
Pyridoxin dependentní epilepsie – kazuistiky
Pyridoxine-dependent Epilepsy – Case Reports
Pyridoxine-dependent epilepsy is a rare autosomal recessive hereditary disorder causing severe intractable epileptic seizures presenting typically in prenatal and neonatal period, rarely in early infancy (age up to 3 years). Pyridoxine-dependent epilepsy, caused by metabolic disturbance of pyridoxine, is associated with mutations in the ALDH7A1 or ALDH4A1 gene. Pyridoxine-dependent epilepsy is successfully treatable using high doses of pyridoxine. The diagnosis is based on biochemical and genetic examinations. Three case reports of patients with a typical clinical course of pyridoxine-dependent epilepsy and genetically detected mutation in the ALDH7A1 gene are presented.
Key words:
pyridoxine – pyridoxal-phosphate – pyridoxine-dependent epilepsy – pyridoxine-dependent seizures
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Chinese summary - 摘要
吡哆素依赖性癫痫 - 病例报告吡哆素依赖性癫痫是一种罕见的常染色体隐性遗传性疾病,导致严重顽固性癫痫发作,典型地出现在产前和新生儿期,很少在婴儿期(3岁以下)。 由吡哆醇代谢紊乱引起的吡哆素依赖性癫痫与ALDH7A1或ALDH4A1基因突变有关。 吡哆素依赖性癫痫可以使用高剂量的吡哆醇成功治疗。 诊断是基于生物化学和遗传学检查。 介绍了典型的吡哆醇依赖性癫痫临床病程和ALDH7A1基因遗传检测突变患者的病例报告。
关键词:
吡哆醇 - 吡哆醛 - 磷酸 - 吡哆素依赖性癫痫 - 吡哆醇依赖性癫痫发作
Autoři:
Š. Aulická 1; L. Fajkusová 2; P. Šilerová 2; L. Elstnerová 3; T. Jimramovský 3; E. Dortová 4; H. Ošlejšková 1
Působiště autorů:
Centrum pro epilepsie Brno, Klinika dětské neurologie LF MU a FN Brno
1; Centrum molekulární biologie a genové terapie, Interní hematoonkologická klinika LF MU a FN Brno
2; Pediatrická klinika LF MU a FN Brno
3; Neonatologické oddělení, FN Plzeň
4
Vyšlo v časopise:
Cesk Slov Neurol N 2017; 80/113(3): 346-349
Kategorie:
Kazuistika
doi:
https://doi.org/10.14735/amcsnn2017346
Tento projekt byl podpořen Lékařskou fakultou Masarykovy Univerzity (číslo grantu ROZV/ 24/ / LF/ 2016).
Souhrn
Pyridoxin dependentní epilepsie je autozomálně recesivně dědičné onemocnění, které se prenatálně, neonatálně a v časném dětství do 3 let projevuje farmakorezistentními epileptickými záchvaty. Jde o dědičné poruchy metabolizmu pyridoxinu asociované s mutacemi v genech ALDH7A1 nebo ALDH4A1. Pyridoxin dependentní epilepsie jsou úspěšně léčitelné vysokými dávkami pyridoxinu. Diagnostika je založena na genetickém a biochemickém vyšetření. Prezentovány jsou kazuistiky tři pacientů s typickým průběhem pyridoxin dependentní epilepsie a geneticky potvrzenou mutací v ALDH7A1 genu.
Klíčová slova:
pyridoxin – pyridoxal-fosfát – pyridoxin dependentní epilepsie – pyridoxin dependentní záchvaty
Úvod
Pyridoxin dependentní epilepsie (PDE) jsou autozomálně recesivně dědičná onemocnění, která se projevují farmakorezistentními epileptickými záchvaty prenatálně, neonatálně a v časném dětství do 3 let věku. Jde o poruchy metabolizmu pyridoxinu asociované s mutacemi v genech ALDH7A1 nebo ALDH4A1. Incidence se uvádí v rozmezí 1 : 276 000 – 1 : 100 000.
Patofyziologie
Pyridoxin je (vedle pyridoxalu a pyridoxaminu) součástí komplexu vitaminu B6. Všechny tyto látky, společně se svými fosfáty, se účastní metabolizmu aminokyselin a sacharidů. Všechny formy vitaminu B6 se vyskytují v potravě a jsou vstřebávány ze střeva. Aktivní formou tohoto vitaminu je pyridoxal-5-fosfát, důležitý kofaktor enzymů.

PDE jsou podmíněny defektem enzymu α-AASA dehydrogenázy (semialdehyd kyseliny α-aminoadipové dehydrogenáza), který je způsoben mutacemi v genu ALDH7A1/ antikvitin. α-AASA dehydrogenáza se zúčastňuje degradace lysinu v mozku: odbourává meziprodukty P6C (piperidin-6-karboxylát) a α-AASA (semialdehyd kyseliny α-aminoadipové) na kyselinu α-aminoadipovou. Při defektu α-AASA dehydrogenázy se v mozku kumuluje P6C, který inaktivuje pyridoxalfosfát. Ve zvýšené míře se tvoří i kyselina pipekolová, která působí jako modulátor kyseliny γ-aminomáselná (GABA).
Podtypem PDE je hyperprolinemie typu II – porucha podmíněná defektem enzymu pyrrolin-5-karboxylát dehydrogenázy, který zabezpečuje konverzi P5C (pyrrolin-5-karboxylát) – přes semialdehyd kyseliny glutamové – na glutamát. Tento enzymový defekt je způsoben mutacemi v genu ALDH4A1.
Chybná degradace lysinu, event. prolinu tedy vede ke kumulaci meziproduktů metabolických drah, které následně inaktivují pyridoxal-fosfát. Tento funkční deficit pyridoxal-fosfátu je možné překonat vysokými dávkami pyridoxinu (ze kterého následně pyridoxal-fosfát vzniká).
Klinický obraz
Pyridoxin dependentní záchvaty (PDS) se v typickém případu projevují jako paroxysmy generalizovaných tonicko-klonických křečí, často prolongovaných, výjimkou není status epilepticus. Vzácněji jsou popisovány paroxysmy atonické, myoklonické a infantilní spazmy. K přidruženým příznakům patří psychomotorická retardace, retardace expresivní složky řeči, iritabilita, obstrukční gastrointestinální příznaky a syndrom dechové tísně.
Lze odlišit dvě formy PDE: klasickou formu a atypickou formu.
Klasická forma PDE
V „klasickém“ případě se PDS objevují již v prvních hodinách po narození. Anamnesticky lze v některých případech zjistit i intrauterinní výskyt záchvatů od pátého měsíce gestace (vnímání klonických trhavých pohybů plodu v trvání 5 – 15 min). Intrauterinní záchvaty často vedou k hypoxii plodu, což v perinatálním období může vést k chybné diagnóze post-hypoxických křečí. Typickým znakem je rezistence na terapii běžnými antiepileptiky (AEDs) a rychlá odpověď na parenterální podání pyridoxinu intravenózně (i.v.) nebo intramuskulárně [1,2].
Atypická forma PDE
V „atypickém“ případě se pyridoxinová dependence projeví později, většinou kolem 18 měsíců věku (vzácněji až kolem 3 let věku). V neonatálním období diagnostiku komplikuje pozitivní efekt AEDs (nejčastěji fenobarbitalu), který se však postupně vytrácí. Pyridoxinový test provedený v tomto období bývá obvykle negativní. U dětí s refrakterními záchvaty do 18 měsíců (resp. až do 3 let věku) je proto vždy potřeba myslet na PDE a pyridoxinový test zopakovat [1,2].
Kritéria pyridoxinové dependence [1,3,4]:
- pozitivní pyridoxinový test – přerušení záchvatu po parenterálním podání pyridoxinu,
- kompletní kontrola nad záchvaty při monoterapii pyridoxinem,
- recidiva záchvatu po vysazení pyridoxinu,
- rezistence na AEDs,
- intrauterinní nebo časné novorozenecké záchvaty,
- pozitivní rodinná anamnéza.
Diagnostika
V diagnostice PDE se uplatňuje anamnéza, klinické vyšetření, EEG, zobrazovací metody (CT, MR mozku), biochemické a genetické vyšetření.
EEG
Elektroencefalografický obraz je bez specifického korelátu pro PDE. Iktálně nacházíme většinou vysokovoltážní bilaterálně synchronní komplexy hrot – pomalá vlna s frekvencí 1 – 4 Hz. Interiktálně je často přítomno difuzní zpomalení základní aktivity.
CT, MR mozku
U pacientů postižených PDE byly zjištěny určité asociace v CT a MR obraze mozku: atrofie šedé a bílé hmoty, hydrocefalus nejasné geneze, dysgeneze/ ageneze corpus callosum, leukoencefalopatie.
Genetické a biochemické vyšetření
Přehled genů, jejichž mutace jsou asociovány s poruchami metabolizmu pyridoxinu a biochemických markerů spojených s PDE, uvádí tab. 1 [3 – 6]. Molekulárně genetická analýza těchto genů byla zahájena v roce 2009 v Centru molekulární biologie a genové terapie, FN Brno.
Terapie a její rizika
PDE je úspěšně léčitelná vysokými dávkami pyridoxinu. V literatuře není jednoznačné doporučení pro dávkování – pohybuje se v širokém rozmezí 20–300 mg/den [1]. Optimální je takové dávkování, při kterém už jsou pod kontrolou epileptické záchvaty, a současně ještě nejsou vyjádřeny nežádoucí účinky u konkrétního pacienta [1]. U pacientů užívajících dlouhodobě vysoké dávky pyridoxinu byly jako nežádoucí účinky léčby popsány polyneuropatie, poruchy citlivosti, ataxie a titubace. V případě bolusového parenterálního podání vysoké dávky pyridoxinu může dojít k útlumu kardiorespiračních a neurologických funkcí se závažnými následky (bradykardie, apnoe, hypotonie) [1]. Bezpečná dávka pyridoxinu u dětí je 100 mg/den, kterou v případě nedostatečného efektu navyšujeme.
Soubor pacientů a výsledky
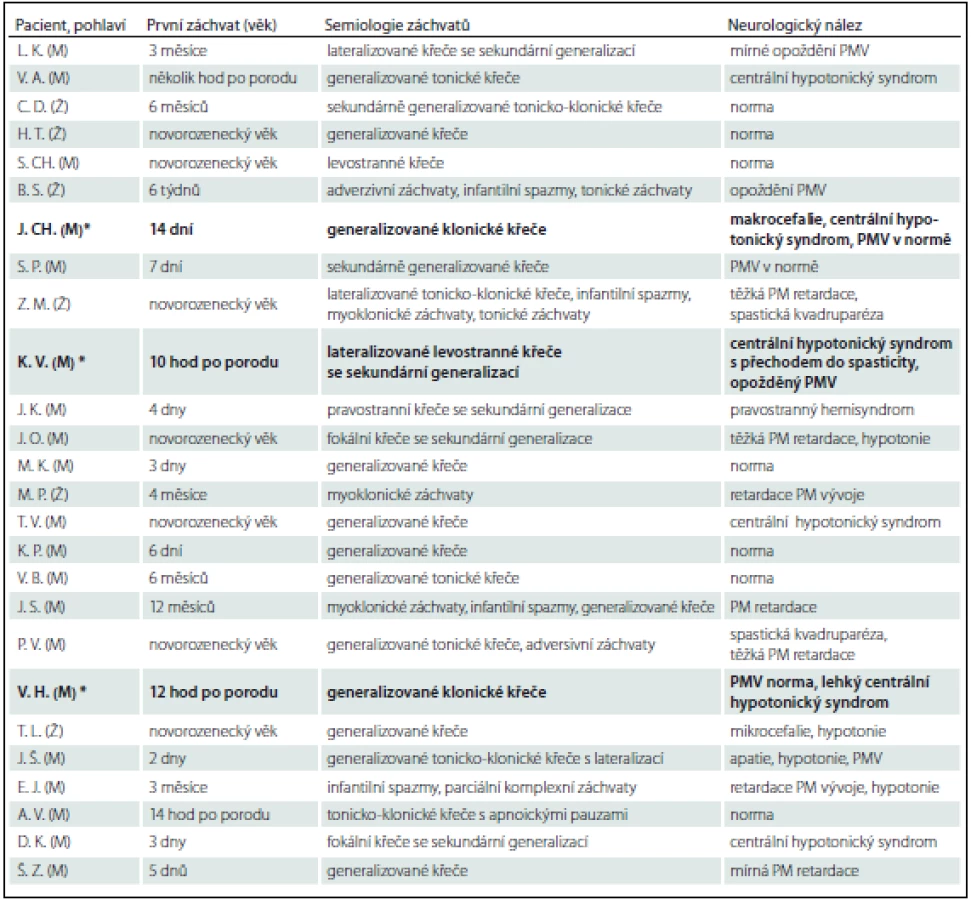
Prezentujeme soubor 25 pacientů, u kterých bylo ve FN Brno v období 5/2009–3/2015 provedeno genetické vyšetření na PDE (tab. 2). Vstupním kritériem pro genetické vyšetření byly refrakterní novorozenecké křeče s pozitivním nebo parciálně pozitivním pyridoxinovým testem. Do souboru byli zařazeni i pacienti s rozvojem křečí do 1 roku života, u kterých byla přítomna rezistence ke konvenční antiepileptické léčbě a nebyla objasněna příčina epilepsie. U 19 pacientů došlo k rozvoji křečové aktivity v novorozeneckém věku (v prvních hodinách až dnech života). V této skupině byla diagnóza PDE potvrzena u tří pacientů (u všech pacientů se jednalo o mutaci v ALDH7A1 genu). U zbylých šesti pacientů ve věku 3 – 12 měsíců byla zvažována atypická forma PDE. Tato diagnóza však nebyla potvrzena u žádného z nich.
V další části práce budeme prezentovat kazuistiky pacientů s potvrzenou diagnózou PDE.
Kazuistika 1
Chlapec J. CH. narozený v roce 2009, rodinná anamnéza bez pozoruhodností, porod spontánně záhlavím ve 37. týdnu. Před porodem pozorována decelerace ozev, během porodu zkalená plodová voda, Apgar skóre 7 – 8 – 9, porodní hmotnost 2 700 g/46 cm. V prvních hodinách po porodu byly přítomny nespecifické symptomy: neklid, třes, a vzhledem k dušnosti a vzedmutému břichu byla zvažována časná novorozenecká sepse, která nebyla potvrzena. V následujících dnech se zdravotní stav postupně stabilizoval a pacient byl 8. poporodní den propuštěn. Čtrnáctý poporodní den došlo k rozvoji generalizovaných klonických křečí s cyanózou a přechodem do status epilepticus. Tento akutní stav byl řešen i.v. fenobarbitalem, současně musel být pacient neodkladně zaintubován, byla zahájena umělá plicní ventilace (UPV). Přes adekvátní dávkování fenobarbitalu nedošlo k plné kontrole křečí, proto byl současně podán bolus pyridoxinu 100 mg i.v., po kterém křeče vymizely. Provedli jsme genetické vyšetření na PDE. Byly nalezeny dvě heterozygotní mutace v ALDH7A1 genu (c.503_506delTCTT a c.1008+3_1008+6delGAGT), čímž byla diagnóza PDE potvrzena. U obou rodičů a sourozence byla nalezena jedna z těchto mutací (u matky mutace c.503_506delTCTT, u otce mutace c.1008+3_1008+6delGAGT a u sourozence c.1008+3_1008+6delGAGT). Z komplementárních vyšetření byla provedena MR mozku a EEG – obojí s normálním nálezem.
Pacient má aktuálně 4,5 roku a je bez křečových projevů na dávce pyridoxinu 80 mg/ kg/ den. Přestože byl iniciálně přítomen centrální hypotonický syndrom, neurologický nález je nyní normální, psychomotorický vývoj odpovídá širší věkové normě.
Kazuistika 2 (pacient z jiného pracoviště, anamnestická data jsou limitována)
Chlapec V. H. narozený v roce 2011. V rodinné anamnéze exitus letalis sourozence na refrakterní status epilepticus v 1,5 roce věku (generalizované klonické záchvaty měl od novorozeneckého věku). Porod proběhl v termínu sekcí. Generalizované klonické křeče se objevily 12 hod po porodu. Vzhledem k rodinné anamnéze byl akutně proveden pyridoxinový test (podáno bolusově 100 mg pyridoxinu i.v.), po kterém křeče hned vymizely. Podezření na PDE bylo následně potvrzeno genetickým vyšetřením – byly nalezeny dvě heterozygotní mutace v ALDH7A1 genu (c.871+2dupT a c.886_887GA>AT, p.(E268I)). U obou rodičů byla zjištěna jedna z těchto mutací (u matky mutace c.871+2dupT, u otce mutace p.(E268I)).
Pacientovi jsou nyní 4 roky a je bez křečových projevů při dávce pyridoxinu 10 mg/ kg/ den. Dávkování je aktuálně navyšováno pro lehké opoždění v psychomotorickém vývoji.
Kazuistika 3
Chlapec K. V. narozený v roce 2014. Rodinná anamnéza bez pozoruhodností, těhotenství bylo rizikové pro suspektní insuficienci placenty. Porod proběhl v termínu spontánně záhlavím, Apgar skóre 9 – 10 – 10, porodní hmotnost 3 670 g/52 cm. Rozvoj lateralizovaných levostranných křečí s postupnou sekundární generalizací a přechodem do status epilepticus s nutností intubace a UPV nastal 10 hod po porodu. V akutním iktálním EEG záznamu zjištěn burst suppression vzorec. Léčba byla zahájena standardně – bolusem fenobarbitalu v dávce 20 mg/ kg i.v. – bez efektu, rovněž midazolam v dávce 0,15 mg/ kg i.v. bez efektu. Vzhledem k neúčinnosti této konvenční antiepileptické léčby bylo následně přistoupeno k provedení pyridoxinového testu: po i.v. bolusu 100 mg pyridoxinu křeče ustoupily a vymizel burst suppression vzorec v EEG. Akutně provedená MR prokázala difuzní vazogenní mozkový edém bílé hmoty s vícečetnými hemoragiemi periventrikulárně bilaterálně. Ve věku 7 měsíců se objevily záchvaty charakteru infantilních spazmů, po navýšení pyridoxinu na dávku 80 mg/ kg/ den se již neopakovaly.
Aktuálně má pacient 9 měsíců a na kombinované terapii fenobarbital (5 mg/ kg/ den) a pyridoxin 80 mg/ kg/ den je bez křečových projevů. EEG bez specifické patologie, kontrolní MR mozku již s normálním nálezem. Iniciálně přítomný centrální hypotonický syndrom přechází do spasticity. Psychomotorický vývoj je opožděn – aktuálně na úrovni přelomu I./ II. trimenonu. Genetickým vyšetřením byly prokázány dvě heterozygotní mutace v ALDH7A1 genu (c.796C>T, p.(R266*) a c.517+2T>A). U obou rodičů byla zjištěna jedna z těchto mutací (u matky mutace p.(R266*), u otce mutace c.517+2T>A). Vzhledem ke klinickému obrazu, iniciálnímu MR nálezu, efektivitě pyridoxinu a pozitivitě genetického vyšetření se tedy nejspíše jedná o kombinaci PDE a symptomatické epilepsie na podkladě periventrikulárního krvácení.
Závěr
Hlavním smyslem diagnostiky PDE je možnost kauzální léčby, která signifikantně snižuje morbiditu a mortalitu pacientů. Předpokládáme, že v České republice se jedná o značně poddiagnostikované onemocnění a že jeho incidence bude s rozvojem nových diagnostických možností narůstat. Tento předpoklad podporuje skutečnost, že od roku 2009 – kdy byla ve FN Brno zavedena genetická diagnostika – jsme diagnózu PDE potvrdili u tří pacientů z celkového počtu 26 vyšetřených.
Seznam použitých zkratek
α-AASA – semialdehyd kyseliny α-aminoadipové
AEDs – antiepileptika
ALDH4A1 gen – aldehyde dehydrogenase 4 family, member A1
ALDH7A1 gen – aldehyde dehydrogenase 7 family, member A1
CSF – mozkomíšní mok
EDTA – kyselina etylendiamintetraoctová
GABA – kyselina γ-aminomáselná
PDE – pyridoxin dependentní epilepsie
PDS – pyridoxin dependentní záchvaty
P5C – pyrrolin-5-karboxylát
P6C – piperidin-6-karboxylát
3-MT – 3-metoxytyrozin
MUDr. Štefánia Aulická, Ph.D.
Centrum pro epilepsie Brno
Klinika dětské neurologie
LF MU a FN Brno
Černopolní 9
613 00 Brno
e-mail: stefania. aulicka@gmail.cz
Přijato k recenzi: 11. 10. 2016
Přijato do tisku: 13. 3. 2017
Zdroje
1. Parish A, Nissen MD, O’Neil C, et al. Vitamin B 6 Dependency Syndromes. eMedicine Paediatrics 2008. [accessed 2017 20 Mar]. Available from URL: http://emedicine.medscape.com/article/985667-overview.
2. Rusnáková Š, Fajkusová L, Jansová E, et al. Pyridoxin dependentní epilepsie – nové trendy v diagnostice a terapii. Neurol Praxi 2010;11(5):322 – 5.
3. Gospe SM. Pyridoxine-dependent seizures: new genetic and biochemical clues to help with diagnosis and treatment. Curr Opin Neurol 2006;19(2):148 – 53.
4. Plecko B, Paul K, Paschke E, et al. Bio chemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene. Hum Mutat 2007;28(1):9 – 26.
5. Baxter P. Pyridoxine-dependent seizures: a clinical and biochemical conundrum. Biochim Biophys Acta 2003;1647(1 – 2):36 – 41.
6. Farrant RD, Walker V, Mills GA, et al. Pyridoxal phosphate de-activation by pyrroline-5-carboxylic acid. Increased risk of vitamin B6 deficiency and seizures in hyperprolinemia type II. J Biol Chem 2001;276(18):15107 – 16.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2017 Číslo 3
Nejčtenější v tomto čísle
- Myotonická dystrofie – jednota v různosti
- Riziko poškození plodu v důsledku rentgenových výkonů u gravidních žen
- Vertebrogenní algický syndrom – medicína založená na důkazech a běžná klinická praxe. Existuje důvod něco změnit?
- Febrilní křeče – méně je někdy více