
Atypický parkinsonizmus a frontotemporální demence – klinické, patologické a genetické aspekty
Atypical Parkinsonism and Frontotemporal Dementia – Clinical, Pathological and Genetic Aspects
Frontotemporal dementia (FTD) is a heterogeneous clinical syndrome including behavioural variant FTD (bvFTD) and primary progressive aphasia (PPA), which are clinically characterized by behavioural changes and impairment of executive functions and speech. Behavioural changes and speech disorders are often accompanied by motor symptomatology, including especially parkinsonism and/or symptoms of the upper and lower motor neuron involvement. These symptoms may be present in various combinations, thereby creating a very wide spectrum of clinical entities, some of which are precisely genetically and pathologically defined. Besides these few defined entities, there is a number of variable clinical phenotypes consisting of different combinations of behavioural and personality changes, speech disorders and symptoms of typical and atypical parkinsonism associated with different mutations and histopathologic findings. These phenotypes cannot be clearly classified according to their clinical picture and instead, they are placed somewhere within the dementia-parkinsonism continuum. In our paper, we present an overview of the current state of knowledge of possible clinical manifestations of parkinsonism and FTD and their genetic and pathological aspects.
Key words:
frontotemporal dementia – primary progressive aphasia – progressive supranuclear palsy – corticobasal syndrome – frontotemporal lobar degenerations
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
K. Menšíková 1; L. Tučková 2; P. Kaňovský 1
Působiště autorů:
Neurologická klinika LF UP a FN Olomouc
1; Ústav klinické a molekulární patologie, LF UP a FN Olomouc
2
Vyšlo v časopise:
Cesk Slov Neurol N 2016; 79/112(3): 275-286
Kategorie:
Přehledný referát
doi:
https://doi.org/10.14735/amcsnn2016275
Souhrn
Frontotemporální demence (FTD) je heterogenní klinický syndrom zahrnující behaviorální variantu FTD (bvFTD) a primární progresivní afázii (PPA). Klinicky jsou tyto syndromy charakterizovány behaviorálními změnami, poruchami exekutivních funkcí a řeči. Behaviorální poruchy i poruchy řeči jsou poměrně často doprovázeny motorickou symptomatikou, zejména parkinsonizmem a/ nebo příznaky postižení horního a dolního motoneuronu. Tyto příznaky mohou být přítomny v nejrůznějších kombinacích, čímž dochází k vytváření velmi širokého spektra klinických jednotek, z nichž jsou některé přesně definovány geneticky a patologicky. Vedle těchto několika definovaných entit je zde řada variabilních klinických obrazů daných nejrůznějšími kombinacemi behaviorálních a osobnostních změn, řečových poruch, příznaků typického a atypického parkinsonizmu asociovaných s různými histopatologickými nálezy a mutacemi, které z klinického obrazu nelze jednoznačně klasifikovat a které vytvářejí spíše jakési kontinuum demence – parkinsonizmus. V naší práci uvádíme přehled současného stavu poznání možných klinických manifestací parkinsonizmu s FTD a jejich genetických a patologických aspektů.
Klíčová slova:
frontotemporální demence – primární progresivní afázie – progresivní supranukleární paralýza – kortikobazální syndrom – frontotemporální lobární degenerace
Parkinsonizmus u frontotemporálních demencí
Frontotemporální demence (FTD) je neurodegenerativní onemocnění charakterizované progredující atrofií frontálních a/ nebo temporálních laloků. Jedná se o heterogenní klinický syndrom zahrnující behaviorální variantu FTD (bvFTD) a primární progresivní afázii (PPA). V rámci PPA jsou dále rozlišovány tři subtypy; non-fluentní varianta PPA (nfvPPA), sémantická varianta PPA (svPPA) a logopenická varianta (lvPPA). Klinicky jsou FTD charakterizovány behaviorálními změnami, poruchami exekutivních funkcí a řeči, které mohou být již v časném průběhu onemocnění doprovázeny příznaky onemocnění motoneuronu (FTD-MND nebo FTD-ALS) či parkinsonizmem, zejména s příznaky progresivní supranukleární paralýzy (PSP) nebo kortikobazálního syndromu (CBS) jako tzv. PSP-like a CBS-like syndromy [1]. Každý ze syndromů FTD je charakterizován specifickým nálezem atrofie při zobrazení mozku (tab. 1). Výzkum prováděný v posledních letech přinesl řadu nových poznatků týkajících se bližší asociace jednotlivých klinických syndromů s genetickými a histopatologickými nálezy (schéma 1). Na základě těchto poznatků byla v roce 2011 revidována současná klinická diagnostická kritéria. Byla doplněna o odpovídající genetické a patologické nálezy a nálezy zobrazovacích vyšetření, s cílem dosáhnout co největší diagnostické přesnosti již v časných stadiích onemocnění [2,3].

Epidemiologie a historie onemocnění
FTD představuje třetí nejčastější příčinu neurodegenerativních demencí, po Alzheimerově nemoci a demenci s Lewyho tělísky, tvoří 5 – 10 % všech patologicky potvrzených případů. Po Alzheimerově nemoci je druhou nejčastější příčinou presenilní demence postihující pacienty mladší 65 let [4]. Údaje o výskytu FTD se různí, nejčastěji udávané hodnoty prevalence se pohybují v rozmezí 15 – 22 případů na 100 000 obyvatel ve věku 45 – 64 let [5 – 7]. Průměrný věk začátku onemocnění je 50 – 60 let, ale přibližně v 10 % dochází k manifestaci onemocnění až po 70. roce [8]. Délka trvání onemocnění je udávána mezi 2 a 20 lety, což je patrně reflexí typu patologického procesu způsobujícího rozvoj onemocnění [9].
Klinický syndrom charakterizovaný progredující afázií, apraxií a behaviorálními změnami spojenými s fokální atrofií frontálních a temporálních laloků poprvé popsal v roce 1892 pražský psychiatr a neurolog Arnold Pick [10]. Patologickým korelátem tohoto klinického syndromu byly zvláštní nafouklé neurony a podivné intraneuronální inkluze, které byly v roce 1911 označeny německým psychiatrem a neuropatologem Aloisem Alzheimerem jako „Pickovy buňky“ a „Pickova tělíska“ [11]. Název Pickova nemoc byl tomuto onemocnění přidělen v roce 1926 japonským neurologem a neuropatologem Onarim Kimurou a německým neuropatologem Hugem Spatzem [12]. Prvním poznatkem z oblasti molekulární patologie bylo v roce 1986 zjištění, že Pickova tělíska obsahují s mikrotubuly asociovaný protein tau [13]. V roce 1994 začalo být různými výzkumnými skupinami zabývajícími se problematikou tohoto neurodegenerativního onemocnění prosazováno označení „frontotemporální demence“ a odpovídající histopatologické změny byly rozlišovány na změny „Pickova typu“ a změny tzv. nespecifické [14]. V posledních letech došlo ke značnému pokroku a rozšíření poznatků zejména v oblasti molekulární patologie a genetiky.
Patologické aspekty FTD
Základní histopatologický obraz FTD, společný pro všechny klinické subtypy vč. amyotrofické laterální sklerózy (ALS), PSP a CBS je označován jako frontotemporální lobární degenerace (FTLD). FTLD je charakterizována selektivní atrofií frontálního a temporálního kortexu, s úbytkem neuronů, gliózou a spongiózou povrchových vrstev, zejména vrstvy II. Nomenklatura histopatologického obrazu onemocnění byla od prvního Pickova a Alzheimerova popisu onemocnění, starého více než 100 let, několikrát změněna. Termín Pickova nemoc je tak v dnešní době používán pouze pro označení patologické diagnózy a je vyhrazen jen pro ty případy FTLD, u nichž se zobrazí intraneuronální argyrofilní inkluze, tzv. Pickova tělíska obsahující 3R izoformu tau proteinu.
FTLD zahrnují značně heterogenní skupinu onemocnění. Jednotlivé typy FTLD se liší typem akumulovaného patologického proteinu, podle kterého jsou označovány a dále děleny. FTLD-tau je charakterizována přítomností inkluzí obsahujících tzv. s mikrotubuly asociovaný protein tau (MAPT), FTLD s tau-negativními, ale ubikvitin a TDP-43 (Transactive response DNA binding Protein 43 kD) pozitivními inkluzemi je označována jako FTLD-TDP, varianta s akumulací tzv. fusion in sarcoma proteinu nese označení FTLD-FUS a případy s ubikvitin pozitivními a TDP-43 negativními inkluzemi jsou označovány jako FTLD-UPS [15].
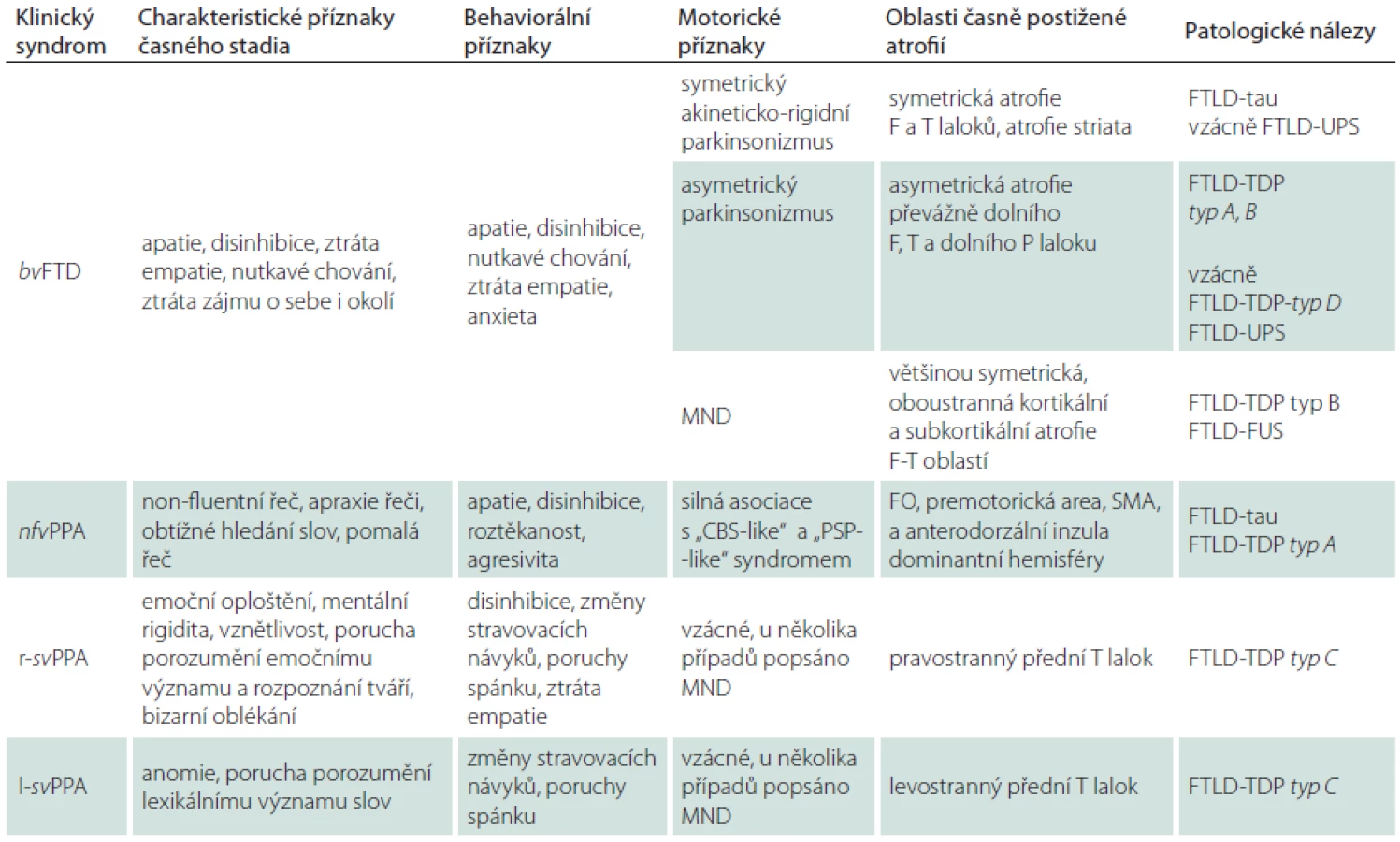
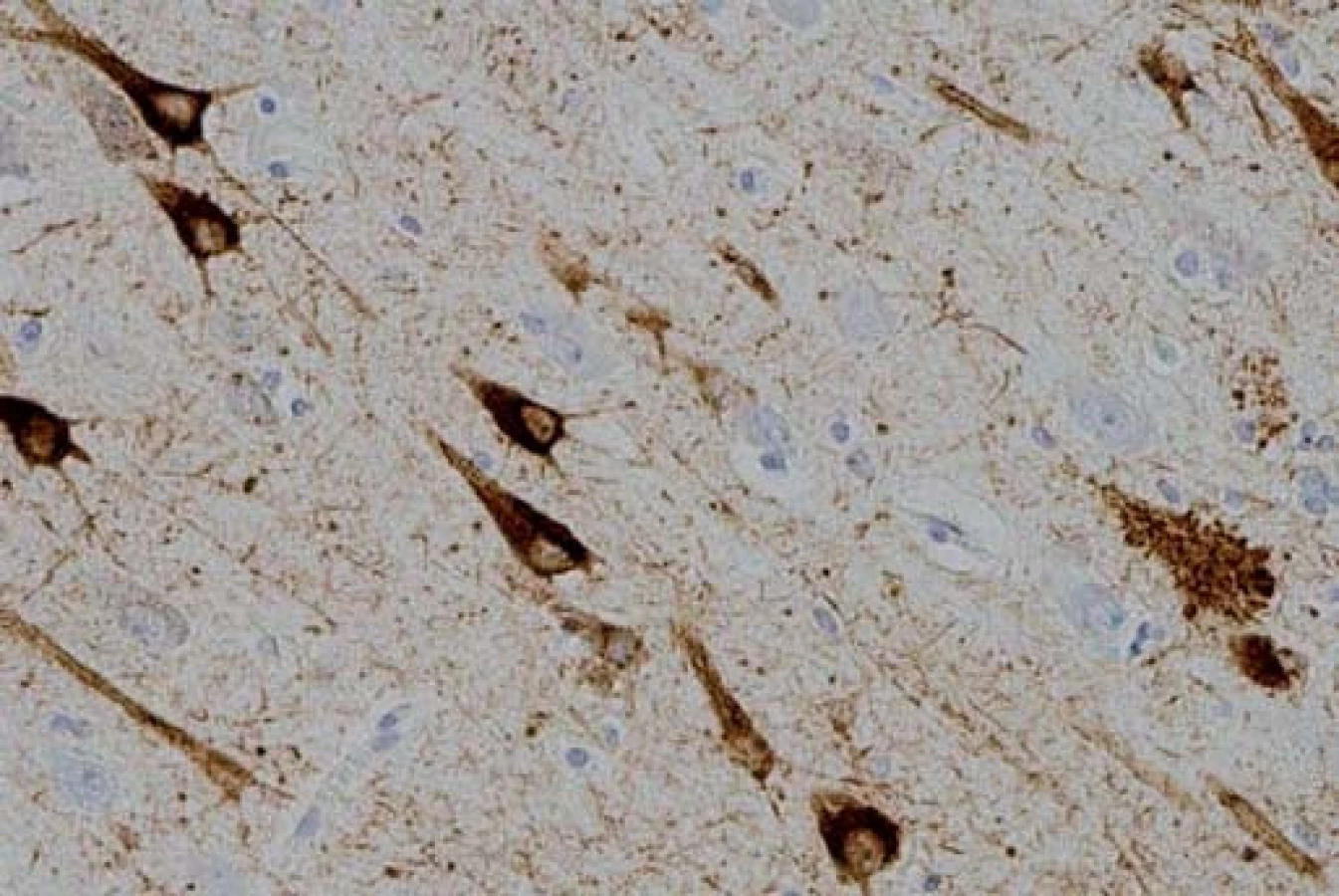
FTLD-tau je nejčastější variantou FTLD. Tau protein je produktem alternativního sestřihu jediného genu označovaného jako MAPT (Microtubule-Associated Protein Tau), který je umístěn na chromozomu 17. Jeho hlavní funkcí je modulace stability axonálních mikrotubulů. V lidské mozkové tkáni je tau protein přítomen v šesti izoformách, které jsou výsledkem alternativního sestřihu MAPT genu v exonech 2, 3 a 10. Tyto izoformy se od sebe liší počtem vazebných domén, jimiž se vážou na mikrotubuly. Tři izoformy obsahují tři vazebné domény, ty jsou označovány jako 3R, další tři obsahují čtyři vazebné domény a jsou označovány jako 4R izoformy tau proteinu [16]. Poměr 3R a 4R izoformy tau proteinu, morfologie a lokalizace inkluzí, jež předurčují výsledný klinický obraz, jsou dány typem MAPT mutace, kterých byla popsána celá řada. Inkluze, které lze nalézt v rámci FTLD-tau, zahrnují Pickova tělíska, neurofibrilární „tangles“ a „pretangles“ lokalizované ve frontálním a temporálním kortexu, v hipokampu a subkortikálních jádrech, někdy rovněž v mezencefalu, mozkovém kmeni, mozečku a míše (obr. 1). Jindy jsou nacházena gliální klubka, tzv. tangles and coiled bodies, lokalizovaná v bílé hmotě (obr. 2) [17]. Mezi klinické jednotky s FTLD-tau patologií jsou řazeny FTD s parkinsonizmem vázané na chromozom 17 (FTDP-17/ MAPT), PSP syndrom, CBS syndrom, některé typy nfvFTD a další dvě vzácné nemoci jako „argyrophilic grain disease“, nemoc s argyrofilními zrny (AGD) a multisystémová tauopatie s demencí (MSTD) [15].
FTLD-TDP, druhá nejčastější varianta FTLD, je charakterizována přítomností ubikvitin-pozitivních inkluzí, jejichž hlavní komponentou je tzv. TAR-DNA-binding protein 43 (Transactive response DNA binding Protein 43 kDa) označovaný jako TDP-43 [18]. Podle morfologie a distribuce inkluzí jsou v rámci FTLD-TDP rozlišovány další čtyři subtypy.
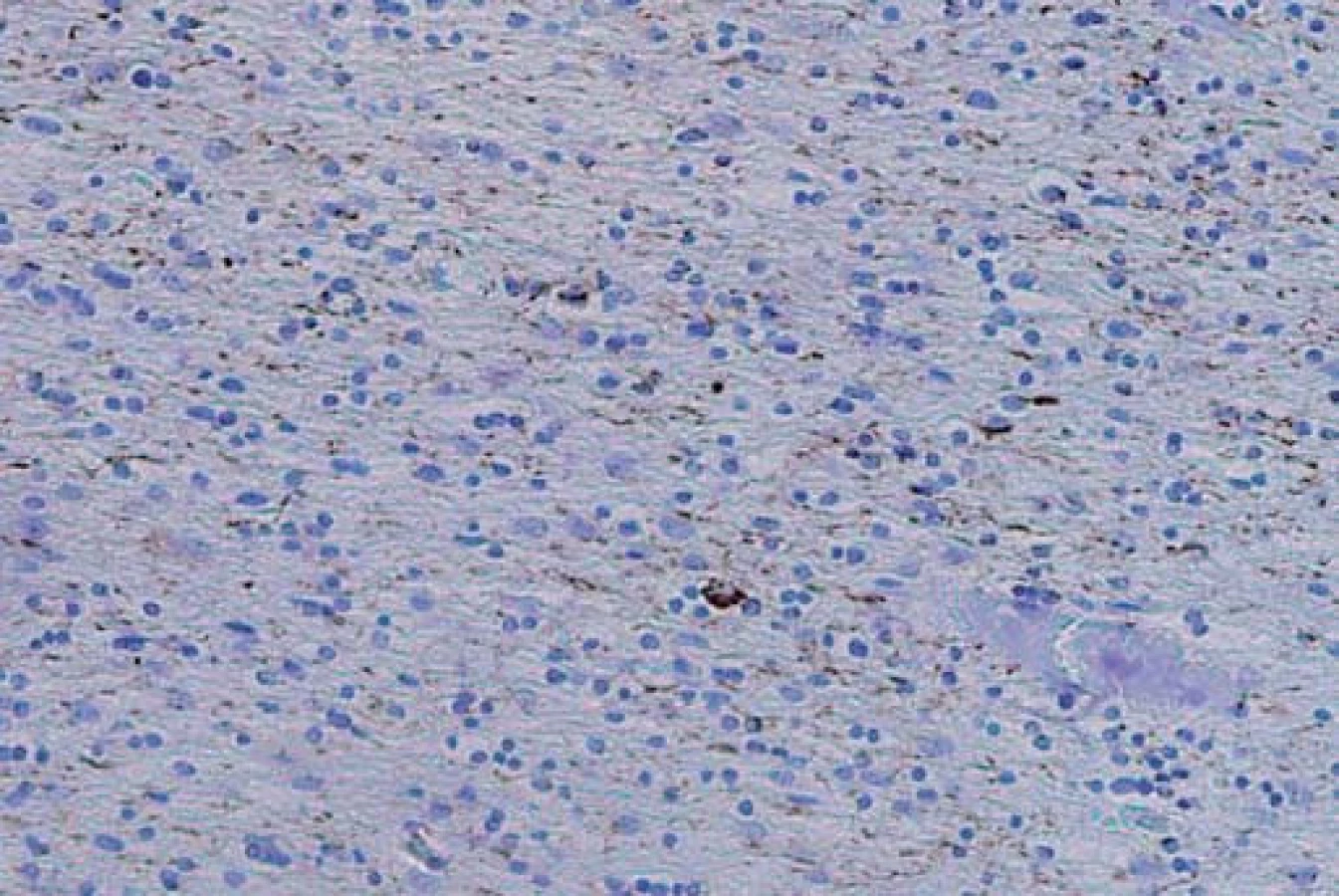
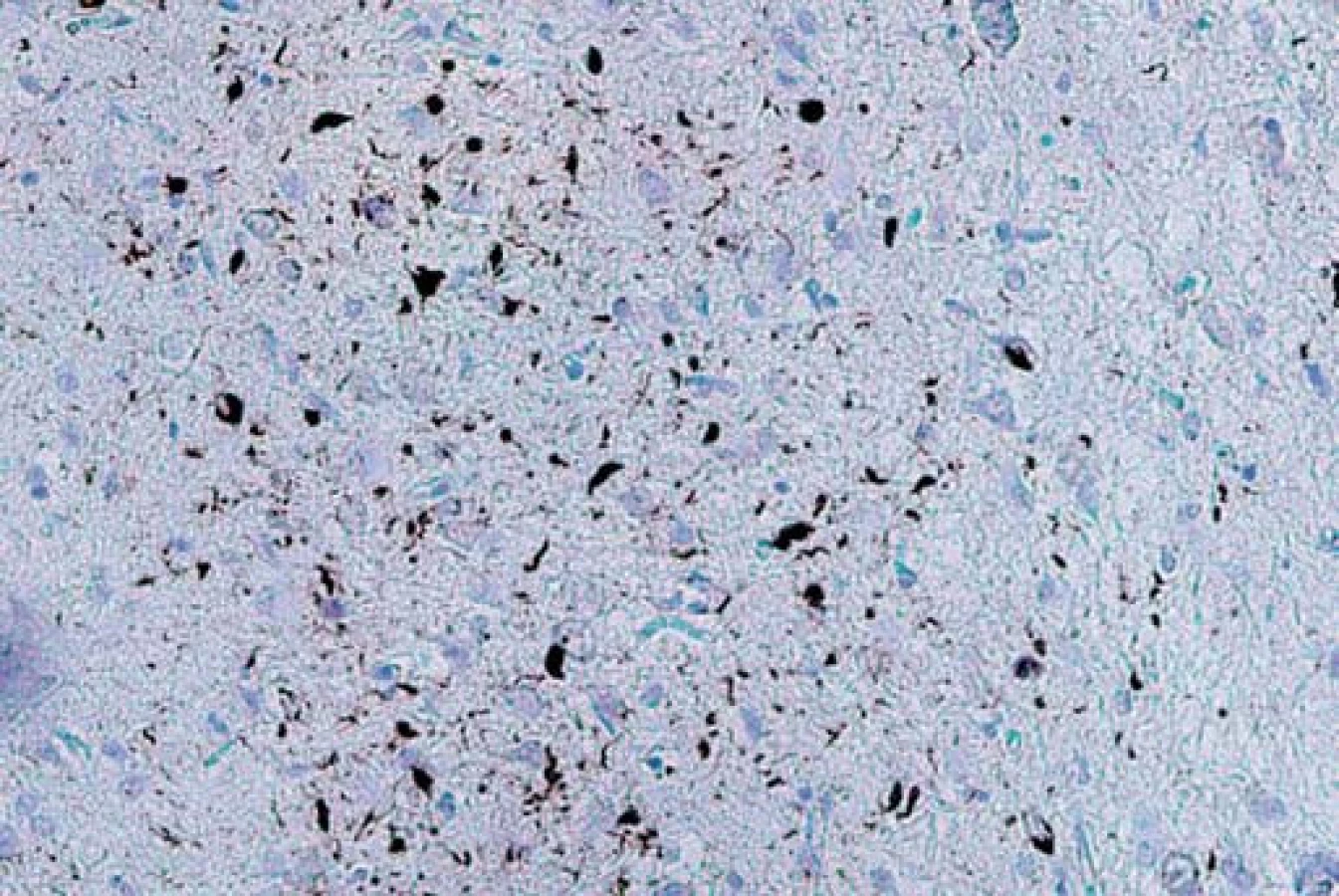
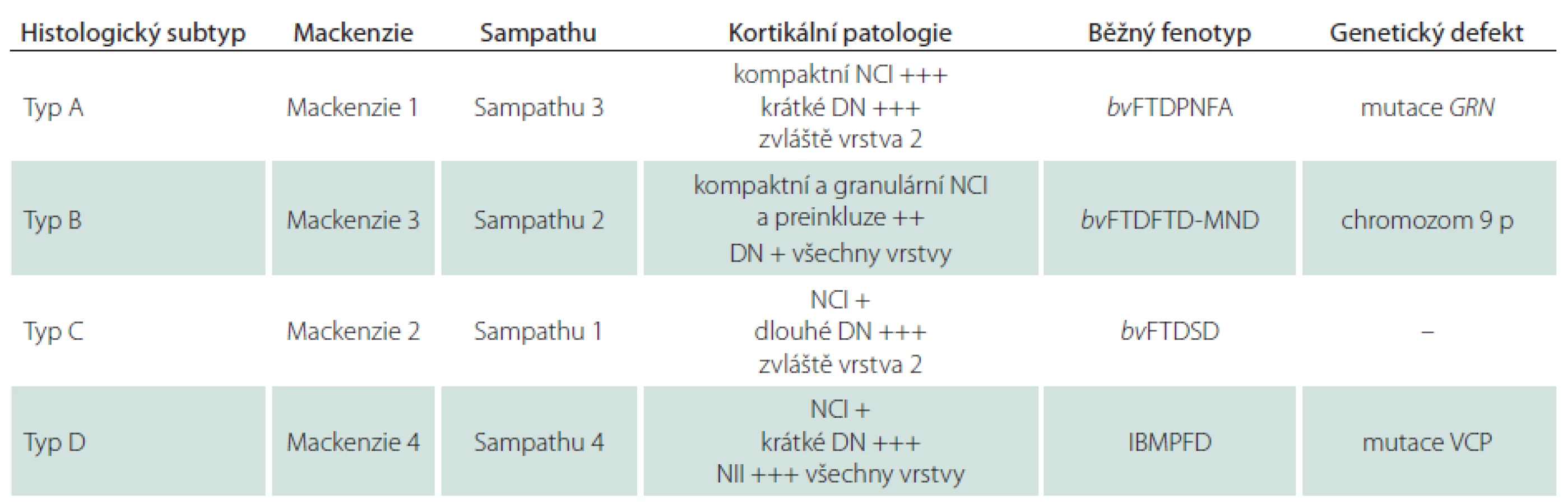
Typ A je charakterizován přítomností četných, krátkých, dystrofických neuritů (DN) a oválných neuronálních cytoplazmatických inkluzí (NCI) soustředěných především ve II. vrstvě neokortexu. Nekonstantním, ale poměrně častým znakem jsou čočkovité neuronální intranukleární inkluze (NII) (obr. 3, 4). Tento typ je asociován s mutací genu pro progranulin (PGRN) lokalizovaného na chromozomu 17. Klinicky se manifestuje jako bvFTD nebo nfvPPA, které jsou mnohdy doprovázeny parkinsonizmem (schéma 1).
Typ B je charakterizován velmi malým množstvím DN a přítomností NCI, jež jsou na rozdíl od předchozího typu méně početné a jsou distribuovány ve všech vrstvách neokortexu. Nejčastější genetickou příčinou je mutace genu C90RF72 na krátkém raménku chromozomu 9, která je klinicky asociována s bvFTD-MND nebo parkinsonizmem. Další, méně častá, je mutace TARDBP genu, klinicky spojovaná častěji s MND než s parkinsonizmem. Vzácnou příčinou může být mutace genu pro dynactin (DCTN1) způsobující Perryho syndrom (schéma 1).
Typ C je charakterizován převahou protáhlých DN zejména ve vyšších kortikálních vrstvách a velmi malým množstvím NCI. Klinicky se manifestuje jako svPPA. Konkrétní mutace odpovídající za tento typický klinickopatologický obraz nebyla dosud popsána (schéma 1).
Typ D je charakterizován četnými krátkými DN, četnými čočkovitými NII a pouze malým množstvím NCI. Tato nejvzácnější varianta FTLD je asociována s mutací genu VCP (Valosin Containing Protein gene) lokalizovaného na 9. chromozomu. Klinicky se manifestuje jako familiární, tzv. Inclusion Body Myopathy with Paget’s disease of bone and Frontotemporal Dementia; IBMPFD. Patologická klasifikace FTLD-TDP s asociovanými klinickými fenotypy a genetickými mutacemi je přehledně uvedena v tab. 2.
U malého počtu případů FTLD s ubikvitin pozitivními inkluzemi (FTLD-U) nebyla pozitivita TDP-43 proteinu prokázána. Tyto případy, které měly značně uniformní klinický i histopatologický obraz, byly označovány jako atypická FTLD-U (aFTLD-U). Teprve v rámci pozdějšího výzkumu byla prokázána imunoreaktivita těchto ubikvitin pozitivních a TDP-43 negativních inkluzí s protilátkou proti FUS (Fused in Sarcoma) proteinu [19]. Spolu s aFTLD-U byly popsány ještě další klinickopatologické jednotky charakterizované v imunohistopatologickém vyšetření přítomností ubikvitin pozitivních, TDP-43 negativních a FUS pozitivních inkluzí. Tato další, méně častá varianta FTLD, dostala označení FTLD-FUS. Podle charakteru inkluzí jsou tedy dnes v rámci FTLD-FUS rozlišovány: Basophilic Inclusion Body Disease (BIBD) [20], neuronal intermediate filament Inclusion Disease (NIFID) [21] a aFTLD-U [22].
V klinickém obraze aFTLD-U je typicky přítomna bvFTD s časným začátkem, průměrně kolem 40 let, s výraznou progredující poruchou chování, změnami osobnosti a psychotickými projevy, zatímco řeč a motorika zůstávají většinou ušetřeny. Onemocnění je sporadické, postižení jedinci mají negativní rodinnou anamnézu a dosud nebyla popsána žádná korespondující mutace. Na MR mozku je kromě frontotemporální atrofie přítomna atrofie hlavy nucleus caudatus [22]. Histopatologicky se aFTLD-U vyznačuje přítomností TDP-43 negativních a ubikvtin a FUS pozitivních, neuronálních jak cytoplazmatických (NCI), tak intranukleárních inkluzí (NII). NCI jsou nejčetnější ve frontálním a temporálním neokortexu a v granulárních a pyramidových buňkách hipokampu. Mohou být přítomny i v bazálních gangliích, thalamu a mozkovém kmeni a navzdory absenci klinických příznaků MND jsou NCI nacházeny i v dolních motoneuronech. Neuronální intranukleární inkluze mají rozmanitou morfologii a bývají nejpočetnější v hipokampu a neokortexu. Ubikvitin a FUS pozitivní inkluze lze kromě neuronů nalézt i v gliálních buňkách (GCI) bílé hmoty postižených regionů [23].
Basophilic Inclusion Body Disease (BIBD) – je termín používaný k označení klinicky a patologicky heterogenních případů, jejichž společným jmenovatelem je přítomnost neuronálních cytoplazmatických ubikvitin a FUS pozitivních inkluzí, které jsou bazofilní při barvení hematoxylinem-eosinem. Tyto bazofilní inkluze jsou nejčetnější v subkortikálních oblastech hlavně v bazálních gangliích a tegmentu mozkového kmene [24,25], recentně byla prokázána jejich přítomnost rovněž v neuronech a gliálních buňkách frontotemporálního kortexu a hipokampu [19]. Z klinických fenotypů asociovaných s tímto histopatologickým nálezem byly popsány bvFTD [24,25], sporadická a familiární ALS [26 – 28] a ALS s demencí [25,29,30].
Neuronal Intermediate Filament Inclusion Disease (NIFID) je vzácná forma FTLD s ubikvitin pozitivními inkluzemi, které jsou rovněž imunoreaktivní s protilátkou proti alfa-internexinu a dalším neurofilamentům. Jak bylo zjištěno později, tyto inkluze vykazují rovněž imunoreaktivitu s protilátkou proti FUS proteinu. Klinický obraz je charakterizován časným nástupem a poměrně rychlou progresí bvFTD s pyramidovou a extrapyramidovou symptomatikou. Vzhledem k tomu, že postižení je podobně jako u všech dalších FTD asymetrické, může klinický obraz odpovídat CBS [31,32].
FTLD-UPS je poslední varianta FTLD charakterizovaná přítomností ubikvitin pozitivních, TDP-43 a FUS negativních inkluzí. V současné době byla v souvislosti s tímto histopatologickým nálezem popsána pouze jedna klinická jednotka. Jedná se o autozomálně dominantně dědičné onemocnění způsobené mutací CHMP2B genu lokalizovaného na 3. chromozomu [33]. Klinický obraz odpovídá bvFTD, která může být doprovázena parkinsonizmem, dystonií, pyramidovými příznaky nebo myoklonem [34].
Genetické aspekty FTD
Pozitivní rodinnou anamnézu lze nalézt u 30–50 % pacientů s bvFTD, zatímco u pacientů s nfvPPA a svPPA je méně častá [35]. Z genetického hlediska je FTLD také značně heterogenní jednotka; poměrně recentně bylo popsáno několik mutací. K nejčastějším patří mutace v MAPT genu, v PGRN genu kódujícím protein progranulin a před několika lety popsaná mutace C9ORF72 [36,37]. Z vzácnějších mutací jsou to mutace v genech VCP (Valosin Containing Protein), CHMP2B (Chromatin Modifying Protein 2B), TARDBP (TAR-DNA Binding Protein) a FUS (Fused in Sarcoma). Jednotlivé typy mutací jsou většinou doprovázeny více či méně charakteristickým histopatologickým a klinickým obrazem.
MAPT gen kódující tzv. s mikrotubuly asociovaný protein tau, podle něhož nese své označení, je lokalizován na chromozomu 17q21.32. V rodinách s familiárním výskytem FTD bylo v tomto genu identifikováno více než 50 mutací. Odhadovaná frekvence MAPT mutace u pacientů s FTD je přibližně 50 % [38]. Mutace v tomto genu jsou odpovědné za akumulaci hyperfosforylovaného proteinu tau v neuronech a/ nebo v gliových buňkách [39]. Jak již bylo uvedeno výše, alternativní sestřih v exonech 2, 3 a 10 má za následek vznik šesti izoforem tau proteinu. Tři izoformy obsahují tři vazebné domény, ty jsou označovány jako 3R, a další tři obsahují čtyři vazebné domény a jsou označovány jako 4R izoformy tau proteinu. Mutace lze rozlišit na missense mutace v exonech 9 – 13 ovlivňující normální funkci tau proteinu při stabilizaci mikrotubulů a mutace v intronech a některých kódujících úsecích, které ovlivňují sestřih exonu 10 na úrovni mRNA, což má za následek změnu poměru 3R a 4R izoforem tau proteinu [40]. FTD způsobená mutacemi v MAPT genu bývá velmi často doprovázena parkinsonizmem a obecně se označuje jako FTD s parkinsonizmem vázaná na chromozom 17q (FTDP-17). Podle převažující klinické symptomatiky je rozlišován jednak demence-dominantní fenotyp s dominujícími behaviorálními změnami s disinhibicí a obsedantně-kompulzivním chováním, jednak parkinsonizmus – dominantní fenotyp s příznaky PSP nebo CBS syndromu [17,41].
PGRN gen je lokalizován na chromozomu 17q21.31, v těsném sousedství MAPT genu. Kóduje protein progranulin, který je hojně exprimován ve specifických neuronálních populacích. Tato mutace je o něco vzácnější než mutace v MAPT genu; odhadovaná frekvence se pohybuje v rozmezí 3 – 26 % [42]. Neuropatologický nález pacientů s touto mutací je charakterizován tau negativními a ubikvitin a TDP-43 pozitivními inkluzemi. Nejčastějšími behaviorálními změnami jsou apatie a sociální zdrženlivost. Přibližně u 25 % pacientů se časně rozvíjí izolovaná řečová dysfunkce připomínající anomickou non-fluentní poruchu spojenou s poruchou chápání významu jednotlivých slov. Často jsou udávány bludy a halucinace [43]. Hojná je rovněž extrapyramidová symptomatika zahrnující zejména CBS s apraxií končetin, asymetrickým parkinsonizmem nebo dystonií [44]. ALS fenotyp je naopak v rámci tohoto genetického spektra velmi vzácný [45].
C9ORF72 (Chromozom 9 Open Reading Frame 72) gen je lokalizován na krátkém raménku 9. chromozomu, lokus 9p21.2. Mutace tohoto genu spočívá v expanzi opakování hexanukleotidu GGGGCC [46]. Za normálních okolností je přítomno pouze několik repetic tohoto hexanukleotidu, nejčastěji 2 – 10 opakujících se jednotek. U jedinců s mutací se počet opakování pohybuje v řádu stovek až tisíců [47]. Je známo, že mutace narušuje normální expresi proteinu kódovaného tímto genem, nicméně přesná funkce tohoto proteinu nebyla dosud jasně popsána. Poměrně nedávno, teprve v roce 2011, byla zjištěna souvislost mezi expanzí hexanukleotidu a FTD, ALS nebo koincidencí obou těchto jednotek, ať už u jednoho jedince nebo v rámci jedné rodiny s familiárním výskytem těchto onemocnění [36,37]. Tato mutace je v současnosti považována za druhou nejčastější mutaci asociovanou s FTD, její frekvence je odhadována na 14 – 48 % [48]. Zatímco různé mutace různých genů jsou spojovány s různými fenotypy FTD, C9ORF72 mutace je spojována konkrétně s bvFTD. Ve vztahu k ALS byla přítomnost C9ORF72 mutace popsána přibližně ve 40 % případů familiární ALS a zhruba v 8–10 % případů sporadické ALS. Je tedy mnohem častější než dosud zvažované SOD1 nebo TARDBP mutace [49]. V neuropatologickém nálezu jsou v souvislosti s C9ORF72 mutací rovněž přítomny TDP-43 pozitivní inkluze, nicméně jedinečnou charakteristikou C9ORF72 expanze je přítomnost inkluzí obsahujících DPR (Dipeptide Repeats Protein) [50].
VCP mutace je vzácná mutace lokalizovaná na 9p13.3 s odhadovanou frekvencí výskytu menší než 1 %. Mutace ve VCP genu jsou spojovány s klinickým syndromem zahrnujícím myopatii s inkluzními tělísky (90 %), Pagetovu chorobou kostí (45 %) a FTD (38 %) označovaným jako familiární IBMPFD (Inclusion Body Myopathy with Paget’s disease of bones and Frontotemporal Dementia) [51].
CHMP2B mutace je rovněž vzácná mutace lokalizovaná na 3p11.2 s frekvencí výskytu menší než 1 %. Jak již bylo zmíněno výše, klinický obraz způsobený touto mutací je tvořen bvFTD, která může být doprovázena parkinsonizmem, dystonií, pyramidovými příznaky nebo myoklonem [34].
TARDBP mutace lokalizovaná na chromozomu 1 se vyskytuje přibližně u 5 % případů familiální ALS, vzácněji bývá asociována s FTD nebo FTD-MND [52].
FUS mutace byla nalezena přibližně v 5 % případů familiární ALS a u několika málo pacientů s bvFTD asociovanou s MND [53,54].
Navzdory významnému pokroku v oblasti genetiky ve vztahu k FTD stále zůstává řada případů s pozitivní rodinnou anamnézou FTD, u nichž nebyla zatím žádná související mutace popsána. Tyto případy se manifestují nejčastěji jako bvFTD s poruchami paměti s příznaky MND nebo bez nich. V patologickém nálezu bývá přítomna TDP-43 proteinopatie a hipokampální skleróza se ztrátou neuronů a gliózou v oblasti cornu ammonis a subicula hipokampu [55].
Klinický obraz FTD
FTD je způsobena selektivní vulnerabilitou specifických neuroanatomických sítí. Postižení začíná v konkrétních strukturách a šíří se v čase napříč specifickými oblastmi, způsobem podobným šíření prionů. Výsledkem jsou specifické a jedinečné klinické charakteristiky v jednotlivých fázích onemocnění [56,57].
Cílem diagnostického procesu by měla být identifikace klinického fenotypu (bvFTD vs. nfvPPA vs. svFTD vs. jiné typy demence nebo psychiatrické poruchy), predikce nejpravděpodobnější proteinopatie a možné genetické mutace. Tento přístup pak může pomoci při stanovení přesnější prognózy onemocnění a v případě objevení specifické léčby i co nejpřesnějšího způsobu terapie [58].
Obecně jsou v klinických obrazech FTD přítomny významné rozdíly v závislosti na pravo - nebo levostranném postižení. Pacienti s dominující pravostrannou atrofií, jako je tomu ve většině případů bvFTD a pravostranné varianty svFTD (r-svFTD), mají tendenci být emočně oploštělí a odtažití, často mají narušeny rodinné vztahy a trpí poruchami chování, které bývají mnohdy špatně interpretovány jako projevy různých psychiatrických onemocnění [59,60]. U pacientů s dominujícím levostranným postižením jsou hlavním příznakem poruchy řeči a jazyka.
bvFTD je nejčastěji se vyskytující syndrom v rámci FTD, tvoří přibližně 70 % všech případů FTD. K rozvoji bvFTD dochází typicky před 65. rokem věku, průměrný věk počátku tohoto nemocnění se pohybuje kolem 58 let. Dominujícím klinickým znakem bvFTD jsou poruchy chování. Jelikož časné neurodegenerativní změny postihují především paralimbické struktury ventromediálního prefrontálního kortexu, přední cingulární kortex a přední insulu, patří k počátečním symptomům zejména sociální disinhibice, snížená motivace, apatie a ztráta empatie [58,59]. Nejbližším okolím jsou tyto příznaky často interpretovány jako ztráta zájmu o rodinu, deprese nebo jiné psychiatrické onemocnění. Projevy sociální disinhibice mohou být vyjádřeny v různé intenzitě, od nepřiměřeného až po výhradně antisociální chování. S progresí onemocnění a selektivním šířením degenerativního procesu do temporálního laloku, zejména pravostranného, dochází k rozvoji mentální rigidity a zvláštních stravovacích návyků. U řady pacientů se vyvine craving neboli „bažení“ ve vztahu k sladkému nebo slanému jídlu. Kompulzivní chování, další z častých projevů bvFTD, může mít charakter od jednoduchých opakujících se pohybů až po těžší poruchy (shromažďování dokumentů, čištění povrchů nebo stravování se konkrétními potravinami v konkrétním čase). Degenerace dorzolaterálního prefrontálního kortexu má za následek rozvoj exekutivní dysfunkce s poruchou pracovní paměti, narušení rozhodovacích procesů a poruchu pozornosti. Epizodická paměť a zrakově-prostorové schopnosti zůstávají relativně dlouho zachovány [61].
„Fenokopie“ bvFTD je označení pro pomalu progredující poruchu postihující převážně muže, která se od klasické bvFTD liší podstatně delším klinickým průběhem, jenž může trvat i několik desetiletí [62]. Klinický obraz je prakticky neodlišitelný od klasické bvFTD, pouze exekutivní dysfunkce bývá méně závažná a stupeň mozkové atrofie mírnější nebo může zcela chybět. Většina těchto pacientů je vedena pod některou z psychiatrických diagnóz, ačkoliv se zdá, že rozvoj tohoto syndromu může být asociován s C9ORF72 mutací [63].
Primární progresivní afázie (PPA) je porucha řeči a jazyka zahrnující poruchu schopnosti mluvit, číst, psát a rozumět tomu, co jiní říkají. Jde o příznaky asociované s časnou atrofií temporálního laloku. PPA reprezentuje spektrum selektivních jazykových poruch, kdy jednotlivé klinické varianty PPA mohou být vodítkem k základnímu patologickému substrátu (schéma 1) [64]. V roce 2011 byla postulována nová diagnostická kritéria, čímž byl učiněn významný krok vpřed stanovením konzistentní terminologie a klasifikace PPA. Podle těchto kritérií je PPA klasifikována na tři klinické subtypy: non-fluentní PPA (nfvPPA nebo PNFA), sémantická PPA (svPPA nebo SD) a logopenická varianta PPA (lvPPA) [65]. Diagnostika PPA by měla být prováděna v několika krocích. Nejprve by měl pacient splňovat základní charakteristiku PPA, kterou je přítomnost postižení jazykových funkcí limitujících jeho každodenní životní aktivity. Následně by měly být posouzeny hlavní jazykové domény, které zahrnují tvorbu slov, opakování, porozumění významu slov, pojmenování, sémantické schopnosti, čtení a pravopis. Nakonec by měla být, na základě specifických řečových a jazykových projevů charakteristických pro jednotlivé subtypy, určena klinická varianta PPA. Tato klasifikace může být dále upřesněna přítomností odpovídajícího nálezu při zobrazení mozku nebo detekcí odpovídající mutace (tab. 1) [66].
nfvFTD je syndrom charakterizovaný postižením struktury a praxe řeči. K hlavním příznakům patří non-fluentní řeč s agramatizmy a fonemickými parafáziemi a apraxie řeči [64,66]. Pacienti nemají problém s porozuměním řeči nebo významu objektů, ale mají potíže s tvorbou složitějších vět. Podkladem je neurodegenerativní postižení neuroanatomických sítí zahrnujících dominantní frontální operculum a jeho spoje se suplementární motorickou areou, premotorickou areou a insulárním kortexem [68]. Časnými příznaky jsou tak zpomalení řeči, obtížné hledání slov, snížení slovního outputu a zkrácení délky frází. Na rozdíl od pacientů s bvFTD jsou si pacienti s nfvPPA vědomi svých potíží a před druhými se snaží uchovat si sociální dekorum. S progresí neurodegenerativního procesu do kontralaterální frontální oblasti dochází nakonec k rozvoji behaviorálních poruch. Finálně může obraz nfvPPA splývat s CBS nebo PSP syndromem [68,69]. Patologicky se jedná nejčastěji o FTLD-tau, vzácněji o FTLD-TDP, typ A (schéma 1).
svPPA je charakterizována poruchou sémantických schopností se zachovalou intaktní fluentní řečí. V časné fázi onemocnění jsou určité rozdíly mezi pravostrannou (r-svPPA) a levostrannou (l-svPPA) variantou svPPA. Časným projevem l-svPPA je porucha porozumění lexikálnímu významu slov, zatímco v případě r-svPPA je přítomna porucha porozumění emočnímu významu a rozpoznání tváří [70]. Tyto příznaky korelují s časnou atrofií přední části temporálního laloku jakožto centra sémantických schopností [57]. Prvními příznaky l-svPPA jsou potíže s vybavováním slov, spíše podstatných jmen než sloves. Pacienti nahrazují konkrétní slova nadřazenými pojmy (např. použijí označení zvíře pro označení kočky) a nakonec všechna podstatná jména nahrazují označením „věc“. V pozdějších fázích mají problémy s rozpoznáním předmětů, které jsou jim ukazovány, a nechápou ani jejich obecný význam. V případě r-svPPA jsou první příznaky behaviorální, které jsou v souladu s pravostranným postižením a mají charakter emočního oploštění a ztráty empatie. Následně dochází k rozvoji poruch rozpoznání známých tváří. V pozdějších stadiích se, v důsledku šíření neurodegenerativního procesu do kontralaterální hemisféry, mohou klinické obrazy obou variant PPA vzájemně překrývat [70,71]. Patologickým podkladem obou variant svPPA je výhradně FTLD-TDP, typ C (schéma 1).
Atypický parkinsonizmus u FTD
Behaviorální poruchy i poruchy řeči jsou u FTD poměrně často doprovázeny motorickou symptomatikou, zejména parkinsonizmem a/ nebo příznaky postižení horního a dolního motoneuronu. Tyto příznaky mohou být přítomny v nejrůznějších kombinacích. Často dochází k jejich vzájemnému překrývání a stanovení klinické diagnózy může být značně obtížné. S přibýváním genetických poznatků, klinickopatologických korelací a pokračujícím rozvojem histopatologických metod tak dochází k rozšiřování spektra FTLD zahrnujících stále narůstající množství různých sporadických a familiárních jednotek, definovaných klinicky, geneticky a patologicky. Vedle několika jednoznačně klinicky, geneticky a patologicky definovaných entit je zde řada variabilních klinických obrazů daných nejrůznějšími kombinacemi behaviorálních a osobnostních změn, řečových poruch, příznaků typického a atypického parkinsonizmu asociovaných s různými histopatologickými nálezy a mutacemi, které z klinického obrazu nelze jednoznačně klasifikovat a které vytvářejí spíše jakési kontinuum demence-parkinsonizmus [72].
Parkinsonizmus může předcházet, rozvíjet se koincidentálně nebo následovat manifestaci behaviorálních změn či řečových a jazykových poruch. Odpovídavost na podávání L-DOPA je většinou nízká, pouze u některých klinických jednotek může parkinsonizmus reagovat na léčbu L-DOPA dobře, nicméně většinou pouze přechodně.
Parkinsonské fenotypy, s nimiž se lze v rámci spektra FTD – parkinsonizmus setkat, zahrnují progresivní supranukleární paralýzu (PSP), kortikobazální syndrom (CBS), progredující akinezi s freezingem chůze (PAGF) a řadu dalších klinických variant reprezentujících odlišné histopatologické nálezy. Patologické obrazy zahrnují nálezy odpovídající PSP, CBD, Alzheimerově nemoci, FTLD-tau, FTLD-TDP či FTLD-UPS [73].
Nejčastějším motorickým fenotypem sporadické FTLD-tau jsou PSP a CBS [73 – 75]. Sporadická FTLD-TDP se obvykle manifestuje fenotypem FTD-MND charakterizovaným rychle progredující FTD, afázií, parkinsonizmem neodpovídajícím na L-DOPA, slabostí končetin a orofaciálního svalstva [74,76].
Dědičná forma FTLD-tau je asociována s FTD a parkinsonizmem vázaným s MAPT mutací na chromozomu 17 (FTDP-17T/ MAPT) [39]. Nejčastější klinickou manifestaci dědičné formy FTLD-TDP představuje FTD s parkinsonizmem vázaná s mutací genu pro progranulin na chromozomu 17 (FTLD-17U/ PGRN) [77]. Vzácnější manifestací dědičné FTLD-TDP je Perryho syndrom, asociovaný s mutací pro dynactin 1[78]. Perryho syndrom je autozomálně dominantně dědičné onemocnění charakterizovné L-DOPA-responzivním parkinsonizmem, parézou vertikálního pohledu, respiračními obtížemi, váhovým úbytkem, depresí a dalšími psychiatrickými symptomy. Respirační obtíže manifestující se hypoventilací (progredující do obrazu těžké respirační insuficience s dušností až apnoe) jsou důležitým klinickým diferenciálně diagnostickým znakem oproti PSP. Byly popsány i případy s prokázanou mutací a negativní rodinnou anamnézou [79].
Z dalších forem FTLD mohou být parkinsonizmem provázeny FTD-3 asociovaná s již výše zmiňovanou mutací CHMP2B (FTD-3/CHMP2B) charakterizovaná bvFTD, která může být doprovázena parkinsonizmem, dystonií, pyramidovými příznaky nebo myoklonem, a dále bvFTD s ALS asociovaná s FUS mutací (ALS-FTD/ FUS) manifestující se ALS a psychózou. Jelikož se v obou případech jedná o velmi vzácné klinické jednotky, nebyl parkinsonizmus zatím blíže charakterizován.
Velmi vzácně lze parkinsonizmus, jako součást klinického obrazu, nalézt u familiární FTD s myopatií s inkluzními tělísky a Pagetovou chorobou kostí (IBMPFD) asociované s mutací genu pro valosin-containing protein (VCP) [80], a u familiární ALS s FTD asociované s TARDBP mutací lokalizovanou na chromozomu 1 [81].
Klinické charakteristiky FTD s parkinsonizmem
FTDP-17/MAPT
Mutace v MAPT genu je přítomna téměř u 50 % případů FTD; dosud bylo v MAPT genu popsáno přes 50 patogenních mutací [82]. Způsob dědičnosti je autozomálně dominantní. Průměrný věk začátku onemocnění je 49 let, ale může kolísat v rozmezí 25 – 76 let. Průměrná délka trvání onemocnění je přibližně sedm let. Jedinci postižení FTDP-17/MAPT mají téměř vždy pozitivní rodinnou anamnézu, penetrance MAPT mutace je uváděna jako téměř 100%. Onemocnění postihuje stejnou frekvencí obě pohlaví [83,84]. Klinický obraz FTDP-17/MAPT může být variabilní, nicméně kardinální příznaky zahrnují osobnostní a behaviorální změny, demenci a parkinsonizmus. Na začátku onemocnění bývá přítomen pouze jeden z těchto symptomů a s progresí onemocnění dochází k postupné manifestaci všech ostatních. Iniciální FTDP-17 fenotyp, který je dán typem MAPT mutace, lze rozlišit na FTD-predominantní nebo parkinsonizmus-predominantní [85]. Parkinsonizmus tedy může být prvním příznakem FTDP-17/ MAPT. V počátečních fázích může rovněž velmi dobře odpovídat na léčbu pomocí L-DOPA. Z tohoto důvodu je u většiny těchto pacientů často mylně diagnostikována Parkinsonova nemoc. Doba manifestace parkinsonizmu ve vztahu k FTD je dána typy jednotlivých mutací v MAPT genu. Parkinsonizmus obvykle zahrnuje těžkou, symetrickou bradykinézu, rigiditu vyjádřenou axiálně i na končetinách a posturální instabilitu. Motorická symptomatika FTDP-17/MAPT může mít rovněž podobu atypického parkinsonizmu, zejména PSP syndromu, který je v tomto případě přítomen ve srovnání s CBS podstatně častěji. V některých případech FTDP-17/MAPT může být parkinsonizmus doprovázen bulbárním syndromem s dysartrií a dysfagií, nebo příznaky postižení kortikospinálního systému, dystonií a myoklonem (schéma 1) [86–88].
FTDP-17/PRGN
Mutace v PRGN genu je odpovědná přibližně za 26 % případů FTD; patogenních mutací bylo dosud popsáno více než 70. Dědičnost tohoto onemocnění je rovněž autozomálně dominantní. Průměrný věk začátku onemocnění je kolem 59 let, tedy o 10 let později než v případě FTDP-17/ MAPT, ale může kolísat v rozmezí od 44 do 83 let [89]. Délka trvání onemocnění je obvykle kratší než v případech asociovaných s MAPT mutací. Klinický obraz FTDP-17/ PRGN zahrnuje široké spektrum kognitivních, behaviorálních a motorických příznaků, nicméně jednotlivé mutace nekorelují s konkrétními klinickými syndromy [90–94]. PRGN mutace jsou obvykle asociované s bvFTD, ale na rozdíl od nosičů MAPT mutací jsou zde často přítomny i příznaky syndromu PPA [89]. Z toho důvodu může být detailní vyšetření řečových funkcí užitečné v předpovězení PRGN mutace u pacientů s FTDP. Podobný význam může mít stanovení sérové hladiny progranulinu u pacientů s FTDP, která bývá v případech s PRGN mutací extrémně nízká [95]. K rozvoji parkinsonizmu dochází většinou v pozdějších stadiích onemocnění, kdy už jsou vyjádřeny kognitivní a behaviorální příznaky, a většinou se jedná o mírnější formu většinou asymetrického, akineticko-rigidního syndromu (tab. 1) [96]. Pouze u malého počtu jedinců s PRGN mutací (p.Leu271LeufsX10) může být parkinsonizmus dominujícím klinickým příznakem [86,93]. V některých případech se parkinsonizmus vyskytuje společně s vizuálními halucinacemi, a klinický obraz se tak může velmi podobat demenci s Lewyho tělísky [87]. Jindy je FTDP-17/ PRGN doprovázena atypickým parkinsonizmem charakterizovaným asymetrickou rigiditou a apraxií a jako taková může připomínat CBS (schéma 1). Tento klinický obraz bývá nejčastěji asociován s mutacemi p.V200GfsX18, pA9D a p.E498DfsX12 [97]. Z tohoto důvodu jsou PRGN mutace taky uváděny jako nejobvyklejší příčina familiární formy CBS [92].
FTD asociované s C9ORF72 expanzí
FTD asociované s C9ORF72 expanzí charakterizovanou expanzí opakování hexanukleotidu GGGGCC představuje přibližně 48 % všech FTD. Tato abnormální expanze je odpovědná za variabilní kombinace klinických fenotypů. Zatím není známo, zda je určitý počet repetic odpovědný za konkrétní klinický obraz [98]. K hlavním klinickým fenotypům asociovaným s C9ORF72 expanzí patří bvFTD, ALS a FTD-MND. Věk začátku a délka trvání onemocnění jsou značně variabilní. Průměrný věk začátku onemocnění se pohybuje kolem 55 let, přičemž může kolísat v rozmezí od 33 do 75 let. Průměrná délka trvání onemocnění je 4,5 roku, s typickým kolísáním v rozmezí 3 – 10 let [99,100]. Parkinsonizmus je popisován přibližně u jedné třetiny případů [100,101]. Obvykle je asociován s bvFTD nebo FTD-MND, u čistých forem ALS nebyl dosud popsán [102,103]. K jeho manifestaci dochází typicky v prvních letech průběhu onemocnění, většinou je charakterizován symetrickým, akineticko-rigidním syndromem a poruchami chůze, přechodně může dobře odpovídat na léčbu L-DOPA [99,104]. Rovněž byly popsány případy s fenotypem CBS a PSP (schéma 1) [105].
FTD-3 (CHMP2B)
Mutace genu CHMP2B lokalizovaného na chromozomu 3p11.2 je vzácná genetická příčina familiárních FTD. V souvislosti s FTD byla poprvé popsána v roce 2005, v jedné velké dánské rodině a v jedné rodině v Belgii [92]. Dosud byly v tomto genu popsány čtyři patogenní mutace. Způsob dědičnosti je autozomálně dominantní. Onemocnění je charakterizováno pozvolným rozvojem a relativně pomalou progresí. Průměrný věk v začátku onemocnění je udáván kolem 58 let. Průměrná délka onemocnění je přibližně 10 let, ale může být značně variabilní [106]. Klinický obraz konstantně zahrnuje bvFTD doprovázenou zejména v pozdějších stadiích parkinsonským syndromem, dystonií, pyramidovými příznaky a myoklonem. Parkinsonizmus se manifestuje většinou v podobě asymetrického, hypokineticko-rigidního syndromu. Vzácněji může být přítomen atypický parkinsonizmus doprovázený supranukleární okohybnou poruchou nebo apraxií [107,108].
IBMPFD
IBMPFD je onemocnění asociované s mutací VCP genu na chromozomu 9p13.3 kódujícího tzv. valosin-containing protein. Mutace v tomto genu byla poprvé popsána v roce 2004. Klinický obraz tohoto onemocnění může být značně variabilní. Jedním z typických projevů jsou behaviorální a kognitivní změny [109]. Fenotyp VCP mutace může také zahrnovat projevy MND ve formě familiární ALS [110]. U několika jedinců z postižených rodin byl rovněž popsán rozvoj parkinsonizmu v podobě symetrické bradykineze, hypokineze a rigidity, k jehož manifestaci došlo zejména v pozdějších stadiích onemocnění [110 – 112]. U několika málo případů byl přítomen klidový třes, poruchy chování v REM spánku, bradykineze a rigidita odpovídající na podávání L-DOPA, tedy příznaky připomínající spíše Parkinsonovu nemoc [109,110,113]. Nicméně asociace parkinsonizmu s VCP mutací je vzácná.
TARDBP mutace
Mutace v genovém lokusu TARDBP byla popsána v roce 2008 v příčinné souvislosti s ALS. Gen je uložen na chromozomu 1p36.22 a dosud bylo popsáno více než 30 patogenních mutací. Nejčastějšími klinickými jednotkami asociovanými s TARDBP mutací jsou familiární ALS, (vzácněji) FTD a parkinsonizmus. V závislosti na typu mutace může docházet k různým kombinacím jednotlivých fenotypů. Onemocnění se tak může manifestovat fenotypem odpovídajícím Parkinsonově nemoci, nebo jako FTD s L-DOPA responzivním parkinsonizmem bez příznaků ALS, či kombinací parkinsonizmu s ALS a FTD. Dominující příznaky parkinsonského syndromu jsou bradykineza a rigidita, které typicky dobře reagují na léčbu L-DOPA [114,115].
FUS mutace
Mutace ve FUS genu byly původně popsány v roce 2009, v příčinné souvislosti s ALS. Podobně jako TARDBP mutace jsou odpovědné za přibližně 4 % případů familiární ALS [116]. Některé typy mutací FUS genu jsou dávány do souvislosti s FTD a esenciálním třesem [117,118]. Rozvoj parkinsonizmu byl popsán v několika rodinách s familiární ALS i u několika případů sporadické ALS asociovaných s FUS mutací [119]. O FTD s parkinsonizmem souvisejících s FUS mutacemi je zatím známo velmi málo.
Závěr
FTLD se vyznačují značnou klinickou, genetickou a patologickou heterogenitou. Klinické fenotypy zaujímají široké spektrum příznaků sahajících od behaviorálních poruch k poruchám řeči a jazyka a jsou různorodě spojovány s extrapyramidovou symptomatikou a MND/ ALS. V posledních 10 letech byl učiněn značný pokrok směrem k odhalení genetických příčin u většiny popsaných forem FTD. Byla popsána řada genů (MAPT, PRGN, C9ORF72, TARDBP, VCP, Dynactin, CHMP2B a FUS), jež jsou zodpovědné až za 60 % familiárních forem FTD. Z neuropatologického hlediska převažují dva typy nálezů. FTLD-tau, související s MAPT mutací a FTLD-TDP, která je asociována s mutacemi PRGN, C9ORF72, Dynactinu, TARDBP a VCP. Vzácněji jsou pak přítomny FTLD-FUS související s FUS mutací a FTLD-UPS asociovaná s CHMP2B mutací. Proč jsou různé geny spojovány s neurodegenerativním procesem postihujícím selektivně frontální a temporální laloky, je stále otázkou.
Vzhledem k rozmanitosti klinického nálezu a vzájemnému překrývání symptomů mezi jednotlivými typy FTD neexistuje jednoznačná korelace mezi typem mutace či neuropatologickým nálezem a konkrétním klinickým syndromem. Neuropatologický obraz se dá předpovědět pouze v případě známé mutace. Lze však nalézt několik klinických nebo neuroanatomických charakteristik, které mají konkrétní molekulární souvislost, a mohou tak pomoci v diferenciální diagnostice FTD. Například pacienti s C9ORF72 mutací mají většinou začátek onemocnění v mladším věku, mají pozitivní rodinnou anamnézu FTD (zejména bvFTD s disinhibicí), která je doprovázena MND/ ALS. MAPT mutace se obvykle manifestuje bvFTD, k jejímuž rozvoji dochází před 50. rokem věku, a je doprovázena symetrickým akineticko-rigidním parkinsonizmem. Naopak v případě PRGN mutace začíná onemocnění později, ve věku kolem 60 let, parkinsonizmus je asymetrický, klinický obraz může často připomínat CBS. Vyšetření TARDBP mutace by mělo být provedeno u všech familiárních i sporadických C9ORF72 negativních případů FTD-MND/ ALS. FTD spojená s myopatií a Pagetovou chorobou kostí svědčí pro VCP mutaci. Perryho syndrom asociovaný s mutací pro Dynactin 1 má své typické klinické charakteristiky, které již byly uvedeny výše.
Z neuroradiologického hlediska, nápadná stranová asymetrie frontotemporální atrofie, se současným postižením parietálního laloku, svědčí pro PRGN mutaci, zatímco relativně symetrické postižení frontotemporálních oblastí je přítomno v případech MAPT mutace. U případů s C9ORF72 mutací je atrofie více generalizovaná a symetrická (tab. 1).
Ve snaze co nejvíce zpřesnit klinickou diagnostiku s možností předpovědět odpovídající proteinopatii či související mutaci bylo provedeno několik pokusů o identifikaci různých biomarkerů. Bylo prokázáno, že extrémně nízká plazmatická hladina progranulinu je spolehlivým ukazatelem mutace v PRGN genu a jí odpovídající neuropatologie FTLD-TDP. Stanovení sérové hladiny progranulinu by tak mělo být provedeno u všech pacientů před sekvenováním PRGN genu. Jako další biomarker k predikci FTLD-TDP patologie by mohl sloužit poměr množství fosfo-Tau181 a celkového množství tau proteinu v mozkomíšním mozku, tzv. phospho-Tau181/ total-Tau (p/ t-Tau) ratio, který vykazuje významné snížení v případech FTLD-TDP ve srovnání s FTLD-tau [120].
Při současném vývoji poznání by se diagnostika FTD měla opírat o detailní popis klinického obrazu zahrnující kromě neurologického nálezu i podrobné vyšetření neuropsychologické a logopedické, zhodnocení vzorce atrofie při zobrazovacím vyšetření mozku, případně vyšetření uvedených biomarkerů. Cílem by pak mělo být předpovězení možné proteinopatie, event. detekce odpovídající mutace. Jedině tak bude v budoucnosti možné zajistit výběr vhodných pacientů do klinických hodnocení testujících účinnost potenciálně léčebných preparátů a v případě, že tyto budou objeveny, zahájit včas správnou terapii.
Práce byla podpořena granty: IGA MZ CR NT-14407, IGA MZ CR NT-12221, IGA-LF-2015-013, AZV MZ CR 15-32715A, and the Institutional Support MZ CR, RVO FNOL-2015.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Kateřina Menšíková, Ph.D.
Neurologická klinika
LF UP a FN Olomouc
I. P. Pavlova 6
775 20 Olomouc
e-mail: katmen@centrum.cz
Přijato k recenzi: 7. 10. 2015
Přijato do tisku: 28. 12. 2015
Zdroje
1. Josephs KA, Petersen RC, Knopman DS, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006;66(1):41–8.
2. Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76(11):1006 – 14. doi: 10.1212/WNL.0b013e31821103e6.
3. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134(9):2456 – 77. doi: 10.1093/brain/awr179.
4. Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry 2002;180 : 140 – 3.
5. Ratnavalli E, Brayne C, Dawson K, et al. The prevalence of frontotemporal dementia. Neurology 2002;58(11):1615 – 21.
6. Harvey RJ, Skelton-Robinson M, Rossor M. The prevalence and causes of dementia in People under the age of 65 years. J Neuro Neurosurg Psychiatry 2003;74(9):1206 – 9.
7. Borroni B, Alberici A, Grassi M, et al. Is frontotemporal lobar degeneratin a rare disorder? Evidence from a preliminary study in Brescia County, Italy. J Alzhemiers Dis 2010;19(1):111 – 6. doi: 10.3233/JAD-2010-1208.
8. Seelaar H, Kamphorst W, Rosso SM, et al. Distinct genetic forms of frontotemporal dementia. Neurology 2008;71(16):1220 – 6. doi: 10.1212/01.wnl.0000319702.37497.72.
9. Hodges JR, Davies R, Xuereb J, et al. Survival in frontotemporal dementia. Neurology 2003;61(3):349 – 54.
10. Pick A. Über die Bezeihungen der senilen Hirnatrophie zur Aphasie. Prager Medicinische Wochenschrift 1892;17 : 165 – 7.
11. Alzheimer A. Über eigenartige Krankheitsfälle des späteren Alters. Z Ges Neurol Psychiatr 1911 : 1914.
12. Onari K, Spatz H. Anatomische Beträge zur Lehre von der Pickschen umschribenen Groβhirnrinden-Atrophie („Picksche Kranheit“). Z Ges Neurol Psychiatr 1926;101 : 470 – 511.
13. Pollock NJ, Mirra SS, Binder LI, et al. Filamentous aggregates in Pick’s disease, progressive supranuclear palsy and Alzheimer’s disease share antigenic determinants with mikrotubule-associated protein Tau. Lancet 1986;328 : 1211.
14. Brun A, Englund B, Gustafson L. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatr 1994;57(4):416 – 8.
15. Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 2010;119(1):1–4. doi: 10.1007/s00401-009-0612-2.
16. Goedert M, Spillantini MG, Jakes R, et al. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer‘s disease. Neuron 1989;3(4):519 – 26.
17. van Swieten J, Spillantini MG. Hereditary frontotemporal dementia caused by tau gene utations. Brain Pathol 2007;17(1):63–73.
18. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314(5796):130 – 3.
19. Mackenzie IR, Neumann M, Cairns NJ, et al. Novel types of frontotemporal lobar degeneration: beyond Tau and TDP-43. J Mol Neurosci 2011;45(3):402 – 8. doi: 10.1007/s12031-011-9551-1.
20. Munoz DG, Neumann M, Kusaka H, et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol 2009;118(5):617 – 27. doi: 10.1007/s00401-009-0598-9.
21. Neumann M, Rademakers R, Roeber S, et al. A new subtype of frontotemporal lobardegeneration with FUS pathology. Brain 2009;132(11):2922 – 31. doi: 10.1093/ brain/awp214.
22. Urwin H, Josephs KA, Rohrer JD, et al. FUS pathology defines the majority of tau - and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol 2010;120(1):33 – 41. doi: 10.1007/s00401-010-0698-6.
23. Neumann M, Roeber S, Kretzschmar HA, et al. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 2009;118(5):605 – 16. doi: 10.1007/s00401-009-0581-5.
24. Munoz-Garcia D, Ludwin SK. Classic and generalized variants of Pick’s disease: a clinicopathological, ultrastructural, and immunocytochemical comparative study. Ann Neurol 1984;16(4):467 – 80.
25. Yokota O, Tsuchiya K, Terada S, et al. Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 2008;115(5):561 – 75.
26. Kusaka H, Matsumoto S, Imai T. An adult-onset case of sporadic motor neuron disease with basophilic inclusions. Acta Neuropathol 1990;80(6):660 – 5.
27. Kusaka H, Matsumoto S, Imai T. Adult-onset motor neuron disease with basophilic intraneuronal inclusion bodies. Clin Neuopathol 1993;12(4):215 – 8.
28. Tsuchiya K, Matsunaga T, Aoki M, et al. Familial amyotrophic lateral sclerosis with posterior column degeneration and basophilic inclusion bodies: a clinical, genetic and pathological study. Clin Neuropathol 2001;20(2):53 – 9.
29. Hamada K, Fukazawa T, Yanagihara T, et al. Dementia with ALS features and diffuse Pick body-like inclusions (atypical Pick’s disease?). Clin Neuropathol 1995;14(1):1 – 6.
30. Ishihara K, Araki S, Ihori N, et al. An autopsy case of frontotemporal dementia with severe dysarthria and motor neuron disease showing numerous basophilic inclusions. Neuropathology 2006;26(5):447 – 54.
31. Cairns NJ, Grossman M, Arnols SE, et al. Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology 2004;63(8):1376–84.
32. Mackenzie IR, Munoz DG, Kusaka H, et al. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol 2011;121(2):207 – 18. doi: 10.1007/s00401-010-0764-0.
33. Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 2005;37(8):806–8.
34. van der Zee J, Urwin H, Engelborghs S, et al. CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum Mol Genet 2008;17(2):313–22.
35. Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009;73(18):1451 – 6. doi: 10.1212/WNL.0b013e3181bf997a.
36. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72(2):245 – 56. doi: 10.1016/j.neuron.2011.09.011.
37. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72(2):257 – 68. doi: 10.1016/j.neuron.2011.09.010.
38. Edwards TL, Scott WK, Almonte C, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 2010;74(2):97 – 109. doi: 10.1111/j.1469-1809.2009.00560.x.
39. Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 59-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998;393(6686):702–5.
40. Goedert M, Spillantini MG, Potier MC, et al. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 1989;8(2):393–9.
41. van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol 2008;7(10):965–74. doi: 10.1016/S1474-4422(08)70194-7.
42. Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 2006;15(20):2988–3001.
43. Le Ber I, Camuzat A, Hannequin D, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 2008;131(3):732–46. doi: 10.1093/brain/awn012.
44. Beck J, Rohrer JD, Campbell T, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 2008;131(3):706–20. doi: 10.1093/brain/awm320.
45. Kelley BJ, Haidar W, Boeve BF, et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging 2009;30(5):739–51.
46. Bigio EH. C9ORF72, the new gene on the block, causes C9FTD/ ALS: new insights provided by neuropathology. Acta Neuropathol 2011;122(6):653–5. doi: 10.1007/s00401-011-0919-7.
47. van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C-ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 2013;12(10):978 – 88. doi: 10.1016/S1474-4422(13)70210-2.
48. Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11(4):323–30. doi: 10.1016/S1474-4422(12)70043-1.
49. Herdewyn S, Zhao H, Moisse M, et al. Whole-genome sequencing reveals a coding non-pathogenic variant tagging a non-coding pathogenic hexanucleotide repeat expansion in C9orf72 as cause of amyotrophic lateral sclerosis. Hum Mol Genet 2012;21(11):2412–9. doi: 10.1093/hmg/ dds055.
50. Mori K, Weng SM, Arzberger T, et al. The C9or72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ ALS. Science 2013;339(6125):1335–8. doi: 10.1126/science.1232927.
51. Kimonis VE, Fulchiero E, Vesa J, et al. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta 2008;1782(12):744 – 8. doi: 10.1016/j.bbadis.2008.09.003.
52. Benajiba L, Le Ber I, Camuzat A, et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 2009;65(4):470 – 3. doi: 10.1002/ana.21612.
53. Kwiatkowski TJ, Bosco DA, Leclerc AL, et al. Mutations in the FUS/ TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323(5918):1205 – 8. doi: 10.1126/science.1166066.
54. Van Langenhove T, van der Zee J, Sleegers K, et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 2010;74(5):366-71. doi: 10.1212/WNL.0b013e3181ccc732.
55. Seelaar H, Kamphorst W, Rosso SM, et al. Distinct genetic forms of frontotemporal dementia. Neurology 2008;71 : 1220 – 6. doi: 10.1212/01.wnl.0000319702.37497.72.
56. Kfoury N, Holmes BB, Jiang H, et al. Transcellular propagation of tau aggregation by fibrillar species. J Biol Chem 2012;287(23):19440 – 51. doi: 10.1074/jbc.M112.346072.
57. Guo CC, Gorno-Tempini ML, Gesierich B, et al. Anterior temporal lobe degeneration produces widespread network-driven dysfunction. Brain 2013;136(10):2979 – 91. doi: 10.1093/brain/awt222.
58. Rosen HJ, Gorno-Tempini ML, Goldman WP, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 2002;58(2):198–208.
59. Liu W, Miller BL, Kramer JH, et al. Behavioral disorders in the frontal and temporal variants of frontotemporal dementia. Neurology 2004;62(5):742–8.
60. Marczinski CA, Davidson W, Kertesz A. A longitudinal study of behavior in frontotemporal dementia and primary progressive aphasia. Cogn Behav Neurol 2004;17(4): 185–90.
61. Kramer JH, Jurik J, Sha SJ, et al. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol 2003;16(4):211–8.
62. Kipps CM, Hodges JR, Hornberger M. Nonprogressive behavioural frontotemporal dementia: recent developments and clinical implications of the ’bvFTD phenocopy syndrome’. Curr Opin Neurol 2010;23(6):628 – 32. doi: 10.1097/WCO.0b013e3283404309.
63. Khan BK, Yokoyama JS, Takada LT, et al. Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry 2012;83(4):358 – 64. doi: 10.1136/jnnp-2011-301883.
64. Grossman M. Primary progressive aphasia: clinicopathological correlations. Nat Rev Neurol 2010;6(2):88–97. doi: 10.1038/nrneurol.2009.216.
65. Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76(111):1006–14. doi: 10.1212/WNL.0b013e31821103e6.
66. Snowden JS, Thompson JC, Stopford C, et al. The clinical diagnosis of early-onset dementias: diagnostic accuracy and clinicopathological relationships. Brain 2011;134(9):2478–92. doi: 10.1093/brain/awr189.
67. Mesulam MM. Primary progressive aphasia. Ann Neurol 2001;49(4):425–32.
68. Catani M, Mesulam MM, Jakobsen E, et al. A novel frontal pathway underlies verbal fluency in primary progressive aphasia. Brain 2013;136(8):2619 – 28. doi: 10.1093/brain/awt163.
69. Ash S, Evans E, O’Shea J, et al. Differentiating primary progressive aphasias in a brief sample of connected speech. Neurology 2013;81(4):329 – 36. doi: 10.1212/WNL.0b013e31829c5d0e.
70. Thompson SA, Patterson K, Hodges JR. Left/ right asymmetry of atrophy in semantic dementia: behavioral-cognitive implications. Neurology 2003;61(9):1196–203.
71. Ioannidis P, Konstantinopoulou E, Maiovis P, et al. The frontotemporal dementias in a tertiary referral center: classification and demographic characteristics in a series of 232 cases. J Neurol Sci 2012;318(1–2):171 – 3. doi: 10.1016/j.jns.2012.04.002.
72. Espay AJ, Litvan I. Parkinsonism and frontotemporal dementia: the clinical overlap. J Mol Neurosci 2011;45(3):343 – 9. doi: 10.1007/s12031-011-9632-1.
73. Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 2009;117(1):15 – 8. doi: 10.1007/s00401-008-0460-5.
74. Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007;114(1):5–22.
75. Kovacs GG, Majtenyi K, Spina S, et al. White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol 2008;67(10):963–75. doi: 10.1097/NEN.0b013e318187a80f.
76. Josephs KA, Knopman DS, Whitwell JL, et al. Survival in two variants of tau-negative frontotemporal lobar degeneration: FTLD-U vs FTLD-MND. Neurology 2005;65(4):645–7.
77. Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006;442(7105):916–9.
78. Farrer MJ, Hulihan MM, Kachergus JM, et al. DCTN1 mutations in Perry syndrome. Nat Genet 2009;41(2):163 – 5. doi: 10.1038/ng.293.
79. Aji BM, Medley G, O‘Driscoll K, et al. Perry syndrome: a disorder to consider in the differential diagnosis of Parkinsonism. J Neurol Sci 2013;330(1 – 2):117 – 8. doi: 10.1016/j.jns.2013.04.008.
80. Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36(4):377–81.
81. Rutherford NJ, Zhang YJ, Baker M, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 2008;4(9):e1000193. doi: 10.1371/journal.pgen.1000193.
82. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol 2013;2 : 609–22.
83. Ludolph AC, Kassubek J, Landwehrmeyea BG, et al. Tauopathies with parkinsonism: clinical spectrum, neuropathologic basis, biological markers, and treatment options. Eur J Neurol 2009;16(3):297 – 309. doi: 10.1111/j.1468-1331.2008.02513.x.
84. Carney RM, Kohli MA, Kunkle BW, et al. Parkinsonism and distinct dementia patterns in a family with the MAPT R406W mutation. Alzheimers Dement 2014;10(3):360 – 5. doi: 10.1016/j.jalz.2013.02.011.
85. Baba Y, Tsuboi Y, Baker MC, et al. The effect of tau genotype on clinical features in FTDP-17. Parkinsonism Relat Disord 2005;11(4):205–8.
86. Puschmann A. Monogenic Parkinson‘s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord 2013;19(4):407–15. doi: 10.1016/j.parkreldis.2013.01.020.
87. Boeve BF, Hutton M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN). Arch Neurol 2008;65(4):460–4. doi: 10.1001/archneur.65.4.460.
88. Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 2004;24(4):277–95.
89. Fujioka S, Wszolek ZK. Clinical aspects of familial forms of frontotemporal dementia associated with parkinsonism. J Mol Neurosci 2011;45(3):359 – 65. doi: 10.1007/s12031-011-9568-5.
90. Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006;442(7105):920–4.
91. Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 2006;15(20):2988–3001.
92. Le Ber I, Camuza A, Hannequin D, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 2008;131(3):732 – 46. doi: 10.1093/ brain/awn012.
93. Benussi L, Ghidoni R, Pegoiani E, et al. Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol Dis 2009;33(3):379 – 85. doi: 10.1016/j.nbd.2008.11.008.
94. Moreno F, Indakoetxea B, Barandiaran M, et al. “Frontotemporoparietal” dementia: clinical phenotype associated with the c.709-1G>A PGRN mutation. Neurology 2009;73(17): 1367 – 74. doi: 10.1212/WNL.0b013e3181bd82a7.
95. Schofield EC, Halliday GM, Kwok J, et al. Low serum progranulin predicts the presence of mutations: a prospective study. J Alzheimers Dis 2010;22(3):981-4.
96. Kelley BJ, Haidar W, Boeve BF, et al. Prominent phenotypic variability associated with mutations in progranulin. Neurobiol Aging 2009;30(5):739 – 51.
97. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003;54(Suppl 5):S15 – 9.
98. van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 2013;12(10):978 – 88. doi: 10.1016/S1474-4422(13)70210-2.
99. Boeve BF, Boylan KB, Graff-Radford NR, et al. Characterization of frontotemporal dementia and/ or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135(3):765 – 83. doi: 10.1093/brain/aws004.
100. Smith BN, Newhouse S, Shatunov A, et al. The C9ORF72 expansion mutation is a common cause of ALSţ/ -FTD in Europe and has a single founder. Eur J Hum Genet 2013;21(1):102 – 8. doi: 10.1038/ejhg.2012.98.
101. Waldo ML, Gustafson L, Nilsson K, et al. Frontotemporal dementia with a C9ORF72 expansion in a Swedish family: clinical and neuropathological characteristics. Am J Neurodegener Dis 2013;2(4):276 – 86.
102. Friedland RP, Shah JJ, Farrer LA, et al. Behavioral variant frontotemporal lobar degeneration with amyotrophic lateral sclerosis with a chromosome 9p21 hexanucleotide repeat. Front Neurol 2012;3 : 136. doi: 10.3389/fneur.2012.00136.
103. Savica R, Adeli A, Vemuri P, et al. Characterization of a family with c9FTD/ ALS associated with the GGGGCC repeat expansion in C9ORF72. Arch Neurol 2012;69(9):1164 – 9. doi: 10.1001/archneurol.2012.772.
104. Yeh TH, Lai SC, Weng YH, et al. Screening for C9orf72 repeat expansions in parkinsonian syndromes. Neurobiol Aging 2013;34(4):1311.e3 – 4. doi: 10.1016/j.neurobiolaging.2012.09.002.
105. Lindquist SG, Duno M, Batbayli M, et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin Genet 2013;83(3):279 – 83. doi: 10.1111/j.1399-0004.2012.01903.x.
106. Arvanitakis Z. Update on frontotemporal dementia. Neurologist 2010;16(1):16 – 22. doi: 10.1097/NRL.0b013e3181b1d5c6.
107. Gydesen S, Brown JM, Brun A, et al. Chromosome 3linked frontotemporal dementia (FTD-3). Neurology 2002;59(10):1585 – 94.
108. Stokholm J, Teasdale TW, Johannsen P, et al. Cognitive impairment in the preclinical stage of dementia in FTD-3 CHMP2B mutation carriers: a longitudinal prospective study. J Neurol Neurosurg Psychiatry 2013;84(2):170 – 6. doi: 10.1136/jnnp-2012-303813.
109. Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin--containing protein. Nat Genet 2004;36(4):377 – 81.
110. Spina S, Van Laar AD, Murrell JR, et al. Phenotypic variability in three families with valosin-containing protein mutation. Eur J Neurol 2013;20(2):251 – 8. doi: 10.1111/j.1468-1331.2012.03831.x.
111. van der Zee J, Pirici D, Van Langenhove T, et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology 2009;73(8):626 – 32. doi: 10.1212/WNL.0b013e3181b389d9.
112. Mehta SG, Khare M, Ramani R, et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/ or frontotemporal dementia. Clin Genet 2013;83(5):422 – 31. doi: 10.1111/cge.12000.
113. Chan N, Le C, Shieh P, et al. Valosin-containing protein mutation and Parkinson‘s disease. Parkinsonism Relat Disord 2012;18(1):107 – 9. doi: 10.1016/j.parkreldis.2011.07.006.
114. Rayaprolu S, Fujioka S, Traynor S, et al. TARDBP mutations in Parkinson‘s disease. Parkinsonism Relat Disord 2013;19(3):312 – 5. doi: 10.1016/j.parkreldis.2012.11.003.
115. Mosca L, Lunetta C, Tarlarini C, et al. Wide phenotypic spectrum of the TARDBP gene: homozygosity of A382T mutation in a patient presenting with amyotrophic lateral sclerosis, Parkinson‘s disease, and frontotemporal lobar degeneration, and in neurologically healthy subject. Neurobiol Aging 2012;33(8):1846.e1 – 4. doi: 10.1016/j.neurobiolaging.2012.01.108.
116. Kwiatkowski TJ, Bosco DA, LeClerc AL, et al. Mutations in the FUS/ TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323(5918):1205 – 8. doi: 10.1126/science.1166066.
117. Gao K, Zheng W, Deng X, et al. Genetic analysis of the fused in sarcoma gene in Chinese Han patients with Parkinson‘s disease. Parkinsonism Relat Disord 2014;20(1):119 – 21. doi: 10.1016/j.parkreldis.2013.09.010.
118. Labbe C, Rayaprolu S, Soto-Ortolaza A, et al. Investigating FUS variation in Parkinson‘s disease. Parkinsonism Relat Disord 2014;20(Suppl 1):S147 – 9. doi: 10.1016/S1353-8020(13)70035-X.
119. Van Langenhove T, van der Zee J, Van Broeckhoven C. The molecular basis of the frontotemporal lobar degeneration - amyotrophic lateral sclerosis spectrum. Ann Med 2012;44(8):817 – 28. doi: 10.3109/07853890.2012.665471.
120. Boroni B, Benussi A, Archetti S, et al. CSF p-tau181/ tau ratio as biomarker for TDP pathology in frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener 2015;16(1 – 2):86 – 91. doi: 10.3109/21678421.2014.971812.
121. Mackenzie IR, Neumann M, Baborie A et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122(1):111–3. doi: 10.1007/s00401-011-0845-8.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2016 Číslo 3
Nejčtenější v tomto čísle
- Schwannom nejspíše z krčního sympatiku – kazuistika
- Validační studie české verze Bostonského testu pojmenování
- Klinický standard pro diagnostiku a léčbu pacientů s ischemickou cévní mozkovou příhodou a s tranzitorní ischemickou atakou – verze 2016
- Pre-motorické a non-motorické príznaky Parkinsonovej choroby – taxonómia, klinická manifestácia a neuropatologické koreláty