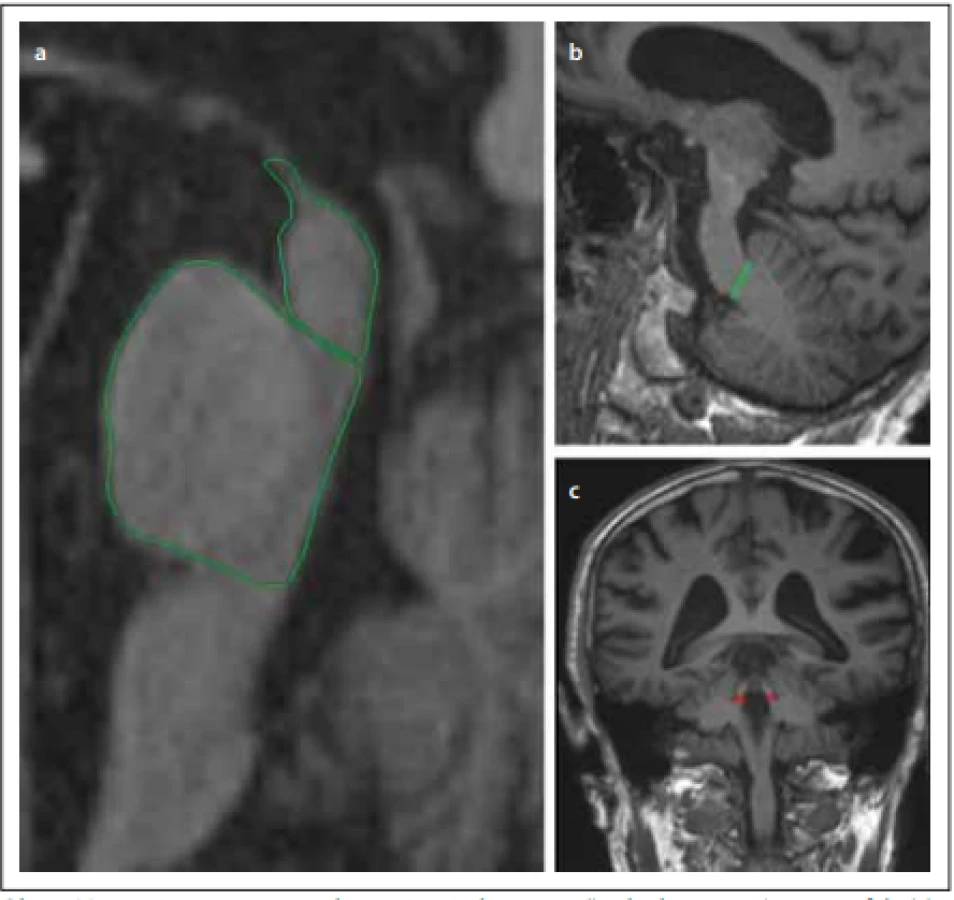
Na sagitálním řezu kmenem můžeme pozorovat atrofii mezencefala v podobě obrazu kolibříka / stojícího tučňáka („hummingbird sign“, „penguin-silhouette sign“), který vzniká oploštěním až konkavitou dorsum mesencephali při relativním ušetření pontu. Na axiálních řezech kmene v úrovni colliculi superiores je patrná atrofie mezencefala jako tzv. příznak svlačce („morning glory sign“), respektive myšáka („Mickey mouse sign“), který vzniká na základě konkavity laterálních částí tegmenta mesencephali (obr. 1). Jednoznačná atrofie mezencefala nicméně nemusí být v počátku nemoci patrná [31]. Při diagnostických pochybnostech je tedy vhodné MR zopakovat dle vývoje klinických příznaků s odstupem 1–2 let. Ze všech variant PSP je atrofie mezencefala nejvýraznější u Steel-Richardsonova syndromu PSP (PSP-RS), což je spojeno s nižší senzitivitou MR u jiných subtypů PSP. Navíc je pacientů s ostatními variantami méně a jejich diagnózy často nejsou histopatologicky potvrzené, takže u těchto fenotypů máme méně relevantních dat (tab. 1). Komplikací rovněž je, že atrofie mezencefala může být v menší míře přítomna i u normotenzního hydrocefalu, kortikobazální degenerace, Wilsonovy nemoci a dalších vzácných onemocnění [21].
Fig. 1. Magnetic resonance parkinsonism index – ratio of the area of the pons/midbrain
(a) multiplied by the ratio of the widths of the middle cerebellar peduncle (b) / superior
cerebellar peduncle (c).
Užitečnější než pouhé kvalitativní rozlišení mohou být kvantitativní parametry s posuzováním např. anterio-posteriorního rozměru tegmenta, celkové plochy a průřezu mezencefala nebo šíře pedunculli cerebellares superiores. Významným prediktorem se zdá být také cerebelární interpedunkulární úhel [21,30].
Jednou z možností standardizovaného kvantitativního hodnocení jsou i tzv. kritéria MRPI (magnetic resonance parkinsonism index). Jedná se o poměr ploch pontu/mezencefala násobený poměrem šíře pedunculli cerebellares medii/superiores, které mohou pomoci odlišení PSP od pacientů s vaskulárním postižením (obr. 2) [32]. Pro další zvýšení senzitivity a specificity byl uveřejněn i index MRPI2 (MRPI násobený šíří III. komory/šíří frontálních rohů). Výsledky indexu byly ve studii signifikantně vyšší u PSP než u PN, s dobrým rozlišením i oproti MSA [33]. Tato metoda má toho času relativně dobrou podporu v provedených studiích [34]. Zároveň se zdá být dobrým markerem progrese při hodnocení po 2 letech [35].
Fig. 2. MRI. (a) Atrophy of the lateral edges of the midbrain on the axial plane (Mickey mouse sign / morning glory sign); (b) dilatation
of the IIIrd cerebral ventricle; (c) atrophy of the cranial and dorsal part of the midbrain on the sagittal plane (hummingbird sign).

V odlišování PSP od PN může být užitečným i tzv. „Akdenizův index“, který je založen na výpočtu poměru ploch pontu/mezencefala násobeného interpedunkulárním úhlem / 180° [36].
Další experimentální metody zahrnují pokročilejší techniky MR založené na vážení difuzí (diffusion weighted imaging; DWI). Mezi základní difuzní metriku patří hodnocení průměrné difuzivity (mean diffusivity; MD) a frakční anizotropie (FA). Pomocí těchto technik lze prokázat snazší difuzi volné vody ve tkáni v důsledku mikrostrukturálních neurodegenerativních změn, respektive úbytku neuronů, myelinizovaných vláken a gliózy. U PSP tak můžeme nalézt převážně subkortikální snížení frakční anizotropie a zvýšení difuzivity vykazující vyšší míru symetrie. Popsané změny jsou nejvíce patrné v mezencefalu a pedunculli cerebellares superiores a mohou pomoci v rozlišení oproti jiným parkinsonským syndromům [21].
V současnosti jsou též publikovány práce s využitím bi-tenzorového difuzního zobrazování. Jedná se zde o zobrazování volné vody (free water imaging), které svědčí pro nárůst objemu volné vody v ncl. caudatus, putamen, nucleus subthalamicus, mezencefalu, pedunculli cerebellares, ncl. dentatus, vermis cerebelli a lobuli V, VI, talamu a corpus callosum u pacientů s PSP, zatímco u pacientů s PN pouze v substantia nigra [37].
Nadále existuje snaha o nalezení dalších podpůrných markerů pomocí PET se značením 18F-flurodeoxyglukózy a se značením ligandů tau proteinu, kde se ukazuje redukce vychytávání v mezencefalu a ve frontálních oblastech, zejména gyrus cinguli, které však prozatím nejsou dostatečně specifické pro rutinní využití v klinické praxi.
Pomocná laboratorní vyšetření
Ultrasonografie
Na rozdíl od PN je u PSP zpravidla patrná normoechogenita substantia nigra a zároveň je přítomné rozšíření III. postranní komory a hyperechogenita ncl. lenticularis. Ncl. lenticularis ale může být hyperechogenní i u MSA [38]. Při kombinaci analýzy echogenit všech výše zmíněných parametrů bylo u PSP dosaženo až 99% specificity [39].
Videookulografie
U pacientů s PSP dominují snížení rychlostí vertikálního pohledu, narušení antisakád, anomálie sledovacích pohybů a sakadické intruze. Pro MSA je příznačné narušení sledovacích pohledů s tzv. „catch up“ – záchytnými sakádami. U PN vídáme sakadické intruze [40].
Posturografie
Při posturografickém vyšetření jsou u PSP popisovány výchylky těžiště především v předozadním plánu v souvislosti s typickou axiální rigiditou. Vzorec stability se tak liší od pacientů s MSA, kde se objevují výchylky těžiště jak v anteroposteriorním, tak mediolaterálním plánu v rámci ataktické komponenty. Stabilita je oproti pacientům s PN výrazněji limitována [41].
Pozorování vedou k předpokladu chybného motorického plánování a omezení myotatických napínacích reflexů při spinálním postižení, event. postižení vestibulární složky [42]. Mimo jiné svou úlohu nejspíš hraje i abnormální centrální integrace senzorických vstupů [43,44].
Hlasová analýza
V rozlišení od MSA a PN může pomoci i hlasová analýza, která odhalí hypokineticko-spastickou dysartrii u pacientů s PSP. Řeč je pomalá, tichá, s deficity artikulace [45].
Genetika
Supranukleární obrna je klasicky považována za sporadické onemocnění. Publikováno bylo pouze několik raritních případů s možným familiárním výskytem [46,47].
Bylo popsáno několik případů se vzácnou mutací genu pro protein asociovaný s mikrotubuly (mikrotubule associated protein; MAPT), které se fenotypově projevovaly stejně jako PSP [48]. Přestože familiární forma PSP je klinicky i neuropatologicky totožná s formou sporadickou, jedná se formálně o odlišné jednotky [47]. H1 haplotyp genu MAPT [49] (resp. subhaplotyp H1c [48]) je považován za rizikový faktor rozvoje PSP, naopak H2 haplotyp genu MAPT za faktor protektivní [50].
V poslední době také roste zájem o jednonukleotidové polymorfizmy (single nucleotide polymorphisms; SNPs) asociované s PSP. Jedná se o mutace v genech kódujících syntaxin 6 (STX6), R-like ptoteinkinázu endoplazmatického retikula (EIF2AK3) a myelinový bazický protein asociovaný s oligodendrocyty (MOBP), které jsou v současnosti rovněž pokládány za rizikové faktory onemocnění [2,8,25]. Zmíněné geny se různou mírou účastní na procesu apoptózy nebo aktivace mikroglie. Stále přibývají nové polymorfizmy SNP asociované s PSP i s kortikobazální degenerací (např. SLCO1A2, DUSP10, SLCO1A, RUNX2) [2].
Klinický obraz
Klinický obraz PSP byl popsán v roce 1964 jako kombinace supranukleární pohledové parézy, dysartrie, dysfagie, axiální rigidity, dystonie, bradykineze, nestability při chůzi a demence [51]. Od té doby se ukázalo, že klinický obraz může být významně pestřejší.
Poruchy chůze a posturální instabilita
Jedním z prvních příznaků bývá posturální instabilita s neprovokovanými pády (nejčastěji nazad), které se rozvíjejí již během prvních 3 let progrese nemoci [52,53]. Narušení posturální stability se může časně projevit v pull testu. Tyto poruchy můžeme pozorovat i při chůzi, která je typicky vrávoravá, toporná, případně o široké bázi [54]. Při otočkách mají pacienti s PSP tendenci k otáčení celého těla „en block“. Takovéto otáčení vzhledem k nekontrolovatelnosti a neobratnosti pohybu snadno končí pádem nazad. U PSP-RS (známého taktéž jako syndrom Steel-Richard-Olszewského) se při chůzi typicky objevuje extenze trupu a kolenou s mírnou abdukcí končetin, na které se podílí axiální dystonie a rigidita.
V další progresi přispívá k nestabilitě a pádům axiální rigidita se změnou postavení těžiště, akineze, freezing při chůzi a později i retrocollis [52]. Svou roli sehrává i chybná senzorická integrace při postižení mozkového kmene. K pádům mimo jiné přispívá i kognitivní deficit frontálního typu, který je spojen s apraxií chůze a také s impulzivitou související s neopatrným chováním pacientů, resp. se zbrklostí vedoucí k pádům. Mobilitu nemocných komplikuje i okohybná porucha omezující zorné pole a orientaci v prostoru. Pacienti rovněž popisují subjektivní „závrativý“ stav, někteří jej dokonce považují za nejvíce obtěžující příznak. Líčí přitom neustálé motání hlavy spíše rotačního charakteru, nezávislé na poloze a činnosti a přetrvávající po většinu dne. Na zmíněných stavech se mohou podílet okohybné poruchy s omezením akomodace při poškození ncl. intestitialis Cajali a porucha centrální senzorické integrace. O přímém postižení vestibulárních funkcí se však nadále vedou diskuse [55].
Parkinsonský syndrom
Parkinsonský syndrom bývá u PSP symetrický s axiální převahou. Typické je od počátku pomalu progredující snížení rychlosti a rozsahu pohybů (bradykineze, hypokineze), porucha iniciace volních pohybů (akineze) a rigidita. Současně se objevují hypomimie, hypofonie, dysartrie a omezená pohyblivost respiračního svalstva. Tremor není častý a nebývá dominantní subjektivní obtíží, nicméně v jedné ze studií byl v určité podobě přítomen až u 42 % pacientů s PSP. Třes je nejčastěji posturální, kinetický, méně pak klidový a při progresi nemoci se mohou vzájemně kombinovat [56].
Okohybné poruchy
U PSP se okohybné poruchy běžně vyskytují do 3 let od prvních příznaků nemoci, nicméně se mohou objevit i později (vzácně i po 10 letech progrese). Obvykle začínají narušením sakadických pohybů ve vertikální rovině, a to především směrem dolů. Později následují obtíže i ve směru horizontálním. To může být dočasně překonatelné pomocí okulocefalického reflexu. S postupující degenerací kmene však může být reflex ztracen. Dále se vyskytují sakadické intruze, tedy nepravidelné mimovolní oční pohyby narušující cílenou fixaci pohledu. Mohou být viditelné i při pokusu o hladké sledovací pohyby, kdy je označujeme jako tzv. macro square-wave jerks [57]. Při vyšetření sledovacích pohybů ve vertikální rovině se může objevit neschopnost provedení sakadického pohybu podél vertikální linie. Při tomto pohybu dochází k přechodnému vybočení horizontálním směrem („round the house sign“) [58]. Narušeny jsou i sledovací pohyby, které jsou zpomalené, trhavé, hypometrické a s poruchou fixace. Později se objevuje i paréza pohledu, opět nejdříve ve vertikálním směru. Okohybné dysfunkce zpravidla progredují v čase, někdy až do obrazu úplné plegie. Pacienti si mohou stěžovat i na fotofobii, zhoršení zrakové ostrosti a diplopii v rámci poruchy konvergence. Vzácněji se může vyskytovat i bilaterální internukleární oftalmoplegie [59].
Dystonie
Časný rozvoj dystonie není u pacientů s PSP vzácný. Její charakter je zde spíše setrvalého až fixního rázu nežli povahy repetitivních dystonických pohybů, přičemž může postihovat kteroukoli svalovou skupinu. Obvykle začíná v kraniofaciální oblasti. Pro pacienta bývá limitující blefarospazmus, eventuálně v asociaci s apraxií otevírání víček. Dystonická retrakce víček, omezené mrkání, popř. dystonie m. procerus, podmiňující vertikální vrásky (tzv. procerus sign) a kontrakce m. frontalis (reptilian sign), přispívají ke strnulému a jakoby „vytřeštěnému“ pohledu nemocných umožňujícímu diagnostický odhad na první pohled [60].
Dystonie orofaciálního a laryngeálního svalstva způsobuje dysartrii, dysfonii a přímo ohrožuje pacienta rozvojem dysfagie i ztíženou respirací. Dystonie dále postihuje krční oblast (retrocollis) a na trupu se nejčastěji objevuje jako hyperextenze s možnou laterální deviací. Zejména v pozdějších stadiích může být přítomna i končetinová dystonie (specifickým příznakem je zde tzv. „pointing gun“ s nataženým palcem a ukazováčkem). V pokročilých stadiích vznikají až fixní kontraktury [61]. Senzorické triky u tohoto typu dystonie většinou nepomáhají.
Poruchy řeči a dysfagie
U PSP se objevuje hypokineticko-spastická dysartrie, případně s ataktickou komponentou, mnohdy progredující až do podoby anartrie s redukcí hlasového projevu na setřelé nesrozumitelné mručení [62]. Charakteristickým projevem je nepřesná, setřelá artikulace a hypofonie z důvodu omezené podpory respiračního svalstva. Výjimkou nejsou ani hesitace nebo tachyfemie [63]. Ve srovnání s pacienty s PN bývá řeč u nemocných s PSP pomalejší, s delšími pauzami a monotónním projevem při redukci intonační variability [64]. Typicky se objevují snížená spontaneita řečového projevu, zhoršený výkon ve verbální fluenci, palilálie nebo echolálie.
Na rozdíl od PN nastupuje dysfagie tekutin i pevné stravy u PSP mnohem časněji a náhled na tuto poruchu bývá zachován. Objevuje se do 3–4 let od začátku nemoci, přičemž dřívější nástup je spojen s horší prognózou. Významněji bývá porušena fáze orální (až u 76 % pacientů) nežli faryngeální. Orální fáze je typická narušením linguálních pohybů (např. omezením retrakce báze jazyka) a dlouhým převalováním bolusu v ústech, kde poté zůstávají i četná rezidua. Iniciace faryngeální fáze je opožděna, avšak hyolaryngeální exkurze umožňující clearance bolusu bývá relativně zachována [65], proto jsou pacienti zpravidla dříve ohroženi malnutricí než aspiracemi. Situaci dále zhoršuje fakt, že u PSP je narušen i reflexní kašel, a pacienti se tak nejsou schopni uspokojivě zbavit potravy z dýchacích cest. Polykací akt může být komplikován pro retrocollis, hyperextenzi trupu zvyšující riziko aspirace, popřípadě i dystonii žvýkacího svalstva omezující otevírání úst. Vzácností není ani apraxie postihující volní fáze polykacího aktu. Dysfagie pacienty ohrožuje aspirací, záněty dýchacích cest, malnutricí a dehydratací.
Ostatní poruchy hybnosti
Pacienti s PSP mohou mít i příznaky pyramidové léze, které se vyskytují jako nativní příznak nemoci při postižení cervikálních interneuronů, nebo jako následek cervikální myelopatie v důsledku dystonie. U některých variant PSP se mohou vyskytovat i známky postižení horního a dolního motoneuronu [66–68].
Spánek
Pacienty obtěžuje prodloužená latence usínání a přerušovaný spánek s výraznou redukcí fáze non-REM (non rapid eye movement), která vede k dlouhodobé spánkové deprivaci bez možnosti úplného zotavení. K tomuto problému mohou vést poruchy hybnosti znemožňující úpravu polohy na lůžku i další nonmotorické příznaky zahrnující poruchy nálady, úzkost apod. Poruchy chování v REM spánku či porucha atonie, které jsou typické pro synukleinopatie, se objevují jen vzácně [69]. Zmíněné obtíže jsou vysvětlitelné poškozením pontinního tegmenta, a především cholinergního pedunkulopontinního jádra důležitého pro generování REM spánku a spánkových vřeten [70,71].
Urologické dysfunkce
Příznaky autonomní dysfunkce nejsou u PSP ani časté ani specifické. Urologické obtíže se zpravidla vyskytují později v průběhu nemoci a jejich časný nástup je pokládán za negativní prediktor přežití [72,73]. Vyskytují se zde různé poruchy jímacích i vyprazdňovacích funkcí, jakými jsou hyperaktivita detruzoru (s nebo bez detruzoro-sfinkterové dyssynergie), nebo močová retence v důsledku nedostatečné funkce detruzoru. Urologické dysfunkce jsou klasicky považovány za méně závažné v porovnání s PN a MSA [62]. Nicméně jedna ze studií upozorňuje, že obtíže jímací fáze mohou být srovnatelné s těmi u PN. Ještě významnější se pak ve stejné studii zdály obtíže vyprazdňovací způsobené nedostatečnou kontrakcí detruzoru, a to v podobné míře jako u pacientů s MSA. Poruchy mikce jsou podobně jako u MSA spojeny s atrofií jader mozkového kmene se zasažením jak mikčního centra, tak i s úbytkem neuronů v oblasti Onufova jádra [74]. Po urologických dysfunkcích je na místě cíleně pátrat, popřípadě je objektivizovat pomocí urodynamického vyšetření, neboť mohou vést k problémům s hygienou, dekubitům a fatálním infekčním komplikacím.
Kognitivní poruchy
Kognitivním deficitem je dříve či později postižena většina pacientů s PSP. Kognitivní poruchy bývají přítomny velice brzy a často progredují až do obrazu demence, přičemž postižení je výraznější a časnější než u MSA nebo PN [75].
U PSP se objevuje především subkortikální typ postižení s narušením frontostriatálních okruhů v kombinaci s postižením kortikálním. Subkortikální složka zahrnuje poruchy pozornosti, zpomalení psychomotorického tempa, exekutivní dysfunkce a poruchy iniciace a perseverace s následnou redukcí fonemické i kategoriální verbální fluence.
Kortikální postižení zahrnuje zejména apraxii, afázii a vizuospaciální či vizuokonstruktivní deficity [76,77]. Poruchy vizuospaciálních funkcí souvisejí i s okulomotorikou [78]. Verbální učení je více a časněji zasaženo než u PN vzhledem k poruchám pozornosti a pracovní paměti (při dysexekutivním syndromu) [79], ale oddálená výbavnost je u PSP o něco lepší. Poruchy dlouhodobé paměti nejsou u PSP významné. Nepřevládá tedy hipokampální forma deficitu [80]. Omezení okamžité výbavnosti slov pak může být částečně způsobeno i neschopností pokračovat v zadaném úkolu pro nedostatek iniciace a schopnosti udržení nastavení [81].
Míra deficitu jednotlivých domén závisí, stejně jako všechny ostatní klinické příznaky, na konkrétním fenotypu PSP, resp. na rozložení tau proteinu. Frontální symptomatika je tak bezpochyby nejvíce vyjádřena u pacientů s variantou PSP-F, kde může být obtížně rozlišitelná od FTLD. Akcentace frontální poruchy zde může vyústit až ve frontální afázii s poruchou fluence při zachovalém pojmenování opakování a porozumění [82]. Neuropatologicky souvisí obraz FTLD s depozity tau proteinu v gyrus frontalis suprerior, gyrus frontalis medius, orbitofrontálním kortexu a gyrus temporalis inferior, a to jak v šedé, tak v bílé hmotě [83].
Výše uvedené poruchy kognitivních funkcí jsou často zjevné při administraci skríningových testů, jakými jsou Montreal Cognitive Assessment (MoCA) a Frontal Assessment Battery (FAB) [77,84]. V těchto testech jsou zahrnuty zkoušky exekutivních funkcí a jsou tedy citlivější pro diagnostiku poškození frontálních a souvisejících subkortikálních oblastí mozku.
Špatné výsledky testů všech výše zmíněných kognitivních domén mohou být rovněž významně prohloubeny pro současné behaviorální problémy ve smyslu apatie a abulie, a také symptomatickou terapií (anticholinergika, levodopa, amantadin) [76,77].
Poruchy chování
Až 95 % pacientů je postiženo apatií, která může být paradoxně doprovázena impulzivitou [85], iritabilitou, popřípadě i verbální a fyzickou agresí. Pacienti na tyto poruchy zpravidla nemají náhled, proto je při podezření na jejich rozvoj nutná cílená explorace od pečovatele. Apatie může mít negativní vliv na motorický výkon a celkový funkční stav pacienta [85].
Stejně tak může být apatie nesprávně vyhodnocena jako deprese, přestože pacienti vnímají svou náladu jako dobrou [86]. Nicméně až 60 % pacientů vykazuje určitou míru depresivní symptomatiky [86]. Anxiózní poruchy se vyskytují méně často (34 %) a nezávisle na depresi [87].
V souvislosti s postižením frontálních laloků se běžně objevuje rovněž spastický pláč nebo smích.
Psychotická produkce a deliriózní stavy se obvykle vyskytují v souvislosti s pokročilým kognitivním deficitem, nežádoucími účinky léčby a infekcí.
Diagnostika
Klinická diagnostika
Uvažovat o diagnóze PSP můžeme začít v případě sporadického progresivního onemocnění nastupujícího po 40. roce života.
Podle revidovaných diagnostických kritérií z roku 2017 [53] jsou vytyčeny 4 základní pilíře klinického obrazu onemocnění, kterými jsou posturální instabilita spolu s akinezí, okohybnou poruchou a kognitivním deficitem [53]. Může se stát, že v úvodu onemocnění některé – nebo i všechny ze zmíněných pilířů – chybí. Sám o sobě je každý z hlavních příznaků velice nespecifický a může se objevit i v průběhu mnoha jiných neurodegenerativních nebo sekundárních extrapyramidových onemocnění. Pro stanovení správné diagnózy je však určující kombinace příznaků. Zároveň bychom měli pátrat i po podpůrných kritériích zahrnujících fotofobii, časný nástup dysartrie, dysfagie a neuspokojivou odpověď na vysoké dávky levodopy (více než 1 g).
Revidovaná diagnostická kritéria jsou o něco širší a benevolentnější než kritéria původní NINDS-SPSP (National Institute of Neurological Disorders and Stroke and the Society for PSP) [88], čímž bylo dosaženo zvýšení senzitivity s přijatelným poklesem specificity. Nicméně jejich použití v klinické praxi skýtá jisté komplikace. Diagnostická kritéria jsou obsažena v 5 komplexních tabulkách, přičemž orientace v nich může být zpočátku poměrně časově náročná. Dalším úskalím je fakt, že pomocí těchto kritérií můžeme na základě splnění více diagnostických domén diagnostikovat několik možných/pravděpodobných typů PSP současně.
Tabulka diagnostických kritérií (tab. 2) obsahuje hlavní zahrnující a vylučující kritéria, která jejich uživatele vedou na prvním místě k širší diferenciálně diagnostické rozvaze. Pouze pokud pacient prošel sítem první tabulky, může se vyšetřující posunout ve svém diagnostickém pátrání do dalšího oddílu.

CSF – mozkomíšní mok; DWI – difuzí vážené obrazy; FLAIR – fl uid attenuated inversion recovery; PSP– progresivní supranukleární obrna; PSP-CBS
– PSP s kortikobazálním syndromem
Tabulka 3 je jednou z nejkratších a uživatelsky zřejmě i nejpřínosnějších částí diagnostických kritérií. Obsahuje 4 základní diagnostické pilíře rozdělené do 3 hladin jistoty podle míry/varianty jejich vyjádření. První hladina jistoty je považována za nejpravděpodobnější obraz PSP, se zvyšujícím se číslem pak míra jistoty klesá.
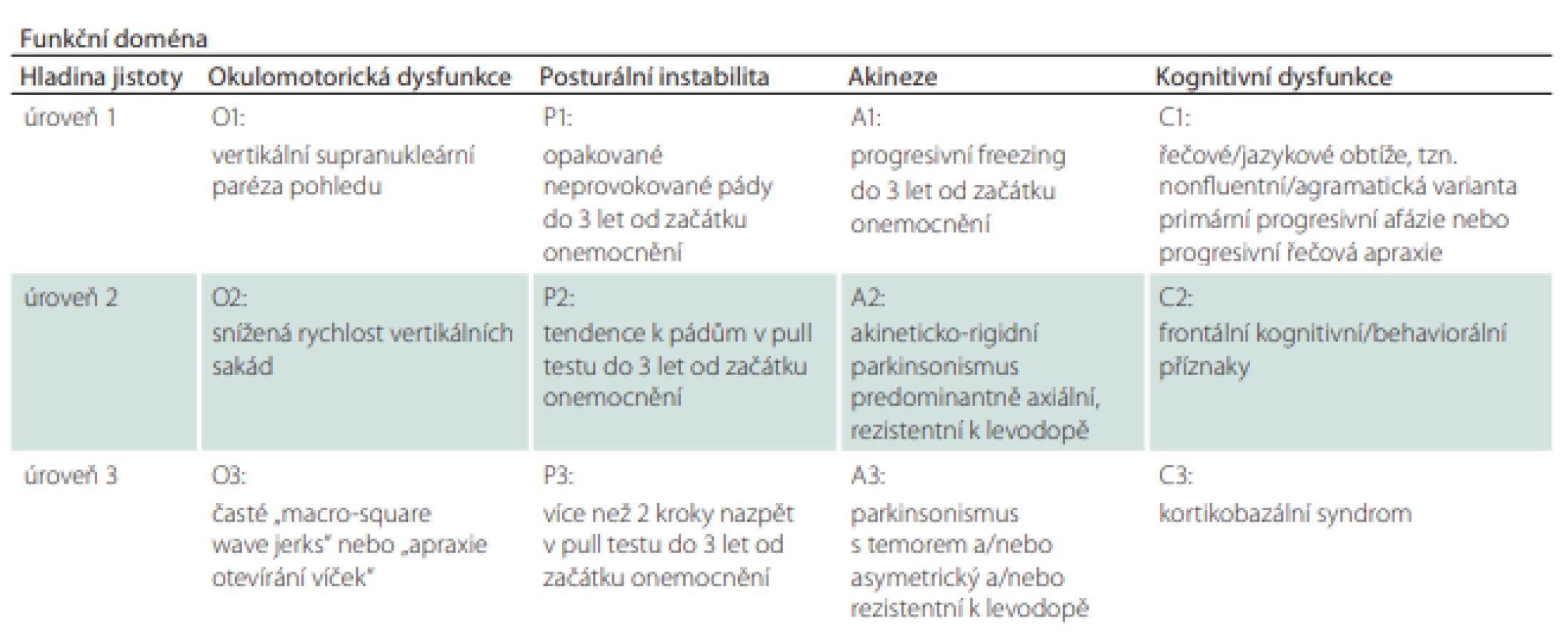
Tabulka 4 shrnuje podporující klinická a zobrazovací kritéria.

Tabulka 5 je pouze podrobným vysvětlením základních klinických kritérií z tab. 3. Je tedy vhodné do ní nahlédnout v případě, že si vyšetřující není jistý jejich přesným významem.
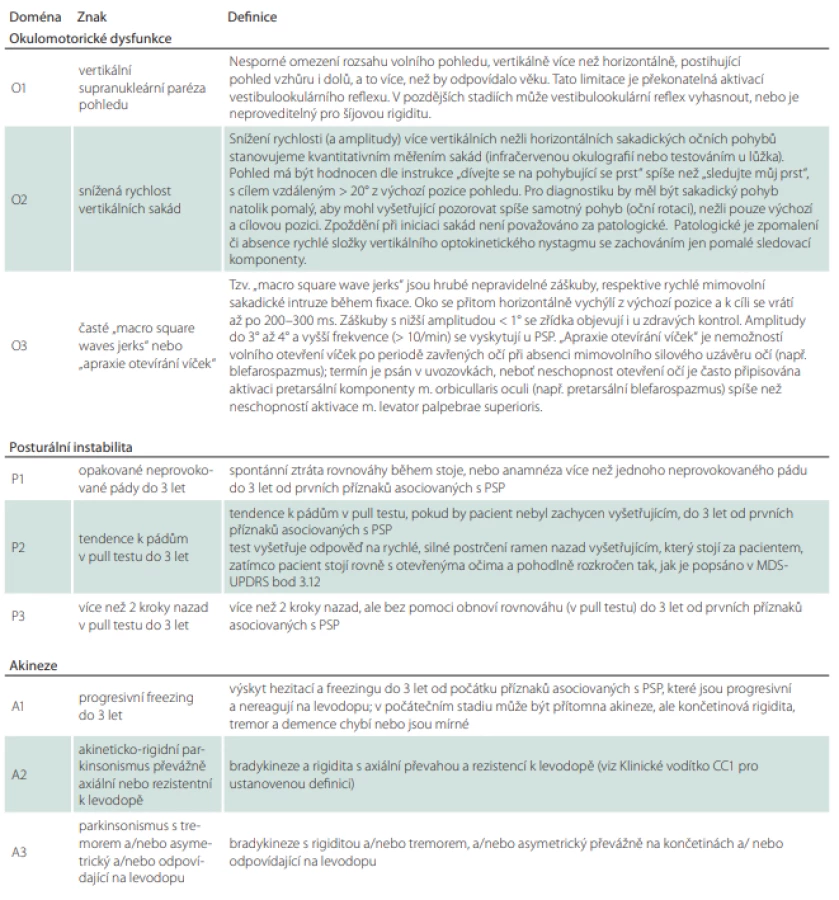

Tabulka 6 nám ukazuje, který typ PSP (popř. i na jaké hladině jistoty) jsme odhalili podle kombinace splněných kritérií z tabulky druhé. Vyšetřujícímu za ideálních okolností stačí pouze dosadit konkrétní kombinaci nalezených klinických kritérií do 3. sloupce. Mimo jiné je možné na základě této tabulky zjistit, zda je pacient vhodným adeptem pro zařazení do klinických studií.
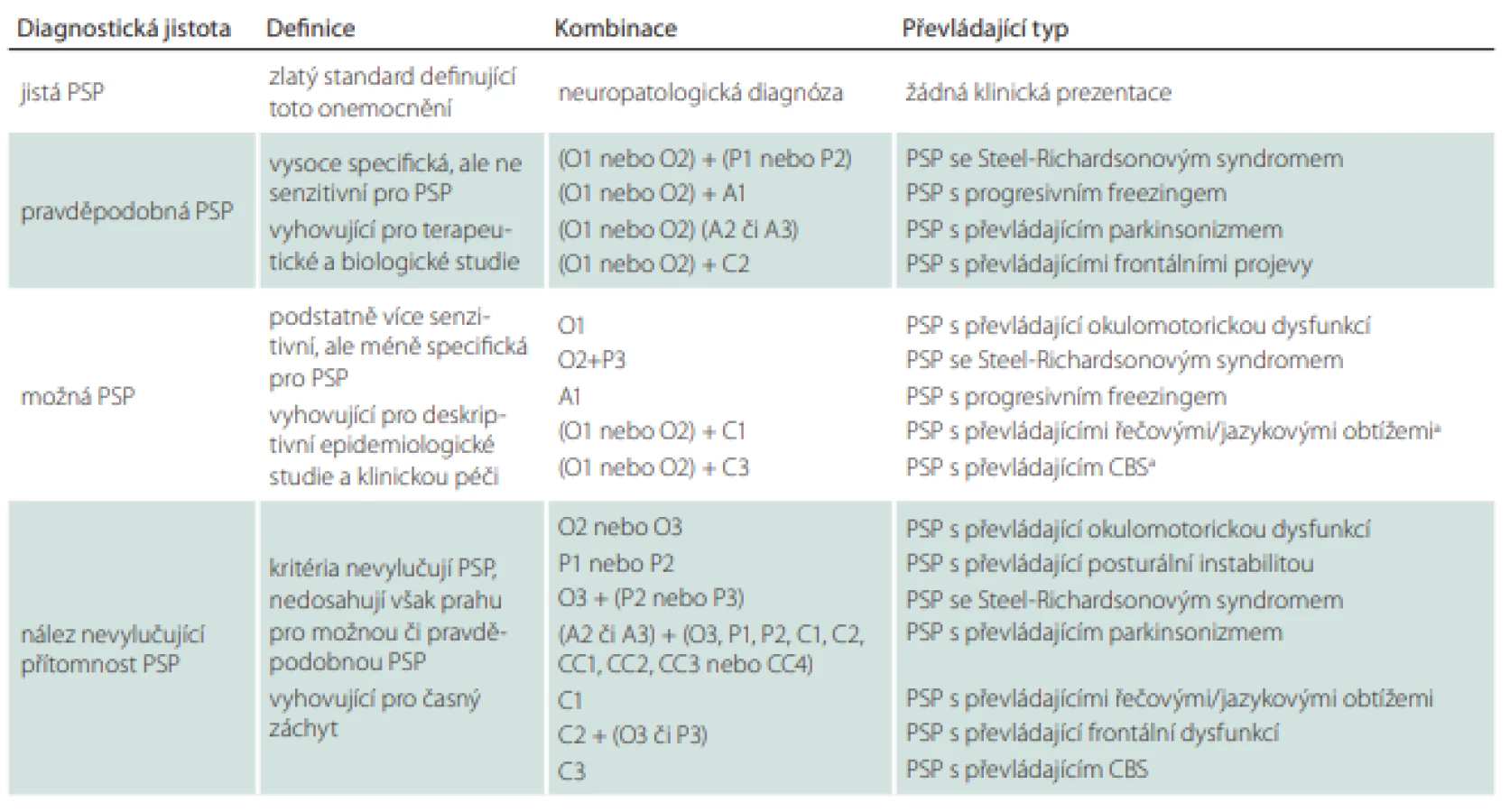
PSP – progresivní supranukleární obrna; CBS – kortikobazální syndrom
Obtíže při používání těchto kritérií mohou nastat za situace, kdy pacient splňuje klinické domény pro více možných/pravděpodobných typů PSP zároveň (např. pacient s klinikou PSP-RS vykazuje rovněž klinickou manifestaci PSP-P a PSP-SL). Pro utřídění překrývajících se diagnóz byla vytvořena upřesnění diagnostických kritérií omezující mnohotná zařazení (Multiple Allocations eXtinction; MAX1-3) (tab. 7). Ta doporučují upřednostnit především klinická kritéria vykazující v danou chvíli vyšší míru jistoty. Nestačí-li k našemu rozhodnutí 1. pravidlo, upřednostníme příznaky, které se vyskytovaly nejdříve (pokud pozdější příznaky jasně nedominují). Panuje-li nejistota i při uplatnění 2. pravidla, je vhodné řídit se jakousi klinickou hierarchií, která klade na první místo okohybnou dysfunkci a posturální instabilitu [89].

Klinické varianty progresivní supranukleární parézy a jejich specifika
V současnosti máme kromě původně popsaného PSP-RS ještě několik dalších známých variant, které mohou po celou dobu onemocnění probíhat odlišně, nebo progredovat do obrazu klasické varianty PSP-RS. Znalost níže uvedených variant může přispět k brzkému stanovení diagnózy.
V současnosti rozlišujeme kromě varianty PSP-RS formy PSP s dominantní poruchou okulomotoriky (PSP-OM), instabilitou (PSP-PI), progresivním freezingem při chůzi (PSP-PGF), parkinsonizmem (PSP-P), PSP-CBS, PSP-F a PSP-SL jako primární řečová apraxie (AOP) nebo primární nonfluentní afázie (non-fluent progressive aphasia, nfPA).
Ze zmíněných variant můžeme vyčlenit formy převážně kortikální: PSP-CBS, PSP-F, PSP-SL, a naopak subkortikální: PSP-PI, PSP-P, PSP-PGF, PSP-OM [90].
Další popsané typy s dominujícím cerebelárním postižením (PSP-C) a s primární laterální sklerózou (PSP-PLS) nebyly zahrnuty do revidovaných diagnostických kritérií z roku 2017, a to vzhledem k riziku znepřehlednění diagnóz oproti amyotrofické laterální skleróze (ALS) a MSA. PSP-CBS a PSP-SL splňují také kritéria pravděpodobné 4R tauopatie pro vyšší míru pravděpodobnosti přechodu do jiného klinického obrazu v průběhu vývoje onemocnění [53].
PSP varianta Steel-Riachardsonova syndromu (PSP-RS)
K prvním klinickým projevům tohoto syndromu patří posturální instabilita s náhlými pády směrem nazad následovaná okohybnou poruchou. Dalším typickým projevem je časný rozvoj kognitivního deficitu. Postupně se rozvíjí axiální parkinsonský syndrom (především bradykineze a rigidita) a retrocollis, které jsou rezistentní k dopaminergní terapii [51]. Pro tuto variantu je příznačná časná dysartrie a dysfagie [47]. Zastoupení PSP-RS oproti ostatním subtypům je asi 24 % [91]. Čím rychleji se vyvine kompletní obraz onemocnění, tím horší je prognóza. Jako negativní prediktory přežití byly uvedeny zejména pády do 1 roku od začátku nemoci zvyšující riziko úrazu a následné imobilizace, rozvoj inkontinence, kognitivního deficitu a především dysfagie, neboť aspirační pneumonie bývá nejčastější příčinou smrti [92,93]. Tento typ je spojen s rychlou progresí onemocnění s trváním zhruba 6–8 let [94].
PSP s dominantním parkinsonizmem (PSP-P)
Tito pacienti vykazují parkinsonský syndrom vč. třesu s asymetrickým postižením končetin. Parciální odpovídavost na levodopu bývá přechodně zachována. Příznaky typické pro variantu PSP-RS, zahrnující okohybnou poruchu, posturální instabilitu a kognitivní deficit, se obvykle objevují až po 5–6 letech progrese onemocnění [91]. Z důvodu pomalé progrese a částečné dopaminergní odpovídavosti může být tento typ často zaměňován za PN. K odlišení od PN může pomoci i pseudobulbární postižení s dysartrií a dysfagií rozvíjející se u PSP-P dříve [94,95]. Medián přežití těchto pacientů je uváděn v rozmezí 7–12 let [94]. Některé studie uvádějí u PSP-P nejdelší dobu přežití ze všech variant (s trváním nemoci až 28 let) [96].
PSP s progresivními zárazy při chůzi (PSP-PGF)
Hlavním příznakem této varianty je rozvoj akineze a zárazů při chůzi (freezing) v prvním roce onemocnění, kdy se mohou objevit startovací hesitace, festinace, pulze a eventuálně zadrhávání v řeči, psaní a jiných motorických činnostech [91]. Freezing a akineze mohou být dlouho jedinými příznaky této varianty. Rozvinout se mohou i bradykineze a rigidita končetin bez zlepšení po podání levodopy. Teprve později se přidávají instabilita s pády a okohybná porucha. Vzhledem k tomu, že pacienti mnohdy dospějí do klasického obrazu PSP-RS i po více než 10 letech, nebo do něj nikdy nedospějí, zdá se být tato varianta o něco příznivější nežli ostatní.
PSP s dominantním okulomotorickým postižením (PSP-OM)
Poruchy očních pohybů zde mohou být dlouho jediným příznakem. Okulomotorické dysfunkce jsou vyjádřeny zpomalením vertikálních (později i horizontálních) sakád, popřípadě jsou přítomny sakadické intruze, porucha optokinetického nystagmu a další specifické obtíže (viz výše okohybná porucha). Dle studií provedených v minulosti se tato varianta týká asi 7 % pacientů, patří tedy k nejvzácnějším [96].
PSP s kortikobazálním syndromem (PSP-CBS)
Kortikobazální varianta se projevuje apraxií končetin, dystonií, poruchou kortikálních senzorických funkcí a syndromem cizí končetiny („alien hand syndrom“). Zmíněné příznaky bývají velice často asymetrické v korespondenci s asymetrickou kontralaterální hemisferální atrofií. Dále se vyskytují apraxie řeči, obličeje, myoklonus a příznaky postižení pyramidového systému. Přítomny jsou i apatie a abulie [1]. Délka onemocnění může být opět mezi 2 a 6 lety [94].
PSP s dominujícím řečovým/jazykovým postižením (PSP-SL)
Tato varianta se projevuje jako apraxie řeči nebo primární nonfluentní afázie. V této fázi onemocnění může být klinický obraz kompatibilní jak s onemocněním PSP, tak i s kortikobazální degenerací [97]. Apraxie řeči se zpomalením, ztrátou prozódie, zhoršenou výbavností a chybami při slovní produkci může být rovněž způsobena na podkladě FTLD [94,98]. Pacienti PSP s primární nonfluentní afázií mohou být oproti kortikobazální degeneraci odlišeni na základě těžké dysartrie, relativně větší atrofie bílé hmoty a rozsáhlejšího kmenového postižení u PSP [99].
PSP s dominantní posturální instabilitou (PSP-PI)
U této varianty může být po dlouhou dobu jediným dominujícím příznakem posturální instabilita. Instabilita se může projevit v pull testu – více než 2 kroky nazad do 3 let od prvních příznaků nemoci. Vyšší míru jistoty pak představuje reference neprovokovaných pádů nazad [47]. Klasickou supranukleární pohledovou parézu však pacienti vyvinou až ve velice pozdním stadiu onemocnění, nebo ji nikdy nevyvinou, stejně jako ostatní motorické příznaky a kognitivní či behaviorální poruchy.
PSP s dominantním frontálním postižením (PSP-F)
Nejdůležitějšími příznaky u PSP-F jsou poruchy kognitivních funkcí a chování předcházející motorickou manifestaci. Ty zahrnují sociální desinhibici, impulzivitu, perseverace, abulii/apatii, oploštění emocí a ztrátu empatie. Kognitivní stránka je vyjádřena především subkortikálním deficitem spojeným s úbytkem frontostriatálních spojů projevujícím se dysexekutivním syndromem a bradyfrenií. Pokud se neobjeví okohybná porucha, je tato varianta prakticky nerozlišitelná od jiných příčin FTLD. Z dalších neurologických příznaků se mohou časně rozvíjet bradykineze, tremor a pády. Medián přežití je zde okolo 8 let [94].
PSP s dominantním cerebelárním postižením (PSP-C)
U tohoto typu po několik let dominuje mozečkový syndrom, ke kterému se až později přidávají časné pády/instabilita, supranukleární paréza pohledu, dysfagie, retrocollis, rigidita a frontální symptomy. U PSP-C se obvykle neobjevuje nystagmus. Případy byly popsány převážně u japonské populace, kde bývá průměrná délka přežití kolem 7 let [94].
PSP s dominantní primární laterální sklerózou (PSP-PLS)
Jedná se o kombinaci příznaků PSP s primární laterální sklerózou vyznačující se poruchou horního motoneuronu, ale vzácně se mohou objevit i případy s postižením dolního motoneuronu. Iniciálně se může vyskytovat dysartrie. Z příznaků podporujících diagnózu PSP u popsaných pacientů dominují axiální i končetinová rigidita, posturální instabilita, poruchy chůze, bradykineze a hypofonie. Méně často jsou přítomné okohybná porucha, končetinová apraxie, dystonie, dysfagie, kognitivní deficit a změny osobnosti [100]. Délka přežití se u sledovaných pacientů pohybovala od 3 do 12 let [94]. Stejně jako předchozí varianta je i PSP-PLS vyňata z recentních diagnostických kritérií pro možnost záměny s ALS [53].
Kromě možnosti rozdílné klinické manifestace v rámci zmíněných subtypů existuje PSP i ve formě komorbidity k jiné neurodegeneraci [11]. Byly popsány koexistence PSP a ALS [10], současný výskyt s AN, nemocí s argyrofilními zrny s nebo s FTLD-TDP [9,101], kde existuje předpoklad sdílené patogeneze a genetických podkladů [102]. Popsán byl i patologický překryv PSP a MSA [11,103].
Zmíněná nejrůznější klinická vyjádření syndromu mohou korelovat s lokalitou a množstvím akumulace tau proteinu. Existuje tedy předpoklad existence preklinických stadií a poté překryvu jednotlivých typů v průběhu času s možným přechodem do PSP-RS spolu s šířením tau proteinu v CNS [104].
Diferenciální diagnostika
V diferenciální diagnostice zvažujeme v zásadě 3 okruhy, kterými jsou neurodegenerativní onemocnění, sekundární parkinsonské syndromy a infekční/autoimunitní encefalitidy.
Při diagnostice je nutné brát v potaz tzv. mandatorní vylučující kritéria vyjadřující hlavní známky svědčící pro jiná onemocnění (tab. 2). Jsou jimi významná ztráta epizodické paměti, autonomní dysfunkce, nevysvětlitelné vizuální halucinace a fluktuace lucidity, končetinová ataxie, multisegmentální postižení horního a dolního motoneuronu, náhlý nástup či skokovitá progrese, jiné vysvětlení pádů, anamnéza encefalitidy nebo strukturální změny na MR charakteristické pro jinou patologii.
Parkinsonský syndrom
V první řadě je nutno vyloučit PN ověřením odpovědi na adekvátní dopaminergní terapii a léčitelné příčiny parkinsonizmu zahrnující normotenzní hydrocefalus, eventuálně vzácné autoimunitní encefalitidy a metabolické encefalopatie. Pro PN je typický asymetrický parkinsonský syndrom s dobrou odpovědí na dopaminergní medikaci. Posturální instabilita a kognitivní deficit se zde vyskytují zpravidla až později v průběhu nemoci.
Pro multisystémovou atrofii svědčí časnýrozvoj posturální instability s pády a parkinsonský syndrom v kombinaci s autonomní dysfunkcí. Eventuálně se zde objevuje cerebelární a pyramidová symptomatika. Kognitivní deficit se zde rovněž rozvíjí až v pozdějších stadiích.
Kortikobazální syndrom zahrnuje progresivní asymetrickou končetinovou apraxii, dystonii, syndrom alien hand, parkinsonismus a frontální kognitivní deficit. Kromě kortikobazální degenerace a progresivní supranukleární obrny může být podkladem tohoto syndromu i AN nebo FTLD. K odlišení může pomoci MR ukazující větší podíl frontální asymetrie u kortikobazální degenerace a eventuálně kognitivní profil nemocných [105].
Byly popsány i případy nemoci s Lewyho tělísky s vertikální parézou pohledu a akinezí. Pro nemoc s Lewyho tělísky jsou zásadní fluktuace kognitivního stavu a poruch chování (komplexní vizuální halucinace). Časně se objevují i kolapsové stavy a pády, které mohou být zapříčiněny jak ortostatickou hypotenzí, tak i posturální instabilitou nebo fluktuacemi psychického stavu [106].
Časné pády s posturální instabilitou, obdobný profil frontálního kognitivního deficitu, močovou inkontinenci a oploštění mezencefala na MR může zapříčinit i normotenzní hydrocefalus. V případě diagnostických rozpaků lze využít kvantitativních metod MR (např. indexu MRPI) [107].
Vícečetné vaskulární postižení mozku se může projevovat apraxií chůze, posturální instabilitou s pády, kognitivním deficitem a případně frontálními poruchami chování. Důležitý je zde nález ischemických změn na CT a/nebo MR. Vaskulární postižení i normotenzní hydrocefalus se přitom mohou kombinovat s různými neurodegenerativními procesy. Vyloučit bychom měli i léky indukovaný parkinsonský syndrom (zejména při dlouhodobém užívání antipsychotik a jejich derivátů).
Poruchy okulomotoriky
Okohybné poruchy mohou být příznakem kortikobazální degenerace, Whippleovy nemoci, Niemann-Pickovy nemoci typu C (NPC) a postižení mezencefala. Supranukleární pohledová paréza může být způsobena také ischemií v povodí a. cerebri posterior, tumorem epifýzy, paraneoplazií, prionovým onemocněním nebo autoimunitní encefalitidou. Opatrnost je na místě u izolovaného mírného omezení amplitudy směrem vzhůru, které může být fyziologicky přítomno u starších pacientů [108]. U Whippleovy nemoci je v neurologickém obraze typicky přítomna triáda kognitivního deficitu, supranukleární oftalmoplegie a myoklonu, s eventuálním doprovodným parkinsonským syndromem. Zhruba u pětiny pacientů se vyskytují též okulomastikatorní myorytmie, které jsou pro tuto nemoc patognomické.
Supranukleární pohledová paréza se může objevit i v rámci pozdní adultní formy NPC [109,110]. Jedná se o lysozomální onemocnění se střádáním lipidů zapříčiněné genetickou mutací genu pro sfingomyelin fosfodiesterázu 1. K odlišení od PSP může pomoci přítomnost cerebelární ataxie nebo významné končetinové dystonie komplikující chůzi [111]. Mimo zmíněné obtíže lze u NPC vidět známky kognitivního úpadku až demence, behaviorálních obtíží, dysartrie a dysfagie.
Zpomalení vertikálních sakád je vzácněji vyjádřeno i u Gaucherovy nemoci, což je další z řady lysosomálních střádavých onemocnění s obtížemi typu hepatosplenomegalie, anémie a trombocytopenie. Zde na podkladě deficitu enzymu glukocerebrosidázy při mutaci genu pro beta-glukocerebrosidázu. Gaucherova nemoc je však mnohem častěji spojena s parézou horizontálního pohledu [112].
Léčba
U PSP se v některých případech může parkinsonský syndrom částečně zlepšit po nasazení levodopy, a to zejména u varianty PSP-PN (tab. 8). Klinický efekt je obvykle zřejmý až při dávkách vyšších než 1 g levodopy (1 500–2 000 mg/den). Na rozdíl od PN nastupují pozitivní účinky s několikadenním až týdenním odstupem a efekt je často nedostatečný a krátkodobý. Proto je nutné léčbu levodopou zahájit co nejdříve a zvyšovat dávku do co největšího možného dobře tolerovatelného množství. Pokud se pacient dlouhodobě nelepší ani při dávkách 1 500–2 000 mg, v léčbě nepokračujeme. V ojedinělých případech pacienti částečně profitují z dávek převyšujících 2 g/den. Přínos léčby lze ověřit náhlým vysazením celkové denní dávky. V případě, že v následujících 7–10 dnech nenastane zhoršení klinického stavu, pacient s jistotou z léčby neprofituje. Při užívání dopaminergní medikace je taktéž třeba brát v potaz možný rozvoj nežádoucích účinků, jakými jsou např. vředová choroba gastroduodena a dekompenzace psychického stavu [56,113,114]. Jako prevenci rozvoje nauzey a arteriální hypotenze je vhodné podávat v úvodu léčby domperidon 3× 10 mg denně. Agonisté dopaminu se pro nedostatečný účinek u PSP nedoporučují [3].
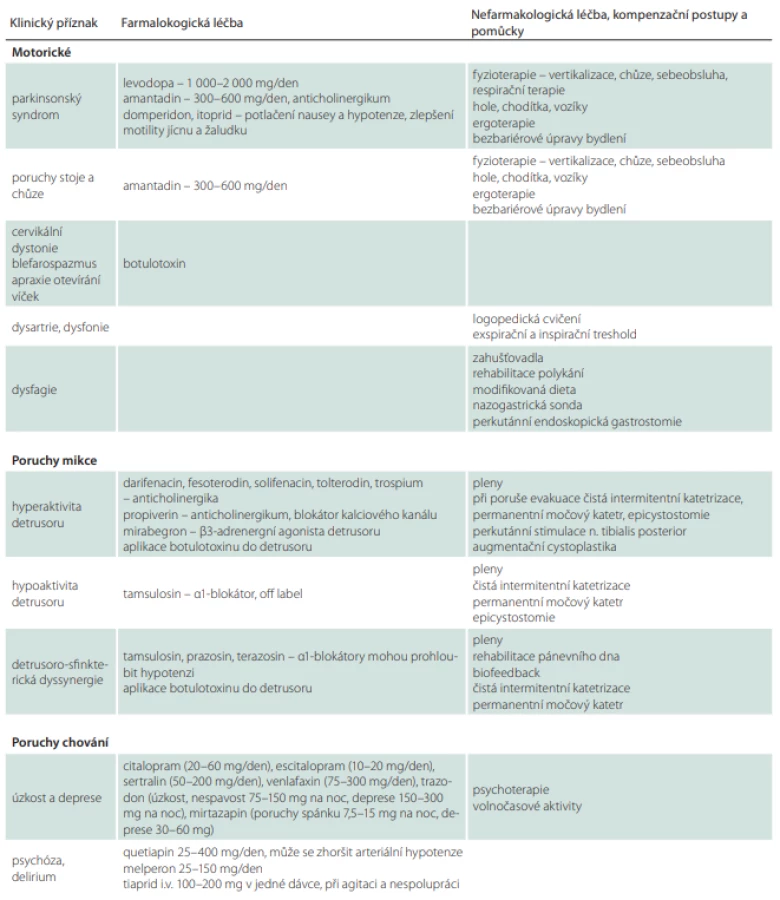
Dále se využívá amantadin, který může mít příznivý efekt na poruchy stability, chůze a dystonie [76,115]. V praxi jsou většinou používány dávky kolem 400–600 mg (např. v rozložení 200–200–100 mg). Amantadin může způsobovat zhoršení kognitivního deficitu a rozvoj poruch chování (iritabilitu, agitaci, psychózu, delirium) [116,117].
Fokální dystonické syndromy, zejména retrocollis nebo blefarospasmus, se mohou zlepšit po botulotoxinu. Při aplikaci do krčních svalů je kvůli možnému zhoršení dysfagie nutná opatrnost. Botulotoxin může mít dobrý efekt i na apraxii otevírání víček. Může být taktéž aplikován do slinných žláz v případě sialorhey, na tuto indikaci však nemá úhradu. Produkci slin lze dále snížit a zahustit pomocí anticholinergik (scopolamin, amitriptylin, 1% kapky atropinu) [3]. Anticholinergní preparáty mohou zhoršovat psychické funkce.
V minulosti bylo u některých pacientů pozorováno mírné zlepšení motorických funkcí při užívání amitriptylinu v dávkách 75–150 mg, při užívání této medikace však existuje vysoké riziko nežádoucích účinků ve smyslu sedace, další deteriorace kognitivních funkcí a poruch chování [116,118].
V případě podezření na apatii či depresi a při výskytu úzkostné symptomatiky je žádoucí provést terapeutický pokus s antidepresivní medikací. Iritabilitu, agresi, psychotické a deliriózní stavy léčíme symptomaticky antipsychotiky. Iritabilita a agresivní projevy se mohou zlepšit po podání inhibitorů zpětného vychytávání serotoninu. Ke zlepšení psychického stavu může přispět redukce polypragmazie, zejména pak dopaminergní medikace a léků s anticholinergními účinky.
V případě močové urgence (tab. 8) lze použít spasmolytik, nicméně je třeba uvážit, že anticholinergní medikace může zhoršit funkci detruzoru s možným rozvojem akutní retence [3]. K empirickému nasazení medikace by se tedy nemělo přistupovat bez zhodnocení vyprazdňovacích funkcí pomocí uroflowmetrie [74].
Existují i ojedinělé případy, kdy byly okulomotorika a další motorické funkce na přechodnou dobu zlepšeny použitím GABA agonisty zolpidemu v denní dávce 10–25 mg [3,119–122].
Při fotofobii je vhodné použít stínění alespoň v podobě slunečních brýlí [123].
Poruchy řeči a polykání je vhodné konzultovat s logopedem zaměřeným na tuto problematiku. Kontakty na logopedy lze získat na webové adrese [124]. Respirační terapie zlepšuje nejen ventilační kapacitu, ale i artikulaci, fonaci a polykání. Logoped podle typu a závažnosti poruchy polykání indikuje modifikaci konzistence stravy a tekutin. V případě, že nelze zajistit bezpečný nebo dostatečný orální příjem (25–30 kcal/kg/d, protein 1,2–1,5 g/kg/d), je vhodné zvážit včasné zavedení perkutánní gastrostomie. Poruchy výživy je možné konzultovat s nutričním terapeutem nebo nutricionistou, jejichž kontakty jsou dostupné na adrese společnosti pro klinickou výživu a intenzivní metabolickou péči [125].
Do nefarmakologické terapie řadíme fyzioterapii s možností omezení pádů pomocí nácviku stability a rehabilitací chůze [126]. Určité zlepšení bylo pozorováno při zařazení vyrovnaného protahování, posilování, balančního a chůzového tréninku [120,127–129]. Nicméně výsledky dosavadních studií poskytují pouze nízkou evidenci, proto v praxi postupujeme empiricky, podobně jako u pacientů s PN. Kvůli očekávanému kognitivnímu zhoršování je vhodné zařadit nefarmakologické postupy co nejdříve. Zapomínat by se nemělo ani na podstatnou úlohu paliativní péče [130]. Pacienti a jejich pečovatelé mohou získat užitečné informace na webových stránkách spolku pro atypické parkinsonské syndromy [131].
Experimentální léčba
Existuje i několik případů stimulace pedunkulopontinního jádra s cílem ovlivnění stability, nicméně prozatím neexistuje dostatek dat, které by ověřily dlouhodobý efekt [61,132]. Substituce pomocí koenzymu Q10 ve vysokých dávkách (5 mg/kg 3× denně), která byla zkoušena pro narušení mitochondriálního metabolizmu a oxidativní fosforylace, se ukázala být neúčinnou [133]. Na základě teorie prion-like transmise proteinu tau se v současnosti zkouší protilátky proti tau proteinu. Je nutné provedení dalších klinických testů a posouzení efektu [17,134,135].
Grantová podpora
Podpořeno grantem Karlovy Univerzity PROGRES Q27/LF1, projektem Evropské referenční sítě pro vzácná neurologická onemocnění Project ID No 739510
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem práce nemají žádný konflikt zájmů.
Poděkování
Autoři děkují za možnost konzultace vybraných oblastí a cenné rady prof. MUDr. Janu Rothovi, CSc., MUDr. Petru Duškovi, Ph.D., Mgr Haně Růžičkové, Ph.D. a PhDr. Olze Klempířové, Ph.D.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
doc. MUDr. Jiří Klempíř, Ph.D.
Centrum extrapyramidových onemocnění Neurologická klinika a Centrum klinických neurověd 1. LF UK a VFN v Praze
Kateřinská 30
120 00 Praha
e-mail jiri.klempir@vfn.cz
Zdroje
1. Ling H. Clinical approach to progressive supranuclear palsy. J Mov Disord 2016; 9 (1): 3–13. doi: 10.14802/jmd.15060.
2. Stamelou M, Giagkou N, Höglinger GU. One decade ago, one decade ahead in progressive supranuclear palsy. Mov Disord 2019; 34 (9): 1284–1293. doi: 10.1002/mds.27788.
3. McFarland, NR. Diagnostic approach to atypical parkinsonian syndromes. Continuum (Minneap Minn) 2016; 22 (4 Movement Disorders): 1117–1142. doi: 10.1212/CON.0000000000000348.
4. Wenning GK, Litvan I, Tolosa E. Milestones in atypical and secondary parkinsonisms. Mov Disord 2011; 26 (6): 1083–1095. doi: 10.1002/mds.23713.
5. Baba Y, Putzke JD, Whaley NR et al. Progressive supranuclear palsy: phenotypic sex differences in a clinical cohort. Mov Disord 2006; 21 (5): 689–692. doi: 10.1002/mds.20769.
6. Litvan I, Lees PS, Cunningham CR et al. Environmental and occupational risk factors for progressive supranuclear palsy: case-control study. Mov Disord 2016; 31 (5): 644–652. doi: 10.1002/mds.26512.
7. Steele JC, Capparos-Lefebvre D, Lees AJ et al. Progressive supranuclear palsy and its relation to pacific foci of the parkinsonism-dementia complex and Guadeloupean parkinsonism. Parkinsonism Relat Disord 2002; 9 (1): 39–54. doi: 10.1016/s1353-8020 (02) 00043-3.
8. Shoeibi A, Olfati N, Litvan I. Frontrunner in translation: progressive supranuclear palsy. Front Neurol 2019; 10: 1125. doi: 10.3389/fneur.2019.01125.
9. Mimuro M, Yoshida M. Chameleons and mimics: Progressive supranuclear palsy and corticobasal degeneration. Neuropathology 2020; 40 (1): 57–67. doi: 10.1111/neup.12590.
10. Fujita K, Matsubura T, Miyamoto R et al. Co-morbidity of progressive supranuclear palsy and amyotrophic lateral sclerosis: a clinical-pathological case report. BMC Neurol 2019; 19 (1): 168. doi: 10.1186/s12883-019-1402-7.
11. Ando S, Kanazawa M, Onodera O. Progressive supranuclear palsy with predominant cerebellar ataxia. J Mov Disord 2019; 13 (1): 20–26. doi: 10.14802/jmd.19061.
12. Vogels T, Leuzy A, Cicognola C et al. Propagation of tau pathology: integrating insights from postmortem and in vivo studies. Biol Psychiatry 2019; 87 (9): 808–818. doi: 10.1016/j.biopsych.2019.09.019.
13. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol 2017; 133 (5): 665–704. doi: 10.1007/s00401-017-1707-9.
14. Zhang CC, Xing A, Tan MS et al. The role of MAPT in neurodegenerative diseases: genetics, mechanisms and therapy. Mol Neurobiol 2016; 53 (7): 4893–4904. doi: 10.1007/s12035-015-9415-8.
15. Huang Y, Wu Z, Zhou B. Behind the curtain of tauopathy: a show of multiple players orchestrating tau toxicity. Cell Mol Life Sci 2016; 73 (1): 1–21. doi: 10.1007/s00018-015-2042-8.
16. Pérez MJ, Jara C, Quintanilla RA. Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front Neurosci 2018; 12: 441. doi: 10.3389/fnins.2018.00441.
17. Colin M, Dujardin S, Schraen-Maschke S et al. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol 2020; 139 (1): 3–25. doi: 10.1007/s00401-019-02087-9.
18. Narasimhan S, Changolkar L, Riddle DM et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J Exp Med 2020; 217 (2): e20190783. doi: 10.1084/jem.20190783.
19. Komori T. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick‘s disease. Brain Pathol 1999; 9 (4): 663–679. doi: 10.1111/j.1750-3639.1999.tb00549.x.
20. Ling H, De Silva R, Massey LA et al. Characteristics of progressive supranuclear palsy presenting with corticobasal syndrome: a cortical variant. Neuropathol Appl Neurobiol 2014; 40 (2): 149–163. doi: 10.1111/nan.12037.
21. Sakurai K, Tokumaru AM, Shimoji K et al. Beyond the midbrain atrophy: wide spectrum of structural MRI finding in cases of pathologically proven progressive supranuclear palsy. Neuroradiology 2017; 59 (5): 431–443. doi: 10.1007/s00234-017-1812-4.
22. Oyanagi K, Tsuchiya K, Yamazaki M et al. Substantia nigra in progressive supranuclear palsy, corticobasal degeneration, and parkinsonism-dementia complex of Guam: specific pathological features. J Neuropathol Exp Neurol 2001; 60 (4): 393–402. doi: 10.1093/jnen/60.4.393.
23. MacLaren DA, Ljungberg TL, Griffin ME et al. Pedunculopontine tegmentum cholinergic loss leads to a progressive decline in motor abilities and neuropathological changes resembling progressive supranuclear palsy. Eur J Neurosci 2018; 48 (12): 3477–3497. doi: 10.1111/ejn.14212.
24. Landwehrmeyer B, Palacios JM. Alterations of neurotransmitter receptors and neurotransmitter transporters in progressive supranuclear palsy. J Neural Transm Suppl 1994; 42: 229–246. doi: 10.1007/978-3-7091-6641-3_18.
25. Respondek G, Kurz C, Arzberger T et al. Which ante mortem clinical features predict progressive supranuclear palsy pathology? Mov Disord 2017; 32 (7): 995–1005. doi: 10.1002/mds.27034.
26. Schirinzi T, Sancesario GM, Di Lazzaro G et al. Clinical value of CSF amyloid-beta-42 and tau proteins in progressive supranuclear palsy. J Neural Transm (Vienna) 2018; 125 (9): 1373–1379. doi: 10.1007/s00702-018-1893-1.
27. Wang SY, Chen W, Xu W et al. Neurofilament light chain in cerebrospinal fluid and blood as a biomarker for neurodegenerative diseases: a systematic review and meta-analysis. J Alzheimers Dis 2019; 72 (4): 1353–1361. doi: 10.3233/JAD-190615.
28. Donker Kaat L, Meeter LH, Zheng Chiu W et al. Serum neurofilament light chain in progressive supranuclear palsy. Parkinsonism Relat Disord 2018; 56: 98–101. doi: 10.1016/j.parkreldis.2018.06.018.
29. Kataoka H, Sugie K. Serum adiponectin levels between patients with Parkinson‘s disease and those with PSP. Neurol Sci 2020; 41 (5): 1125–1131. doi: 10.1007/s10072-019-04216-4.
30. Eraslan C, Acaer A, Guneyli S et al. MRI evaluation of progressive supranuclear palsy: differentiation from Parkinson‘s disease and multiple system atrophy. Neurol Res 2019; 41 (2): 110–117. doi: 10.1080/01616412.2018.1541115.
31. Mueller C, Hussl A, Krismer F et al. The diagnostic accuracy of the hummingbird and morning glory sign in patients with neurodegenerative parkinsonism. Parkinsonism Relat Disord 2018; 54: 90–94. doi: 10.1016/j.parkreldis.2018.04.005.
32. Mostile G, Nicoletti A, Cicero CE et al. Magnetic resonance parkinsonism index in progressive supranuclear palsy and vascular parkinsonism. Neurol Sci 2016; 37 (4): 591–595. doi: 10.1007/s10072-016-2489-x.
33. Quattrone A, Morelli M, Nigro S et al. A new MR imaging index for differentiation of progressive supranuclear palsy-parkinsonism from Parkinson‘s disease. Parkinsonism Relat Disord, 2018. 54: 3–8. doi: 10.1016/j.parkreldis.2018.07.016.
34. Constantinides VC, Paraskevas GP, Stamboulis E et al. Simple linear brainstem MRI measurements in the differential diagnosis of progressive supranuclear palsy from the parkinsonian variant of multiple system atrophy. Neurol Sci 2018; 39 (2): 359–364. doi: 10.1007/s10072-017-3212-2.
35. Quattrone A, Morelli M, Quattrone A et al. Magnetic resonance parkinsonism index for evaluating disease progression rate in progressive supranuclear palsy: a longitudinal 2-year study. Parkinsonism Relat Disord 2020; 72: 1–6. doi: 10.1016/j.parkreldis.2020.01.019.
36. Oktay C, Özkaynak SS, Eseroğlu E et al. Contribution of the mesencephalon indices to differential diagnosis of parkinsonian disorders. Can Assoc Radiol J 2020; 71 (1): 100–109. doi: 10.1177/0846537119888411.
37. Mitchell T, Archer DB, Chu WT et al. Neurite orientation dispersion and density imaging (NODDI) and free-water imaging in parkinsonism. Hum Brain Mapp 2019; 40 (17): 5094–5107. doi: 10.1002/hbm.24760.
38. Richter D, Katsanos AH, Schroeder C et al. Lentiform nucleus hyperechogenicity in parkinsonian syndromes: a systematic review and meta-analysis with consideration of molecular pathology. Cells 2019; 9 (1): 2. doi: 10.3390/cells9010002.
39. Alonso-Canovas A, Ferrairó JI, Martínez-Torres I et al. Transcranial sonography in atypical parkinsonism: How reliable is it in real clinical practice? A multicentre comprehensive study. Parkinsonism Relat Disord, 2019. 68: 40–45. doi: 10.1016/j.parkreldis.2019.09.032.
40. Vintonyak O, Gorges M, Müller HP et al. Patterns of eye movement impairment correlate with regional brain atrophy in neurodegenerative parkinsonism. Neurodegener Dis 2017; 17 (4–5): 117–126. doi: 10.1159/000454880.
41. Panyakaew P, Anan C, Bhidayasiri R. Posturographic abnormalities in ambulatory atypical parkinsonian disorders: Differentiating characteristics. Parkinsonism Relat Disord 2019; 66: 94–99. doi: 10.1016/j.parkreldis.2019.07.016.
42. Ondo W, Warrior D, Overby A et al. Computerized posturography analysis of progressive supranuclear palsy: a case-control comparison with Parkinson‘s disease and healthy controls. Arch Neurol 2000; 57 (10): 1464–1469. doi: 10.1001/archneur.57.10.1464.
43. Dale ML, Horak FB, Wright WG et al. Impaired perception of surface tilt in progressive supranuclear palsy. PLoS One 2017; 12 (3): e0173351. doi: 10.1371/journal.pone.0173351.
44. Kammermeier S, Maierbeck K, Dietrich L et al. Qualitative postural control differences in idiopathic Parkinson‘s disease vs. progressive supranuclear palsy with dynamic-on-static platform tilt. Clin Neurophysiol 2018; 129 (6): 1137–1147. doi: 10.1016/j.clinph.2018.03.002.
45. Tykalova T, Rusz J, Klempir J et al. Distinct patterns of imprecise consonant articulation among Parkinson‘s disease, progressive supranuclear palsy and multiple system atrophy. Brain Lang 2017; 165: 1–9. doi: 10.1016/j.bandl.2016.11.005.
46. de Yébenes JG, Sarasa JL, Daniel SE et al. Familial progressive supranuclear palsy. Description of a pedigree and review of the literature. Brain 1995; 118 (Pt 5): 1095–1103. doi: 10.1093/brain/118.5.1095.
47. Ali F, Josephs K. The diagnosis of progressive supranuclear palsy: current opinions and challenges. Expert Rev Neurother 2018; 18 (7): 603–616. doi: 10.1080/14737175.2018.1489241.
48. Im SY, Kim YE, Kim YJ. Genetics of progressive supranuclear palsy. J Mov Disord 2015; 8 (3): 122–129. doi: 10.14802/jmd.15033.
49. Ingelsson M, Ramasamy K, Russ C et al. Increase in the relative expression of tau with four microtubule binding repeat regions in frontotemporal lobar degeneration and progressive supranuclear palsy brains. Acta Neuropathol 2007; 114 (5): 471–479. doi: 10.1007/s00401-007-0280-z.
50. Borroni B et al. Genetic bases of progressive supranuclear palsy: the MAPT tau disease. Curr Med Chem 2011; 18 (17): 2655–2660. doi: 10.2174/092986711795933722.
51. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964; 10: 333–359. doi: 10.1001/archneur.1964.00460160003001.
52. Kammermeier S, Dietrich L, Maierbeck K et al. Postural stabilization differences in idiopathic parkinson‘s disease and progressive supranuclear palsy during self-triggered fast forward weight lifting. Front Neurol 2017; 8: 743. doi: 10.3389/fneur.2017.00743.
53. Hoglinger GU, Respondek G, Stamelou M et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord 2017; 32 (6): 853–864. doi: 10.1002/mds.26987.
54. Burn DJ, Lees AJ. Progressive supranuclear palsy: where are we now? Lancet Neurol 2002; 1 (6): 359–369. doi: 10.1016/s1474-4422 (02) 00161-8.
55. Cronin T, Arshad Q, Seemungal BM. Vestibular deficits in neurodegenerative disorders: balance, dizziness, and spatial disorientation. Front Neurol 2017; 8: 538. doi: 10.3389/fneur.2017.00538.
56. Fujioka S, Algom AA, Murray ME et al. Tremor in progressive supranuclear palsy. Parkinsonism Relat Disord 2016; 27: 93–97. doi: 10.1016/j.parkreldis.2016.03.015.
57. Anagnostou E, Karavasilis E, Potiri I et al. A cortical substrate for square-wave jerks in progressive supranuclear palsy. J Clin Neurol 2020; 16 (1): 37–45. doi: 10.3988/jcn.2020.16.1.37.
58. Quinn N. The „round the houses“ sign in progressive supranuclear palsy. Ann Neurol 1996; 40 (6): 951. doi: 10.1002/ana.410400630.
59. Matsumoto H, Inaba T, Kakumoto T et al. Progressive supranuclear palsy with wall-eyed bilateral internuclear ophthalmoplegia syndrome: authors‘ second case. Case Rep Neurol 2019; 11 (2): 205–208. doi: 10.1159/000501394.
60. Batla A, Nehru R, Vijay T. Vertical wrinkling of the forehead or Procerus sign in progressive supranuclear palsy. J Neurol Sci 2010; 298 (1–2): 148–149. doi: 10.1016/j.jns.2010.08.010.
61. Marsili L, Bologna M, Kjovic M et al. Dystonia in atypical parkinsonian disorders. Parkinsonism Relat Disord 2019; 66: 25–33. doi: 10.1016/j.parkreldis.2019.07.030.
62. Boxer AL, Lang AE, Grossman M et al. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol 2014; 13 (7): 676–685. doi: 10.1016/S1474-4422 (14) 70088-2.
63. Kim JH, McCann CM. Communication impairments in people with progressive supranuclear palsy: a tutorial. J Commun Disord 2015; 56: 76–87. doi: 10.1016/j.jcomdis.2015.06.002.
64. Skodda S, Visser W, Schlegel U. Acoustical analysis of speech in progressive supranuclear palsy. J Voice 2011; 25 (6): 725–731. doi: 10.1016/j.jvoice.2010.01.002.
65. Clark HM, Stierwalt JA, Tosakulwong N et al. Dysphagia in progressive supranuclear palsy. Dysphagia 2020; 35 (4): 667–676. doi: 10.1007/s00455-019-10073-2.
66. Iwasaki Y, Yoshida M, Hashizume Y et al. Widespread spinal cord involvement in progressive supranuclear palsy. Neuropathology 2007; 27 (4): 331–340. doi: 10.1111/j.1440-1789.2007.00787.x.
67. Vitaliani R, Scaravilli T, Egarter-Vigl E et al. The pathology of the spinal cord in progressive supranuclear palsy. J Neuropathol Exp Neurol 2002; 61 (3): 268–274. doi: 10.1093/jnen/61.3.268.
68. Gawel M, Jamrozik Z, Szmidt-Salkowska E et al. Electrophysiological features of lower motor neuron involvement in progressive supranuclear palsy. J Neurol Sci 2013; 324 (1–2): 136–139.
69. Nomura T, Inoue Y, Takigawa H et al. Comparison of REM sleep behaviour disorder variables between patients with progressive supranuclear palsy and those with Parkinson‘s disease. Parkinsonism Relat Disord 2012; 18 (4): 394–396. doi: 10.1016/j.parkreldis.2011.10.018.
70. Walsh CM, Ruoff L, Walker K et al. Sleepless night and day, the plight of progressive supranuclear palsy. Sleep 2017; 40 (11), zsx154. doi: 10.1093/sleep/zsx154.
71. Yousaf T, Pagano G, Wilson H et al. Neuroimaging of sleep disturbances in movement disorders. Front Neurol 2018; 9: 767. doi: 10.3389/fneur.2018.00767.
72. Oliveira MC, Ling H, Lees AJ et al. Association of autonomic symptoms with disease progression and survival in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 2019; 90 (5): 555–561. doi: 10.1136/jnnp-2018-319374.
73. Yamamoto T, Tateno F, Sakakibara R et al. Urinary dysfunction in progressive supranuclear palsy compared with other parkinsonian disorders. PLoS One 2016; 11 (2): e0149278. doi: 10.1371/journal.pone.0149278.
74. Kim KJ, Jeong SJ, Kim JM. Neurogenic bladder in progressive supranuclear palsy: a comparison with Parkinson‘s disease and multiple system atrophy. Neurourol Urodyn 2018; 37 (5): 1724–1730. doi: 10.1002/nau.23496.
75. Sulena, Gupta D, Sharma AK et al. Clinical profile of cognitive decline in patients with parkinson‘s disease, progressive supranuclear palsy, and multiple system atrophy. J Neurosci Rural Pract 2017; 8 (4): 562–568. doi: 10.4103/jnrp.jnrp_154_17.
76. Rittman T, Coyle-Gilchrist IT, Rowe JB. Managing cognition in progressive supranuclear palsy. Neurodegener Dis Manag 2016; 6 (6): 499–508. doi: 10.2217/nmt-2016-0027.
77. Burrell JR, Hodges JR, Rowe JB. Cognition in corticobasal syndrome and progressive supranuclear palsy: a review. Mov Disord 2014; 29 (5): 684–693. doi: 10.1002/mds.25872.
78. Smith DT, Archibald N. Spatial working memory in progressive supranuclear palsy. Cortex 2020; 122: 115–122. doi: 10.1016/j.cortex.2018.07.004.
79. Santangelo G, Cuoco S, Pellecchia MT et al. Comparative cognitive and neuropsychiatric profiles between Parkinson‘s disease, multiple system atrophy and progressive supranuclear palsy. J Neurol 2018; 265 (11): 2602–2613. doi: 10.1007/s00415-018-9038-x.
80. Sitek EJ, Wieczorek D, Konkel A et al. The pattern of verbal, visuospatial and procedural learning in Richardson variant of progressive supranuclear palsy in comparison to Parkinson‘s disease. Psychiatr Pol 2017; 51 (4): 647–659. doi: 10.12740/PP/OnlineFirst/62804.
81. Barker MS, Nelson NL, O’Sullivan JD et al. Energization and spoken language production: Evidence from progressive supranuclear palsy. Neuropsychologia 2018; 119: 349–362. doi: 10.1016/j.neuropsychologia.2018.09.004.
82. Robinson GA, Spooner D, Harrison WJ. Frontal dynamic aphasia in progressive supranuclear palsy: Distinguishing between generation and fluent sequencing of novel thoughts. Neuropsychologia 2015; 77: 62–75. doi: 10.1016/j.neuropsychologia.2015.08.001.
83. Sakae N, Josephs KA, Litvan I et al. Neuropathologic basis of frontotemporal dementia in progressive supranuclear palsy. Mov Disord 2019; 34 (11): 1655–1662. doi: 10.1002/mds.27816.
84. Fiorenzato E, Antonini A, Camparini V et al. Characteristics and progression of cognitive deficits in progressive supranuclear palsy vs. multiple system atrophy and Parkinson‘s disease. J Neural Transm (Vienna) 2019; 126 (11): 1437–1445. doi: 10.1007/s00702-019-02065-1.
85. Lansdall CJ, Coyle-Gilchrist IT, Simon Jones P et al. Apathy and impulsivity in frontotemporal lobar degeneration syndromes. Brain 2017, 140 (6): 1792–1807. doi: 10.1093/brain/awx101.
86. Schrag A, Sheikh S, Quinn NP et al. A comparison of depression, anxiety, and health status in patients with progressive supranuclear palsy and multiple system atrophy. Mov Disord 2010; 25 (8): 1077–1081. doi: 10.1002/mds.22794.
87. Belvisi D, Berardelli I, Suppa A et al. Neuropsychiatric disturbances in atypical parkinsonian disorders. Neuropsychiatr Dis Treat 2018; 14: 2643–2656. doi: 10.2147/NDT.S178263.
88. Litvan I, Agid Y, Calne D et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996; 47 (1): 1–9. doi: 10.1212/wnl.47.1.1.
89. Grimm MJ, Respondek G, Stamelou M et al. How to apply the movement disorder society criteria for diagnosis of progressive supranuclear palsy. Mov Disord 2019; 34 (8): 1228–1232. doi: 10.1002/mds.27666.
90. Armstrong MJ. Progressive supranuclear palsy: an update. Curr Neurol Neurosci Rep 2018; 18 (3): 12. doi: 10.1007/s11910-018-0819-5.
91. Lopez G, Bayulkem K, Hallett M. Progressive supranuclear palsy (PSP): Richardson syndrome and other PSP variants. Acta Neurol Scand 2016; 134 (4): 242–249. doi: 10.1111/ane.12546.
92. Litvan I, Mangone CA, McKee A et al. Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry 1996; 60 (6): 615–620. doi: 10.1136/jnnp.60.6.615.
93. Arena JE, Weigand SD, Whitwell JL et al. Progressive supranuclear palsy: progression and survival. J Neurol 2016; 263 (2): 380–389. doi: 10.1007/s00415-015-7990-2.
94. Respondek G, Höglinger GU. The phenotypic spectrum of progressive supranuclear palsy. Parkinsonism Relat Disord 2016; 22 (Suppl 1): S34–36. doi: 10.1016/j.parkreldis.2015.09.041.
95. Albert ML, Feldman RG, Willis AL. The “subcortical dementia” of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 1974; 37 (2): 121–130. doi: 10.1136/jnnp.37.2.121.
96. Respondek G, Stamelou M, Kurz C et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord 2014; 29 (14): 1758–1766. doi: 10.1002/mds.26054.
97. Josephs KA, Duffy JR. Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol 2008; 21 (6): 688–692. doi: 10.1097/WCO.0b013e3283168ddd.
98. Dickson DW, Ahmed Z, Algom AA et al. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 2010; 23 (4): 394–400. doi: 10.1097/WCO.0b013e32833be924.
99. Santos-Santos MA, Mandelli ML, Binney RJ et al. Features of patients with nonfluent/agrammatic primary progressive aphasia with underlying progressive supranuclear palsy pathology or corticobasal degeneration. JAMA Neurol 2016; 73 (6): 733–742. doi: 10.1001/jamaneurol.2016.0412.
100. Josephs KA, Katsuse O, Beccano-Kelly DA et al. Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol 2006; 65 (4): 396–405. doi: 10.1097/01.jnen.0000218446.38158.61.
101. Storey K, Johanidesová S, Matěj R et al. FTLD-TDP and progressive supranuclear palsy in comorbidity-a report of two cases with different clinical presentations. Neurocase 2017; 23 (1): 5–11. doi: 10.1080/13554794.2016.1264058.
102. Kertesz A, Finger R, Murell J et al. Progressive supranuclear palsy in a family with TDP-43 pathology. Neurocase 2015; 21 (2): 178–84. doi: 10.1080/13554794.2013.878729.
103. Menšíková K, Matěj R, Tučková L et al. Progressive supranuclear palsy phenotype mimicking synucleinopathies. J Neurol Sci 2013; 329 (1–2): 34–37.
104. Boxer AL, Yu JT, Golbe LI et al. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol 2017; 16 (7): 552–563. doi: 10.1016/S1474-4422 (17) 30157-6.
105. Jabbari E, Holland N, Chelban V et al. Diagnosis across the spectrum of progressive supranuclear palsy and corticobasal syndrome. JAMA Neurol 2020; 77 (3): 377–388. doi: 10.1001/jamaneurol.2019.4347.
106. Nakashima H, Terada S, Ishizu H et al. An autopsied case of dementia with Lewy bodies with supranuclear gaze palsy. Neurol Res 2003; 25 (5): 533–537. doi: 10.1179/016164103101201788.
107. Constantinides VC, Paraskevas GP, Velonakis G et al. Midbrain morphology in idiopathic normal pressure hydrocephalus: a progressive supranuclear palsy mimic. Acta Neurol Scand 2020; 141 (4): 328–334. doi: 10.1111/ane.13205.
108. Lloyd-Smith Sequeira A, Rizzo JR, Rucker JC. Clinical approach to supranuclear brainstem saccadic gaze palsies. Front Neurol 2017; 8: 429. doi: 10.3389/fneur. 2017.00429.
109. Salsano E, Umeh C, Rufa A et al. Vertical supranuclear gaze palsy in Niemann-Pick type C disease. Neurol Sci 2012; 33 (6): 1225–1232. doi: 10.1007/s10072-012-1155-1.
110. Kresojević N, Mandić-Stojmenović G, Dobričić V et al. Very late-onset Niemann-Pick type C disease: example of progressive supranuclear palsy look-alike disorder. Mov Disord Clin Pract 2020; 7 (2): 211–214. doi: 10.1002/mdc3.12892.
111. Nadjar Y, Hütter-Moncada AL, Latour P et al. Adult Niemann-Pick disease type C in France: clinical phenotypes and long-term miglustat treatment effect. Orphanet J Rare Dis 2018; 13 (1): 175. doi: 10.1186/s13023-018-0913-4.
112. Bremova-Ertl T, Schiffmann R, Patterson MC et al. Oculomotor and vestibular findings in Gaucher disease type 3 and their correlation with neurological findings. Front Neurol 2017; 8: 711. doi: 10.3389/fneur.2017.00711.
113. Modreanu R, Özdemir G, Buhmann C et al. Levodopa-induced dystonia in a patient with possible progressive supranuclear palsy with progressive gait freezing. J Neurol Sci 2018; 388: 139–140. doi: 10.1016/j.jns.2018.03.023.
114. Vasta R, Nicoletti A, Mostile G et al. Side effects induced by the acute levodopa challenge in Parkinson‘s disease and atypical parkinsonisms. PLoS One 2017; 12 (2): e0172145. doi: 10.1371/journal.pone.0172145.
115. Hiller A, Murchison C, Nichols J et al. The effects of amantadine on the progression of progressive supranuclear palsy (psp). [online]. Available from: https: //n.neurology.org/content/90/15_Supplement/P6.068
116. Stamelou M, Hoglinger G. A review of treatment options for progressive supranuclear palsy. CNS Drugs 2016; 30 (7): 629–636. doi: 10.1007/s40263-016-0347-2.
117. Kompoliti K, Goetz CG, Litvan I et al. Pharmacological therapy in progressive supranuclear palsy. Arch Neurol 1998; 55 (8): 1099–1102. doi: 10.1001/archneur.55.8.1099.
118. Engel PA. Treatment of progressive supranuclear palsy with amitriptyline: therapeutic and toxic effects. J Am Geriatr Soc 1996; 44 (9): 1072–1074. doi: 10.1111/j.1532-5415.1996.tb02940.x.
119. Daniele A, Moro E, Bentivoglio AR. Zolpidem in progressive supranuclear palsy. N Engl J Med 1999; 341 (7): 543–544. doi: 10.1046/j.1468-1331.2002.0354g.x.
120. Zampieri C, Di Fabio RP. Balance and eye movement training to improve gait in people with progressive supranuclear palsy: quasi-randomized clinical trial. Phys Ther 2008; 88 (12): 1460–1473. doi: 10.2522/ptj. 20070302.
121. Dash SK. Zolpidem in progressive supranuclear palsy. Case Rep Neurol Med 2013; 2013: 250865. doi: 10.1046/j.1468-1331.2002.0354g.x.
122. Chang AY, Weirich E. Trial of zolpidem, eszopiclone, and other GABA agonists in a patient with progressive supranuclear palsy. Case Rep Med 2014; 2014: 107064. doi: 10.1155/2014/107064.
123. Cooper AD, Josephs KA. Photophobia, visual hallucinations, and REM sleep behavior disorder in progressive supranuclear palsy and corticobasal degeneration: a prospective study. Parkinsonism Relat Disord 2009; 15 (1): 59–61. doi: 10.1016/j.parkreldis.2008.01.011.
124. Asociace klinických logopedů České republiky. [online]. Dostupné z URL: https: //www.klinickalogopedie.cz/.
125. SKVIMP. Společnost klinické výživy a intenzivní metabolické péče. [online]. Dostupné z URL: http: //www.skvimp.cz/.
126. Intiso D, Bartolo M, Santamato A et al. The role of rehabilitation in patients with progressive supranuclear palsy: a narrative review. PM R 2018, 10 (6): 636–645. doi: 10.1016/j.pmrj.2017.12.011.
127. Slade SC, Finkelstein DI, McGinley JL et al. Exercise and physical activity for people with progressive supranuclear palsy: a systematic review. Clin Rehabil 2020; 34 (1): 23–33. doi: 10.1177/0269215519877235.
128. Tilley E, McLoughlin J, Koblar SA et al. Effectiveness of allied health therapy in the symptomatic management of progressive supranuclear palsy: a systematic review. JBI Database System Rev Implement Rep 2016; 14 (6): 148–95. doi: 10.11124/JBISRIR-2016-2002352.
129. Phokaewvarangkul O, Bhidayasiri R. How to spot ocular abnormalities in progressive supranuclear palsy? A practical review. Transl Neurodegener 2019; 8: 20. doi: 10.1186/s40035-019-0160-1.
130. Wiblin L, Lee M, Burn D. Palliative care and its emerging role in multiple system atrophy and progressive supranuclear palsy. Parkinsonism Relat Disord 2017; 34: 7–14.
131. Spolek pro atypické Parkinsonské syndromy. [online]. Dostupné z URL: https: //aps.estranky.cz/.
132. Brown FS, Rowe JB, Passamonti L et al. Falls in progressive supranuclear palsy. Mov Disord Clin Pract2020; 7 (1): 16–24. doi: 10.1016/j.parkreldis.2016.10.013.
133. Apetauerova D, Scala SA, Hamill RW et al. CoQ10 in progressive supranuclear palsy: a randomized, placebo-controlled, double-blind trial. Neurol Neuroimmunol Neuroinflamm 2016; 3 (5): e266. doi: 10.1212/NXI. 0000000000000266.
134. Chen Z, Chen JA, Shatunov A et al. Genome-wide survey of copy number variants finds MAPT duplications in progressive supranuclear palsy. Mov Disord 2019; 34 (7): 1049–1059. doi: 10.1002/mds.27702.
135. Boxer AL, Qureshi I, Ahlijanian M et al. Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol 2019; 18 (6): 549–558. doi: 10.1016/S1474-4422 (19) 30139-5.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2020 Číslo 6
Nejčtenější v tomto čísle
- Progresivní supranukleární obrna
- Supraskapulární neuropatie
- Endoskopické vs. skríninkové vyšetření polykání a jejich vliv na výsledný stav u pacientů po akutní cévní mozkové příhodě
- Encefalopatie při infekci COVID-19 s odezvou na léčbu intravenózními imunoglobuliny