
Spinocerebelární ataxie typu 6 – kazuistika
Spinocerebellar Ataxia 6 – a Case Report
Spinocerebellar ataxia 6 (SCA6) is a rather rare neurodegenerative disorder, one of autosomal dominant spinocerebelar ataxias. It is caused by expansion of unstable CAG triplet repeats in the gene responsible for the voltage-dependent calcium channel mapped to the chromosome 19. The disease is characterized by progressive paleo- and neocerebellar symptomatology. The onset of the disease is typically in middle- and older-age. The authors describe a case of 62-year-old women with sudden development of gait deterioration. This was first diagnosed as a post-ischemic stroke state. However, clinical symptoms progressed into gait ataxia. No analogical case was found in family history. Genetic testing, performed after all other potential causes of the condition were excluded, indicated SCA6. Our case report emphasises the importance of considering rare conditions as part of differential diagnosis. In this case, this protected the patient from further diagnostic testing as well as, importantly, enabled predictive testing in relatives in risk.
Key words:
spinocerebellar ataxia – cerebellar syndrome – hereditary spinocerebellar degenerations
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
K. Krakorová 1; J. Polívka 1; F. Šlauf 2
Působiště autorů:
LF UK a FN Plzeň
Neurologická klinika
1; LF UK a FN Plzeň
Klinika zobrazovacích metod
2
Vyšlo v časopise:
Cesk Slov Neurol N 2015; 78/111(4): 482-485
Kategorie:
Kazuistika
Souhrn
Spinocerebelární ataxie typu 6 (SCA6) je relativně vzácné hereditární neurodegenerativní onemocnění patřící do skupiny autozomálně dominantních spinocerebelárních ataxií. Příčinou je expanze nestabilních CAG tripletů v genu kódujícím podjednotku napěťově řízeného kalciového kanálu na 19. chromozomu. Onemocnění je charakteristické postupným rozvojem paleo- a neocerebelární symptomatiky s počátkem ve středním a vyšším věku. V následujícím textu prezentujeme případ nyní 63leté pacientky s náhlým rozvojem nespecifických potíží při chůzi, zprvu hodnocených jako stav po proběhlé cévní mozkové příhodě, s postupným vývojem klinického obrazu do výrazné trupové ataxie a instability chůze při zcela němé rodinné anamnéze. Po vyloučení častějších příčin obtíží byla na základě genetického vyšetření diagnostikována SCA6. Kazuistika ukazuje na nutnost pomýšlet i na možnost výskytu vzácné diagnózy, což v tomto případě přináší ukončení opakovaných vyšetření a v neposlední řadě i možnost prediktivního testování u příbuzných v riziku.
Klíčová slova:
spinocerebelární ataxie – mozečkový syndrom – dědičné spinocerebelární degenerace
Úvod
Spinocerebelární ataxie (SCA) jsou geneticky vázaná neurodegenerativní onemocnění charakterizovaná rozvojem paleo- a neocerebelární symptomatiky s postupnou progresí. Na základě typu dědičnosti je lze dělit na autozomálně dominantní spinocerebelární ataxie (AD SCA), autozomálně recesivní spinocerebelární ataxie (AR SCA) a ataxie vázané na X chromozom [1].
Jedná se o onemocnění relativně vzácná, s celkovou prevalencí přibližně 3– 5 případů na 100 000 obyvatel [2]. Ze skupiny AD SCA je celosvětově nejčastější výskyt SCA3, SCA1, SCA2 a SCA6 [1,3]. Není bez zajímavosti, že prevalence jednotlivých typů AD SCA se liší v závislosti na geografické poloze a na etniku [4]. Například v Brazílii, Portugalsku, Číně a Německu dominuje výskyt SCA3, v jižní Africe SCA1, v Japonsku pak SCA6 [1]. V České republice je popsán výskyt SCA1, SCA2, SCA3, SCA7, SCA8, SCA17, z čehož nejčastější jsou SCA2 a SCA1 [5].
Etiopatogeneticky jsou SCA podmíněny mutacemi v různých chromozomálních lokusech, kdy na základě mutace vznikají patologické, elongované proteiny, které se hromadí v intranukleárních či cytoplazmatických inkluzích buněk. Je poškozena proteolýza, dochází k aktivaci apoptotických dějů a k poškození celé funkce buňky [6]. Některé AD SCA jsou způsobeny expanzemi tripletů bazí, ale jsou známy i netripletové mutace, jako například delece a bodové mutace [1].
Skupina AD SCA dle současných poznatků zahrnuje celkem 46 nozologických jednotek, 36 jednotek označovaných SCA a číslem, dentato‑ rubro‑palido‑ luysiánskou atrofii, hypomyelinizační leukoencefalopatii, autozomálně dominantní spinocerebelární ataxii s hluchotou a narkolepsií (ADCADN) a sedm jednotek epizodické ataxie. Společným znakem je trupová ataxie a ataxie chůze, později ataxie končetinová, k ní se často přidává nystagmus a dysartrie. K symptomům provázejícím SCA se v závislosti na typu nekonstantně přidávají i symptomy další, např. oftalmoplegie, demence, retinopatie, amyotrofie, extrapyramidová symptomatika, epileptické záchvaty, mentální retardace, myoklonus, hyperreflexie, což společně s variabilním věkem a různou rychlostí rozvoje prvních příznaků činí z SCA velmi heterogenní skupinu chorob, náročnou na diferenciální diagnostiku [1].
SCA6 je způsobena abnormální expanzí trinukleotidů CAG v genu CACNA1A, který je lokalizován na chromozomu 19p13. Tento gen kóduje podjednotku alfa 1A napěťově řízeného kalciového kanálu a transkripční faktor alfa 1ACT. Napěťově řízený kalciový kanál je ve vysoké míře exprimován v granulárních a Purkyňových buňkách cerebelárního kortexu a podílí se na synaptické transmisi, transkripční faktor pak zesiluje expresi dalších genů. Je známo, že patologicky elongovaný protein nepoškozuje přímo funkci napěťové řízeného kanálu, ale je toxický pro buňky exprimující alfa 1ACT [7,8]. Mutace v genu CACNA1A jsou mimo SCA6 příčinou familiární hemiplegické migrény (FHM) a epizodické ataxie s pozdním začátkem (EA2). SCA6 je charakterizována zejména čistou mozečkovou ataxií a pozdním začátkem, kdy se první příznaky objevují nejčastěji po 50. roku věku, ale mohou se manifestovat prakticky kdykoli mezi druhou až osmou dekádou. Ataxie bývá mírná, pomalu progredující, někdy provázená dysartrií, dysfagií, nystagmem, diplopií, poklesem svalového tonu a min. poškozením vibračního čití a propriocepce. U 25 % pacientů byla pozorována dystonie a blefarospazmus. Charakteristickým symptomem je ataxie chůze, která je vždy prvním příznakem. Šlachookosticové reflexy bývají normální nebo lehce zvýšené, poruchy povrchového čití, oftalmoparéza či poruchy kognice pozorovány nebyly [1,9]. Není bez zajímavosti, že během těhotenství dochází ke zhoršování příznaků, nicméně nebyl pozorován žádný vliv onemocnění na životaschopnost plodu.
Diagnostika vychází z klinického obrazu, nálezu atrofie mozečku na zobrazovacích vyšetřeních, vyloučení jiných příčin cerebelárního syndromu a na molekulárně‑genetickém vyšetření, které diagnózu definitivně potvrdí či vyloučí.
Kauzální léčba onemocnění neexistuje, je možné pouze částečné ovlivnění příznaků, např. epizody exacerbace ataxie může zmírňovat acetazolamid, k tlumení závratí lze zkusit efekt antivertiginózních léčiv. Rehabilitace však zůstává jediným prostředkem, který dlouhodobě prodlužuje mobilitu pacienta. Při postupné progresi onemocnění je nebytné využití kompenzačních pomůcek [1].
Kazuistika
Pacientka narozená v roce 1951 byla vyšetřena na našem pracovišti poprvé v roce 2010 na doporučení spádového neurologa pro asi čtyři měsíce trvající poruchu stability chůze s nutností lehké opory a třes pravé horní končetiny při zátěži. Pacientka uváděla pocit nejistoty při chůzi, zhoršující se úměrně ušlé vzdálenosti. Na cílený dotaz popisovala rychlý až náhlý počátek obtíží.
V osobní anamnéze byla uvedena operace stenózy páteřního kanálu v segmentu L4/ 5s fixací olistézy v roce 2005, z interních pak hypercholesterolemie. Rodinná anamnéza byla bez nápadností. Při vyšetření horních končetin byla popsána mírná instabilita při předpažení vpravo, lehce porušená taxe pravé horní končetiny a povšechné oboustranné zvýšení reflexů C5/ 8. Vyšetření jemné motoriky bylo s normálním nálezem. Na dolních končetinách byl pravostranně pozitivní Mingazziniho příznak při plné svalové síle, ostatní nález byl normální. Řeč byla zcela v normě, nystagmus zachycen nebyl, iritační pyramidové jevy nebyly přítomny, vyšetření stoje a chůze bylo normální. Zkouška posturální stability pull‑ testem byla též negativní.
Vzhledem k anamnéze náhlého začátku potíží, pravostranné lateralizaci v objektivním nálezu a přítomnosti vaskulárních rizikových faktorů bylo zprvu pomýšleno na cévní etiologii potíží. Bylo provedeno ultrasonografické vyšetření extrakraniálního tepenného řečiště, kde byla zachycena oboustranná, přibližně 35% stenóza vnitřní krkavice. Bylo doplněno vyšetření mozku magnetickou rezonancí (MR) s nálezem nespecifického drobného hyperintenzního ložiska subkortikálně vlevo a se známkami atrofie mozku a mozečku (obr. 1). Vzhledem ke klinickému obrazu, anamnéze náhlého začátku potíží, přítomnosti cévních rizikových faktorů a závěrům vyšetření se jako nejpravděpodobnější příčina stavu jevila proběhlá drobná ischemická cévní mozková příhoda. Pacientka byla zajištěna v rámci sekundární prevence a dimitována do domácího prostředí.
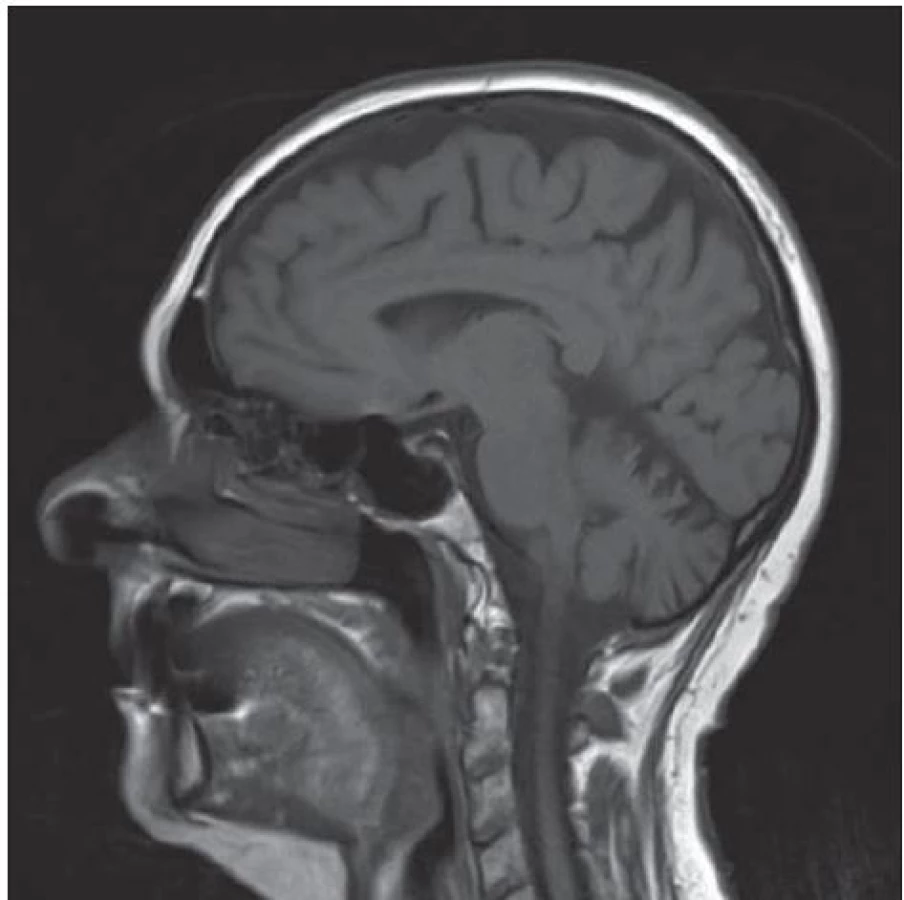
V roce 2013 se pacientka vrátila na naše pracoviště na doporučení spádového neurologa pro postupné zhoršování původních potíží. Subjektivně nemocná uvedla navíc oproti předchozí hospitalizaci slabost dolních končetin a nově třes levé ruky při jakékoli činnosti. V osobní anamnéze přibyl údaj o operaci stenózy bederní páteře v úrovních L3/ 4 a L4/ 5. V klinickém nálezu byla zjištěna mírná ataxie horních končetin a dysdiadochokineza oboustranně, na dolních končetinách stranově souměrná výrazná ataxie. Vyšetření stoje prokázalo titubace až tendence k pádům do všech stran, chůze byla ataktická. Nystagmus přítomen nebyl, dysartrie zachycena nebyla, pyramidové jevy zánikové i iritační nebyly přítomny, nebylo snížení svalové síly ani porucha čití. Pacientka byla schopna samostatné chůze na max. vzdálenost 500 m s pomocí dvou trekingových holí. S úvahou o dědičném neurodegenerativním onemocnění byla cíleně doplněna rodinná anamnéza. Otec pacientky zemřel ve 47 letech, matka v 63 letech, bratr ve 45 letech, všichni na nádorová onemocnění. Druhý bratr pacientky zemřel po opakované recidivě cévní mozkové příhody. Dva synové pacientky ve věku 41 a 43 let jsou zdrávi.
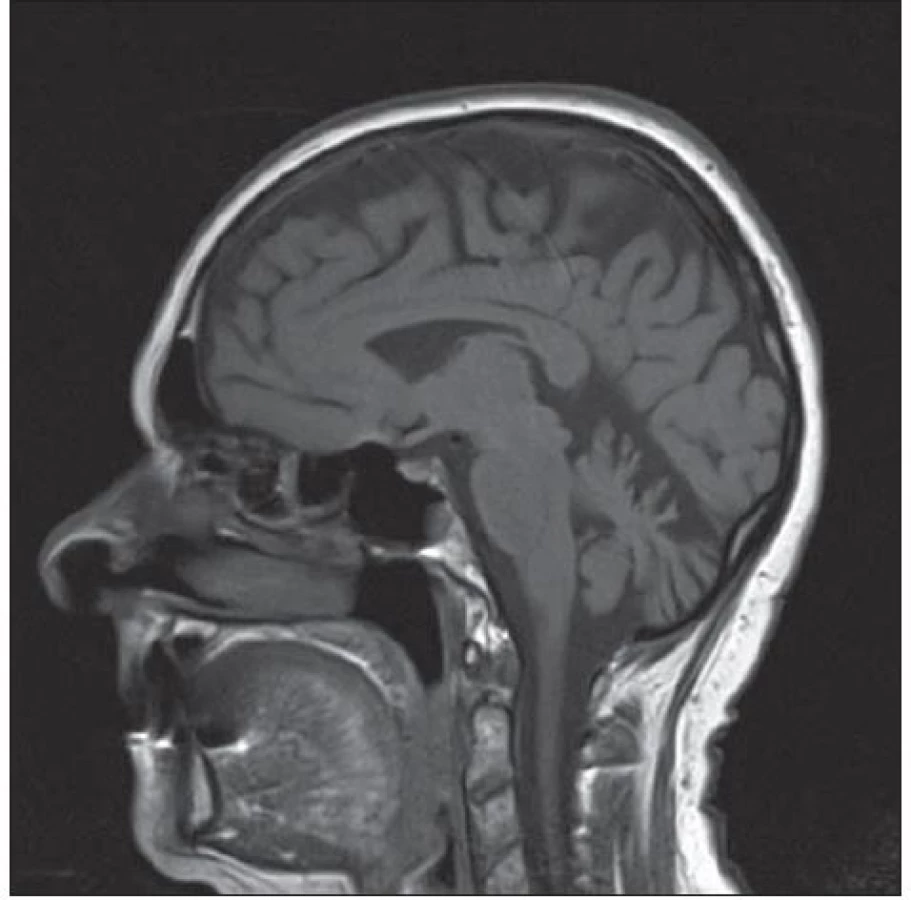
Na provedené MR mozku byla popsána oproti vyšetření z roku 2010 progredující atrofie mozkových hemisfér a mozečku, zejména v oblasti vermis, ostatní nález byl beze změn (obr. 2). Lumbální punkce nebyla provedena vzhledem k rozsáhlým postoperačním změnám v oblasti bederní páteře. Pro trvající suspekci na hereditární neurodegenerativní onemocnění mozečku bylo indikováno genetické vyšetření panelu spinocerebelárních ataxií. Vyšetřeny byly mutace pro SCA1, SCA2, SCA3, SCA6 a SCA7. Výsledek prokázal přítomnost expanze CAG repetice v genu CACNA1A s délkou alel 12/ 22 CAG, což potvrzuje diagnózu SCA6.
Diskuze
SCA6 je neurodegenerativní onemocnění mozečku vykazující autozomálně dominantní typ dědičnosti. Z celé skupiny AD SCA čítá SCA6 přibližně 10– 30 % všech případů. Převažující výskyt SCA6 je zaznamenán v Japonsku a Německu. Klinicky je charakteristická převaha „čistého“ mozečkového syndromu a ostatní výše zmiňované symptomy jsou spíše méně časté. Někteří pacienti zpětně udávali intermitentní intenzivní závrativé stavy před rozvojem ataxie chůze, zatímco u jiných se i ataxie chůze zpočátku objevovala ve formě náhlých zhoršení až záchvatovitého charakteru [3,9,10]. Jak bylo uvedeno výše, mutace v genu CACNA1A je příčinou mimo SCA6 i FHM a EA2. Obě zmiňované nozologické jednotky jsou charakterizovány náhlým začátkem a jak epizodická ataxie typu 2, tak familiární hemiplegická migréna (zejména její „čistá varianta“) jsou typické záchvatovitými stavy, mezi kterými zejména zpočátku bývá zcela normální neurologický nález [11,12].
Při použití zobrazovacích metod je popisována atrofie mozečku, histologické vyšetření prokazuje ztrátu Purkyňových buněk cerebelárního kortexu. U pacientů s SCA6 byla prokázána přítomnost polyglutaminových sluků v jádrech a cytoplasmě Purkyňových buněk [3,9,10].
Rozsah normálních délek alel čítá 5– 19 opakování CAG tripletů, u pacientů s SCA6 je nacházeno 20–25 repetic [9]. U naší nemocné bylo prokázáno 22 CAG repetic, což potvrdilo diagnózu.
U nemocné se jednalo o celkem obvyklý průběh choroby, charakteristický svým pozdním začátkem, přítomností vertiga s čistou cerebelární ataxií bez dalších symptomů a náhlou manifestací příznaků, která zpočátku vedla k mylné diferenciálně diagnostické rozvaze. Ani přes podrobně odebranou rodinnou anamnézu se nepodařilo dohledat příznaky onemocnění u jejích předků a sourozenců. Vzhledem k tomu, že SCA6 je typická svým pozdním začátkem, je mnohem pravděpodobnější, že k úmrtí rodičů i sourozenců došlo ještě před tím, než se mohly manifestovat první příznaky dědičné ataxie. Nemuselo se tedy jednat o první mutaci v rodině. Přítomnost mutace u potomků pacientky lze prokázat genetickým vyšetřením, které se vzhledem k minimálním až žádným terapeutickým možnostem řídí stejnými pravidly jako prediktivní testování např. Huntingtonovy nemoci, tzn. je podmíněno plnoletostí, návštěvou psychiatra, psychologa, genetika a neurologa [13].
Závěr
Skupina spinocerebelárních ataxií je skupina nemocí relativně vzácných, často nediagnostikovaných pro neúplnou diferenciálnědiagnostickou rozvahu a nemožnost průkazu všech typů SCA pomocí dostupného genetického vyšetření. Touto kazuistikou bychom chtěli ukázat na nutnost pomýšlet na tato onemocnění i u pacientů ve vyšším věku, při negativní rodinné anamnéze a při náhlém nástupu obtíží. U naší nemocné se dle dostupné literatury jedná o první popsaný případ SCA6 v České republice.
Podpořeno MZ ČR – RVO (Fakultní nemocnice Plzeň – FNPI, 00669806), projektem Univerzity Karlovy – SVV 260176.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 24. 11. 2014
Přijato do tisku: 27. 4. 2015
MUDr. Kristýna Krakorová
Neurologická klinika
LF UK a FN Plzeň
Alej svobody 80
304 60 Plzeň-Lochotín
e-mail: krakorovak@fnplzen.cz
Zdroje
1. Bird TD. Hereditary Ataxia Overview. Gene Reviews. [online]. Available from URL: http:/ / www.ncbi.nlm.nih.gov/ books/ NBK1138 .
2. Růžička E. Neurodegenerativní onemocnění mozku. In: Bednařík J, Ambler Z, Růzička E (eds). Klinická neurologie, část speciální I. 1. vyd. Praha: Triton 2010: 700– 707.
3. Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics and pathogenesis. Lancet Neurol 2004; 3(5): 291– 304.
4. Sequeiros J, Martins S, Silveira I. Epidemiology and population genetics of degenerative ataxias. Handb Clin Neurol 2012; 103: 227– 251. doi: 10.1016/ B978‑ 0‑ 444‑ 51892‑ 7.00014‑ 0.
5. Musova Z, Sedlacek Z, Mazanec R, Klempir J, Roth J, Plevova P et al. Spinocerebellar ataxias type 8, 12, and 17 and dentatorubro‑pallidoluysian atrophy in Czech ataxic patients. Cerebellum 2013; 12(2): 155– 161. doi: 10.1007/ s12311‑ 012‑ 0403‑ 5.
6. Zumrová A, Kopečková M, Mušová Z, Křepelová A, Apltová L, Paděrova K. Autosomálně dominantní spinocerebelarní ataxie. Neurol Prax 2007; 8(5): 277– 282.
7. Du X, Wang J, Zhu H, Rinaldo L, Lamar KM, Palmenberg AC et al. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 2013; 154(1): 118– 133. doi: 10.1016/ j.cell.2013.05.059.
8. Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW,Amos C et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A‑ voltage‑ dependent calcium channel. Nat Genet 1997; 15(1): 62– 69.
9. Teive HA, Munhoz RP, Raskin S, Werneck LC. Spinocerebellar ataxia type 6 in Brazil. Arq Neuropsiquiatr 2008; 66(3): 691– 694.
10. Soong BW, Paulson HL. Spinocerebellar ataxias: an update. Curr Opin Neurol 2007; 20(4): 438– 446.
11. Baloh RW, Yue Q, Furman JM, Nelson SF. Familiar episodic ataxia: clinical heterogenity in four families linked to chromozome 19p. Ann Neurol 1997; 41(1): 8– 16.
12. Ducros A, Denier C, Joutel A, Vahedi K, Michel A, Darcel F et al. Recurrence of the T666M calcium channel CACNA1A gene mutation in familial hemiplegic migraine with progressive cerebellar ataxia. Am J Hum Genet 1999; 64(1): 89– 98.
13. Guidelines for the molecular genetics predictive test in Hungtington’s disease. International Hungtington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Hungtington’s Chorea. Neurology 1994; 44(8): 1533– 1536.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2015 Číslo 4
Nejčtenější v tomto čísle
- Léčba pudendální neuralgie – klinické zkušenosti po pěti letech
- Význam magnetické rezonance v diagnostice epilepsie
- Experimentální léčba poranění míchy
- Léčba foraminálního výhřezu meziobratlové ploténky u istmické spondylolistézy technikou TLIF