
Dědičné ulceromutilující senzitivní neuropatie – klinická, elektrofyziologická a molekulárně genetická studie tří rodin
Hereditary Ulceromutilating Sensory Neuropathy – Clinical, Electrophysiological and Molecular Genetic Study of Three Families
Aim:
The goal was to clinically and electrophysiologically characterize two types of ulceromutilating hereditary neuropathy CMT2B and HSN1 in three Czech families where molecular genetic cause was confirmed.
Patients:
We describe three families, overall 16 affected patients, with hereditary sensory neuropathy.
Methods:
The diagnosis of sensory, predominantly axonal neuropathy was done on the basis of neurological and electrophysiological examination. Sequencing of the SPTLC1 and RAB7 genes was done in families A, B, C and 24 unrelated patients with clinical suspicion for HSN.
Results:
Hereditary sensory neuropathy type 1 (HSN1) caused by the p.C133Y mutation in the SPTLC1 gene was confirmed in family B and the Charcot-Marie-Tooth type 2B (CMT2B) caused by p.L129F and p.V162M mutations in the RAB7 gene was confirmed in families A and C. All three mutations have been previously described. DNA examination of 24 unrelated patients did not reveal the cause of their disease.
Conclusion:
As in other countries, ulceromutilating hereditary neuropathies CMT2B and HSN1 are rare in the Czech population. However, clinical manifestations are clearly recognizable if correctly obtained genealogical data – family history – is properly taken into account. The three families described here are the only known families with hereditary neuropathies caused by mutations in RAB7 and SPTLC1 in the Czech Republic. Clarification of the cause of ulceromutilating sensory neuropathy is crucial for genetic and clinical prognosis, including targeted genetic prevention, but possibly also for an L-serin therapy in SPTLC1 mutation patients to be tested in a clinical study (Boston, USA).
Key words:
hereditary sensory neuropathy – HSN1 – CMT2B – RAB7 gene – SPTLC1 gene
Autoři:
D. Šafka Brožková 1; R. Mazanec 2; J. Böhm 3; O. Vyšata 4; M. Auer-Grumbach 5; Ch. Windpassinger 6; J. Neupauerová 1; L. Baránková 1; S. Nevšímalová 3; P. Seeman 1
Působiště autorů:
DNA laboratoř Kliniky dětské neurologie 2. LF UK a FN v Motole, Praha
1; Neurologická klinika 2. LF UK a FN v Motole, Praha
2; Neurologická klinika 1. LF UK a VFN v Praze
3; Neurologická klinika LF UK a FN Hradec Králové
4; Department of Orthopaedics, Medical University Vienna
5; Institute of Human Genetics, Medical University of Graz
6
Vyšlo v časopise:
Cesk Slov Neurol N 2014; 77/110(4): 479-486
Kategorie:
Krátké sdělení
Grantová podpora: IGA MZ ČR NT 14348-3, MZ ČR - RVO, FN v Motole 00064203 a grant PRVOUK- P26/LF1/4.
Souhrn
Cíl:
Cílem práce je charakterizovat klinické a elektrofyziologické nálezy dvou typů ulceromutilující dědičné neuropatie CMT2B a HSN1 u tří českých rodin s molekulárně geneticky objasněnou příčinou.
Soubor:
Popisujeme tři rodiny s dědičnou senzitivní neuropatií s celkem 16 postiženými.
Metodika:
Na základě neurologického a elektromyografického vyšetření, výskytu podobných obtíží u příbuzných byla stanovena diagnóza senzitivní, převážně axonální neuropatie. Následně bylo provedeno sekvenování genů SPTLC1 a RAB7 v rodinách A, B, C a u dalších 24 nepříbuzných pacientů s klinickým podezřením na dědičnou senzitivní neuropatii.
Výsledky:
V rodině B byla nálezem mutace p.C133Y v SPTLC1 genu prokázána hereditární senzitivní neuropatie typ 1 (HSN1). V rodinách A a C byla nálezem mutací p.L129F a p.V162M v genu RAB7 prokázána choroba Charcot‑ Marie‑ Tooth typ 2B (CMT2B). Všechny tři mutace již byly dříve popsány a fenotyp odpovídá popisu pacientů z jiných zemí. DNA vyšetření dalších 24 nepříbuzných pacientů však příčinu onemocnění neobjasnilo.
Závěr:
Ulceromutilující dědičné neuropatie CMT2B a HSN1 jsou v české populaci, podobně jako v jiných zemích vzácné, ale klinické projevy jsou jasně poznatelné, pokud jsou zohledněny a správně získány genealogické údaje – rodinná anamnéza. Tyto tři popsané rodiny jsou dosud jediné známé v ČR s objasněnou příčinou dědičné senzitivní neuropatie v důsledku mutací v RAB7 a SPTLC1. Objasnění příčiny ulceromutilující senzitivní neuropatie má význam nejen pro upřesnění genetické a klinické prognózy a pro cílenou genetickou prevenci, ale u pacientů s mutacemi v SPTLC1 genu možná i pro cílenou terapii se substitucí L‑ serinem, která bude testována v klinické studii (Boston, USA)
Klíčová slova:
dědičná senzitivní neuropatie – HSN1 – CMT2B – gen RAB7 – gen SPTLC1
Úvod
Dědičné senzitivní neuropatie patří do rozsáhlé a různorodé skupiny dědičných neuropatií, které se řadí mezi vzácná onemocnění. Dědičné neuropatie jsou nejčastější dědičné nervosvalové onemocnění s výraznou genetickou heterogenitou. Z hlediska různého postižení senzitivních a motorických nervů je používána základní klasifikace dědičných neuropatií na nejčastější, motoricko‑senzitivní (HMSN), kde jsou obdobně postiženy motorické i senzitivní nervy, méně časté motorické (HMN) a vzácné senzitivní (HSN) s postižením převážně motorických nebo převážně a výrazně více senzitivních vláken nervů [1]. Jedno ze základních kritérií pro klasifikaci dědičných motoricko‑senzitivních neuropatií (HMSN) je rychlost vedení vzruchu periferním nervem. Při rychlostech vedení motorickými vlákny n. medianus na předloktí pod 38 m/ s se jedná o neuropatii demyelinizační neboli typ 1 a při vedení nad 38 m/ s o neuropatii axonální neboli typ 2 [2]. Dědičné senzitivní neuropatie se projevují výraznou hypestezií až anestezií pro dotyk, teplo a bolest při relativně zachovalém vibračním čití. Hereditární senzitivní neuropatie jsou také známy pod označeními ulceromutilující dědičně neuropatie, familiární trophoneurosis, familiární syringomyelie [1]. Většinou jsou u pacientů s hereditární převážně senzitivní neuropatií přítomny i motorické výpadky – slabost a atrofie distálních svalů zejména dolních končetin. Pacienti však v důsledku těžké poruchy čití pro bolest a teplo nejvíce trpí kožními defekty po poraněních na dolních končetinách, které mnohdy vedou až k osteomyelitidě a následným amputacím. V elektrofyziologickém nálezu obecně vykazují těžké postižení periferních nervů, dominuje výrazné snížení amplitud převážně senzitivních nervů se zachovalou (aspoň zpočátku) rychlostí vedení svědčící pro axonální typ neuropatie.
Zatím jsou popsány tři geny, jejichž mutace jsou odpovědné za nesyndromové senzitivní neuropatie. První gen spojovaný s HSN1 byl v roce 2001 popsán SPTLC1 [3,4]. Dalším genem spojovaným s axonální převážně senzitivní neuropatií CMT2B byl v roce 2003 publikován RAB7 [5]. Zatím poslední gen, který je spojován se senzitivní autozomálně dominantně dědičnou neuropatií, je gen SPTLC2, jenž byl v souvislosti s HSN publikován v roce 2010 [6]. Protein kódovaný genem SPTLC2 tvoří spolu s další podjednotkou kódovanou genem SPTLC1 funkční enzym podílející se na de novo syntéze sfingolipidů. Gen RAB7 patří do rodiny GTPáz a podílí se na vnitrobuněčném transportu [7,8].
Popisujeme tři české rodiny s mnohočetným výskytem dědičné senzitivní neuropatie s dominantním typem přenosu, a to v důsledku mutací v RAB7 genu ve dvou rodinách a v SPTLC1 genu v jedné velké rodině.
Dále ukazujeme, že česká rodina s mutací p.C133Y v SPTLC1 genu zřejmě není příbuzná s rodinou se stejnou mutací v Rakousku a totéž platí i pro rodiny s mutací p.L129F v RAB7.
Metody a pacienti
Genetická vyšetření
Všichni pacienti podepsali informovaný souhlas s genetickým vyšetřením příčiny dědičných neuropatií. U všech pacientů bylo nejprve provedeno vyšetření na nejčastější formu HMSN – duplikaci CMT1A/ deleci HNPP [9]. Dále byly vyšetřeny geny, jejichž porucha vede k dědičné senzitivní neuropatii. Sekvenováno bylo všech pět kódujících exonů včetně přilehlých intronových sekvencí genu RAB7 vždy u jednoho postiženého člena z rodin A, B i C a dalších 24 nepříbuzných pacientů. U rodiny B bylo sekvenováno všech 14 kódujících exonů včetně přilehlých intronových sekvencí genu SPTLC1. U rodin A, C a 24 nepříbuzných pacientů byl vyšetřen pouze exon 5 genu SPTLC1, kde se nachází mutace u většiny doposud popsaných pacientů a rodin. V případě nálezu mutace byli vyšetřeni i ostatní členové rodin pro průkaz segregace mutace s onemocněním. Pro geny RAB7 a SPTLC1 byly použity tyto referenční sekvence: NM_004637.5 a NM_006415.2.
Vyšetření příbuznosti českých a rakouských rodin se stejnými mutacemi proběhlo porovnáním haplotypů. Haplotypy byly sestaveny na základě vyšetření pomocí mikrosatelitových markerů, které se nacházejí v blízkosti genu RAB7 (čtyři markery) a genu SPTLC1 (16 markerů).
Klinická vyšetření
Neurologické vyšetření bylo, kromě standardního vyšetření, cíleno na vyšetření čití – povrchového – monofilamentum, algického – rozlišení tupých/ ostrých podnětů, termického – teplé/ studené podněty a hlubokého – vibrační čití hodnocené ladičkou.
Pacienti
Rodiny A i B jsou poměrně rozsáhlé rodiny, kde se onemocnění dědičnou neuropatií vyskytuje v několika generacích. To vedlo k podezření na autozomálně dominantně dědičnou neuropatii. V rodině A je postiženo 11 osob, pět mužů a šest žen. V rodině B je postiženo 15 osob, 13 mužů a dvě ženy (obr. 1). V rodině A bylo klinické neurologické a DNA vyšetření provedeno u dvou pacientů. V rodině B proběhlo klinické vyšetření u 10 a DNA vyšetření u 11 osob. V případě rodiny B byli jednotliví členové zpočátku vyšetřováni na různých pracovištích, bez vědomí o mnohočetném familiárním výskytu podobných obtíží a postižení. Díky vyšetření DNA na jediném místě v ČR došlo ke spojení informací přes rodokmen a jména, celá rodina tak mohla být hodnocena a vyšetřena jako jeden celek, a tím nakonec i objasněna.


Rodina C (obr. 1) byla zachycena při vyšetření skupiny 25 nepříbuzných pacientů s klinickým podezřením na HSN. Onemocnění se v této rodině vyskytuje u tří osob, u kterých bylo provedeno klinické i DNA vyšetření. U všech pacientů byla přítomna axonální, převážně senzitivní neuropatie s výskytem kožních, infikovaných, těžko se hojících defektů na nohách.
Výsledky
Molekulárně genetické
V rodině A a C byly prokázány dvě různé mutace v genu RAB7 a to ve 3. a 4. kódujícím exonu; v rodině A záměna aminokyseliny leucin za fenylalanin na pozici 129 (p.L129F) u dvou osob, v rodině Czáměna valinu za methionin na pozici 162 (p.V162M) u tří osob. V rodině B byla prokázána mutace v genu SPTLC1 na pozici kodonu 133, záměna aminokyseliny cystein na tyrosin (p.C133Y), která je příčinou onemocnění v rodině a prokázána byla u 11 osob. Nalezené mutace segregují s projevy senzitivní neuropatie u vyšetřených členů všech rodin.
Obě z nalezených mutací p.C133Y v genu SPTLC1 a p.L129F v genu RAB7 byly již dříve popsány i u dvou rodin z Rakouska, což mohlo svědčit o společném původu obou mutací a o příbuznosti rodin. Porovnání haplotypů z oblasti kolem obou genů ukázalo, že rakouské a české rodiny se stejnými mutacemi nejspíše nejsou navzájem příbuzné, haplotypy se výrazně lišily.
Dovyšetřením všech kódujících exonů RAB7 genu u 25 nepříbuzných pacientů, dříve vyšetřených sekvenováním jen jednoho exonu SPTLC1 a RAB7, byla kauzální mutace nalezena pouze u jedné rodiny a tou byla rodina C.
Celkem byla v těchto třech rodinách prokázána kauzální mutace pro dědičnou senzitivní neuropatii u 16 pacientů. Těchto 16 pacientů představuje v celkovém počtu pacientů s dědičnou neuropatií v ČR, u kterých se podařilo objasnit příčinu na DNA úrovni (aktuálně 1 606 osob v naší databázi) , přibližně 1 %. Jde tedy evidentně o vzácnou příčinu, resp. typ dědičné neuropatie.
Klinické nálezy
V klinickém nálezu u všech tří rodin bylo zjištěno postižení různých modalit citlivosti na končetinách. U postižených v rodině B byla distálně na dolních i na horních končetinách zjištěna výrazná hypestezie až anestezie pro taktilní čití, bolest a teplo, oproti tomu bylo poměrně dobře zachováno vibrační čití. V rodinách A a C byla zjištěna nejen hypestezie až anestezie pro povrchové čití, ale bylo postiženo i hluboké čití – vibrační. V elektromyografickém nálezu převažovala axonální neuropatie s výraznějším postiženým senzitivních vláken. U méně postižených jedinců jsme registrovali snížené amplitudy SNAP i CMAP, jakož i snížené rychlosti vedení senzitivními i motorickými vlákny jako obraz kombinované léze axonu i myelinu. V pokročilých případech už často SNAP i CMAP nebyly výbavné (tab. 1).

U části pacientů z rodiny B byla přítomna také výrazná atrofie distálních svalů s výraznou slabostí dolních i horních končetin (obr. 2).
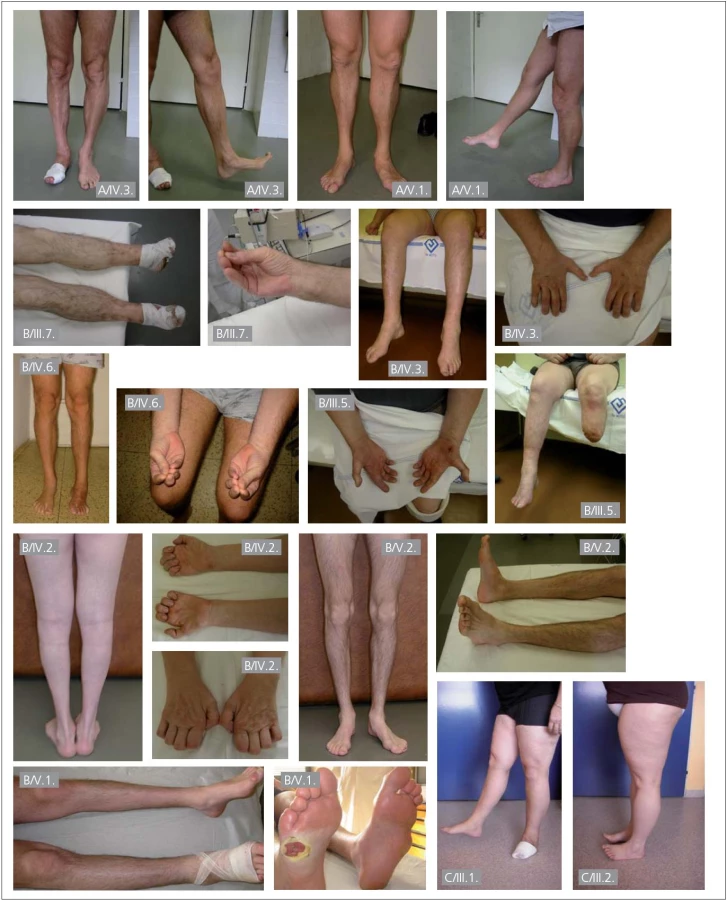
Amputace různého rozsahu na dolních končetinách byly v rodině B; pacient III.5 – amputace levé DK v polovině bérce, III.7 – amputace prstů pravé i levé DK, IV.4 – amputace pravé DK pod kolenem, V.1 – amputace 5. prstu a metatarsu vlevo, hluboký defekt pod hlavičkou 5 metatarsu vpravo (obr. 2).
I pacienti z rodin s mutacemi v genu RAB7 trpí infikovanými a nehojícími se defekty na nohou; v rodině C, pacient III.1 trpí již několik let nehojícím se kožním defektem na levém chodidle a v rodině A měl pacient IV.3 v době vyšetření ulceraci pod metatarzálními klouby na pravém chodidle. Celkem bylo ve třech rodinách klinicky vyšetřeno 11 postižených mužů a čtyři postižené ženy, u mužů se komplikace vyskytují v 54,5 % (šest mužů), ženy jsou všechny bez komplikací. Zdá se tedy, že muži mají těžší postižení periferních nervů než ženy, což při autozomálním typu dědičnosti této nemoci nelze logicky vysvětlit.
Pro lepší přehlednost jsou klinické nálezy vyšetřených pacientů ze všech tří rodin shrnuty v tab. 2.

U pacienta IV.6 z rodiny B byla v jeho 43 letech provedena biopsie surálního nervu. V polotenkých řezech byla patrna totální absence myelinizovaných nervů a mnoho tzv. prázdných kapsiček Schwannových buněk po degenerovaných axonech vyplněných kolagenem. Byla zjištěna i edematózní změna a dilatace jednotlivých axonů a extrémní intersticiální fibróza a nález odpovídal těžké axonální neuropatii.
Diskuze
Popsali jsme tři české rodiny s ulceromutilující dědičnou neuropatií v důsledku mutací v genech RAB7 a SPTLC1. Jde pravděpodobně o první publikované systematické klinické a elektrofyziologické popisy rodin s těmito genovými poruchami v České republice. Tři mutace nalezené u českých rodin s HSN segregují s onemocněním v rodinách a pro jejich kauzalitu svědčí i to, že byly již dříve opakovaně popsány jako patogenní v rodinách z různých zemí. Shodný nález mutací p.L129F (RAB7) a p.C133Y (SPTLC1) u českých a rakouských rodin nás vedl k domněnce, že obě mutace mohou pocházet z minulosti od společného předka, vzhledem ke geografické blízkosti České republiky a Rakouska, společné historii a také anamnéze, podle které jedna rakouská rodina pochází původně z území dnešní České republiky. Ovšem DNA vyšetření a sestavené haplotypy z oblastí genů RAB7, resp. SPTLC1 neukázaly příbuznost a obě mutace, ač stejné, nejspíše vznikly nezávisle.
Společným příznakem dědičné senzitivní neuropatie je výrazné omezení senzitivního vnímání pro teplo, bolest a dotyk. U HSN1, způsobené mutacemi v genu SPLTC1, je relativně zachováno hluboké čití, vyšetřitelné ladičkou. U typu CMT2B způsobeném mutacemi v genu RAB7 je popisováno i snížené vibrační čití. Tyto nálezy jsou v souladu s naším pozorováním, kde v rodině B (gen SPTLC1) bylo relativně zachováno vibrační čití oproti rodinám A a C (gen RAB7), kde bylo vibrační čití sníženo. U obou typů onemocnění mohou být přítomny atrofie svalů končetin různého rozsahu doprovázené svalovou slabostí. Pro HSN1 byly jako důležitý symptom popsány intenzivní neuropatické bolesti (bodavá pronikavá bolest), kterou jsme zjistili i u našich pacientů [10,11].
Výskyt amputací dolních končetin a kožních ulcerací na nohou byl pouze u mužů ve všech třech rodinách (tab. 2). Tento nález odpovídá i dřívějším publikacím, které uvádějí vyšší výskyt ulcerací u mužů s mutacemi v genu RAB7 a celkově těžší a dřívější postižení mužů v rodinách s mutacemi SPTLC1 [12,13]. Vysvětlením může být protektivní efekt druhého X chromozomu u žen, ale pravděpodobný je vliv i jiných, dosud nepoznaných faktorů.
Senzitivní neuropatie jsou relativně vzácné, z 1 606 pacientů, resp. 855 rodin s kauzálními mutacemi v našem registru jsou jen tři rodiny (16 pacientů) s mutacemi, kauzálními pro HSN. Ovšem o závažnosti tohoto onemocnění svědčí to, že jeden z postižených využil již dvakrát preimplantační diagnostiku, při které byly pro těhotenství vybrány pouze embrya bez mutace zodpovědné za onemocnění senzitivní neuropatií v rodině.
V případě senzitivní neuropatie způsobené mutací v genu SPTLC1 již byla zkoušena experimentální léčba L‑ serinem. V přítomnosti mutace dochází ke změně substrátové specifity, kde využívanou aminokyselinou již není serin, ale přednostně alanin a glycin, čímž se tvoří dva neurotoxické metabolity 1- deoxy‑ sfinganin a 1- deoxymetyl‑ sfinganin místo molekuly sfinganinu, která je důležitá pro tvorbu sfingolipidů. Tyto pozměněné metabolity se hromadí v neuronech a při pokusech s buněčnými kulturami senzitivních nervů je v přítomnosti neurotoxických metabolitů prokázána snížená tvorba neuritů [14,15]. Léčba L‑ serinem vychází z výzkumů, že při perorálním podání vyšších dávek L‑ serinu dochází ke snížené tvorbě neurotoxických metabolitů a ke zlepšení projevů neuropatie [16].
Často prvním projevem HSN je poranění nohy, kterého si pacient všimne náhodou, protože pro absenci vnímání bolesti vlastní poranění necítí. Tato poranění nelze podceňovat, natož přehlížet, protože se mohou rozvinout až v osteomyelitidu, která je jen obtížně léčitelná a často končí amputací. Samotné hojení však u pacientů s HSN1, resp. CMT2B postiženo nejspíše není. To jsme měli možnost pozorovat u jednoho člena rodiny B, jenž podstoupil ortopedickou korekční operaci obou nohou pro deformity a pooperačně žádné komplikace zaznamenány nebyly. Při podezření na dědičnou senzitivní neuropatii je zásadní pečlivá rodinná anamnéza a vyšetření přímých příbuzných pro určení familiárního výskytu a typu dědičnosti. Kromě anamnézy je klíčové, zejména v iniciálních stadiích nemoci u mladších jedinců, pečlivé vyšetření citlivosti končetin se zaměřením na poruchu vnímání tepla a bolesti, neuropatickou bolest či nehojící se kožní defekty hlavně na prstech a chodidlech dolních končetin.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Dana Šafka Brožková, Ph.D.
DNA laboratoř Kliniky dětské neurologie
2. LF UK a FN v Motole
V Úvalu 84
150 06 Praha 5
email: dana.brozkova@lf2.cuni.cz
Přijato k recenzi: 20. 2. 2014
Přijato do tisku: 31. 3. 2014
Zdroje
1. Dyck PJ. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF (eds). Peripheral Neuropathy. 3rd ed. Philadelphia: W.B. Saunders 1993: 1065– 1093.
2. Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980; 103(2): 259– 280.
3. Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer‑ Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit‑ 1, cause hereditary sensory neuropathy type I. Nat Genet 2001; 27(3): 309– 312.
4. Bejaoui K, Wu C, Scheffler MD, Haan G, Ashby P, Wu L et al. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet 2001; 27(3): 261– 262.
5. Verhoeven K, De Jonghe P, Coen K, Verpoorten N,Auer‑ Grumbach M, Kwon JM et al. Mutations in the small GTP‑ ase late endosomal protein RAB7 cause Charcot‑ Marie‑ Tooth type 2B neuropathy. Am J Hum Genet 2003; 72(3): 722– 727.
6. Rotthier A, Auer‑ Grumbach M, Janssens K, Baets J,Penno A, Almeida‑ Souza L et al. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I.Am J Hum Genet 2010; 87(4): 513– 522. doi: 10.1016/ j.ajhg.2010.09.010.
7. Nielsen E, Severin F, Backer JM, Hyman AA, Zerial M. Rab5 regulates motility of early endosomes on microtubules. Nat Cell Biol 1999; 1(6): 376– 382.
8. Echard A, Jollivet F, Martinez O, Lacapere JJ, Rousselet A, Janoueix‑ Lerosey I et al. Interaction of a Golgi‑associated kinesin‑like protein with Rab6. Science 1998; 279(5350): 580– 585.
9. Seeman P, Mazanec R, Zidar J, Hrusakova S, Ctvrteckova M, Rautenstrauss B. Charcot‑ Marie‑ Tooth disease type 1A (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP): reliable detection of the CMT1A duplication and HNPP deletion using 8 microsatellite markers in 2 multiplex PCRs. Int J Mol Med 2000; 6(4): 421– 426.
10. Auer‑ Grumbach M, Mauko B, Auer‑ Grumbach P, Pieber TR. Molecular genetics of hereditary sensory neuropathies. Neuromolecular Med 2006; 8(1– 2): 147– 158.
11. Auer‑ Grumbach M, De Jonghe P, Verhoeven K, Timmerman V, Wagner K, Hartung HP et al. Autosomal dominant inherited neuropathies with prominent sensory loss and mutilations: a review. Arch Neurol 2003; 60(3): 329– 334.
12. Houlden H, King R, Blake J, Groves M, Love S, Woodward C et al. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain 2006; 129(2): 411– 425.
13. Manganelli F, Pisciotta C, Provitera V, Taioli F, Iodice R,Topa A et al. Autonomic nervous system involvement in a new CMT2B family. J Peripher Nerv Syst 2012; 17(3): 361– 364. doi: 10.1111/ j.1529‑ 8027.2012.00415.x.
14. Gable K, Gupta SD, Han G, Niranjanakumari S, Harmon JM, Dunn TM. A disease‑ causing mutation in the active site of serine palmitoyltransferase causes catalytic promiscuity. J Biol Chem 2010; 285(30): 22846– 22852. doi: 10.1074/ jbc.M110.122259.
15. Penno A, Reilly MM, Houlden H, Laura M, Rentsch K, Niederkofler V et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 2010; 285(15): 11178– 11187. doi: 10.1074/ jbc.M109.092973.
16. Garofalo K, Penno A, Schmidt BP, Lee HJ, Frosch MP,von Eckardstein A et al. Oral L‑ serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J Clin Invest 2011; 121(12): 4735– 4745. doi: 10.1172/ JCI57549.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2014 Číslo 4
Nejčtenější v tomto čísle
- Vyšetření senzitivity
- Genetická variabilita u poruchy pozornosti s hyperaktivitou (ADHD)
- Spinální arteriovenózní malformace – dvě kazuistiky
- Přínos opakování nepotvrzujícího testu mnohočetné latence usnutí (MSLT) pro stanovení diagnózy narkolepsie