
Neuropsychiatrický pohľad na Huntingtonovu chorobu
Neuropsychiatric View of Huntington‘s Disease
Huntington’s disease is a progressive and fatal neurodegenerative disorder with an incidence of about 5 cases per 100,000 people. There is familial occurrence, with autosomal dominant inheritance. The more family members affected, the greater the burden these families have. A typical phenotype is choreatic hyperkinesia, though rigidity, hypokinesia and gait disturbance appear years later. Cognitive and psychiatric symptoms are typical, often preceding clinical expression of movement disorders. From the clearly defined genotype, a trio of pathological symptoms grows, with considerable phenomenological polymorphic variations from individual to individual. Psychiatric disorders often impair the quality of life more than motor abnormalities and can lead to suicide, the second leading cause of death in individuals with Huntington’s disease. Therefore, the neuropsychiatric view of this disease is not just a quantitative synthesis of findings from neurology and psychiatry, but rather an integral and comprehensive approach is also taken, allowing both a better understanding of the pathogenesis and a mostly superior therapeutic approach that takes into account several aspects of the multifaceted suffering endured by these patients. The article presents an overview of the theory from a neuropsychiatric perspective on Huntington’s disease, as well as two short case studies of patients – siblings who have the disease.
Key words:
Huntington’s disease – chorea – depression –suicide – dementia – executive dysfunction
Autoři:
J. Necpál 1; M. Patarák 2
Působiště autorů:
Neurologické oddelenie, Nemocnica Zvolen a. s.
1; Psychiatrické oddelenie, FNsP F. D. Roosevelta Banská Bystrica
2
Vyšlo v časopise:
Cesk Slov Neurol N 2013; 76/109(4): 438-445
Kategorie:
Přehledný referát
Souhrn
Huntingtonova choroba je progresívne fatálne neurodegeneratívne ochorenie s výskytom okolo 5 prípadov na 100 tisíc obyvateľov. Vyskytuje sa familiárne, s autozomálne dominantou formou dedičnosti. Bremeno v týchto rodinách je o to ťažšie, o koľko viac členov je postihnutých. Typickým fenotypom sú choreatické hyperkinézy, ku ktorým sa po rokoch pridávajú rigidita, hypokinéza a poruchy chôdze. Typické sú kognitívne a psychiatrické symptómy, často predchádzajúce klinické vyjadrenie pohybových porúch. Z jasne definovaného genotypu tak vyrastá patologický trojlístok symptómov, so značnou fenomenologickou polymorfnosťou kolísajúcou od jedinca k jedincovi. Psychiatrické poruchy často zhoršujú kvalitu života viac ako motorické a môžu viesť až k samovražde, ktorá je druhou najčastejšou príčinou smrti u jedincov s Huntingtonovou chorobou. Neuropsychiatrický pohľad na chorobu teda nie je kvantitatívnou syntézou poznatkov z oboch oborov, ale integrálnym a komplexným prístupom, umožňujúcim nielen lepšie pochopenie patogenézy, ale hlavne kvalitnejší terapeutický prístup zohľadňujúci viaceré aspekty mnohorakého utrpenia týchto pacientov. V článku prinášame prehľadnú teóriu neuropsychiatrického pohľadu, ako aj krátke kazuistiky dvoch pacientov ‑ súrodencov s Huntingtonovou chorobou.
Kľúčové slová:
Huntingtonova choroba – chorea – depresia – suicídium – demencia – exekutívna dysfunkcia
Úvod
V roku 1872 čerstvo promovaný 22 - ročný lekár George Huntington napísal do Medical and Surgical Reporter článok „On Chorea“. Jeho hlavnou časťou boli príspevky o reumatickej chorei, no poslednú časť venoval hereditárnej chorei s progresívnym priebehom, zmenami psychického stavu, s počiatkom v strednom veku a dedenej z generácie na generáciu [1]. Prvý opis pacienta s Huntingtonovou choreou však pochádza už z roku 1842 od Watersa [2]. Chorea (choreia, tanec, gr.) či v anglosaskej literatúre označenie „dancing mania“ je charakterizovaná mimovoľnými, nepravidelnými, neúčelnými, náhlymi a rýchlymi pretrvávajúcimi pohybmi, nepredvídateľnými v čase, smere a distribúcii. Niekedy ich pacienti dokážu „zamaskovať“ do poloúčelových pohybov, tzv. parakinézií. Choreu často sprevádza neschopnosť udržať trvalú kontrakciu na konštantnej úrovni (tzv. motorická imperzistencia alebo negatívna chorea), čoho prejavom je napríklad neschopnosť trvalého stisku rúk (tzv. stisk dojičky, milkmaid’s grip) či udržať jazyk v protrúzii [3]. Keďže chorea nie je jedinou manifestáciou Huntingtonovej chorey, ochorenie sa premenovalo na Huntingtonovu chorobu (HCH) [4]. Ide o zriedkavé, progresívne a fatálne dedičné ochorenie s počiatkom v dospelosti, typicky medzi 35. až 50. rokom života, no môže sa vyskytovať od skorého detstva až do 80 rokov. Smrť zvyčajne nastáva 15 až 20 rokov od začiatku ochorenia. Jeho prevalencia v USA a väčšine európskych krajín je približne 5 : 100 tisíc obyvateľov, kým v Japonsku, Číne, Fínsku a Afrike je oveľa nižšia [5]. Ochorenie je spôsobené expanziou CAG tripletov v géne IT15 (Interesting Transcript 15) na 4. chromozóme, kódujúcom proteín huntingtín [6,7]. To vyúsťuje do rozsiahlej degenerácie neurónov preferenčne v striate, no s jeho progresiou postihuje aj ďalšie oblasti mozgu [8]. Klinicky je charakteristické triádou motorických, kognitívnych a psychiatrických symptómov [4].
Etiopatogenéza
HCH je monogénové autozomálne dominantné ochorenie spôsobené expanziou trinukleotidu CAG v géne na 4. chromozóme kódujúcom proteín huntingtín. Potomkovia postihnutého rodiča majú 50-percentné riziko vzniku ochorenia. Čím väčší je počet opakovaní CAG (repetícií), tým skôr sa ochorenie prejaví. Definitívne sa manifestuje pri ich počte 40 a viac, v rozmedzí 36 – 39 vedie k inkompletnej penetrancii a pri intermediárnom počte 27 – 35 ochorenie nevzniká, avšak v budúcnosti môže dôjsť k jeho expanzii [2,4]. Zvýšený počet repetícií vznikajúci počas spermiogenézy bol zistený v prípade paternálneho prenosu. Niekedy môže dôjsť k špecifickému fenoménu zvanému anticipácia, kedy v každej ďalšej nasledujúcej generácii vzniká ochorenie skôr a je závažnejšie. [9].
Najvýraznejšie neuropatologické zmeny vznikajú v striate, ktoré obsahuje ostnaté projekčné neuróny dvoch druhov: tie, ktoré projikujú do globus pallidus externus a formujú tzv. nepriamu dráhu, a tie, ktoré projikujú do globus pallidus internus a vytvárajú tzv. priamu dráhu. Neuróny nepriamej dráhy potláčajú kortikálnu selekciu nežiaducich pohybov a chorea vzniká pri dysfunkcii nepriamej dráhy, čo má za následok hyperkinézu (obr. 1, 2) [10].
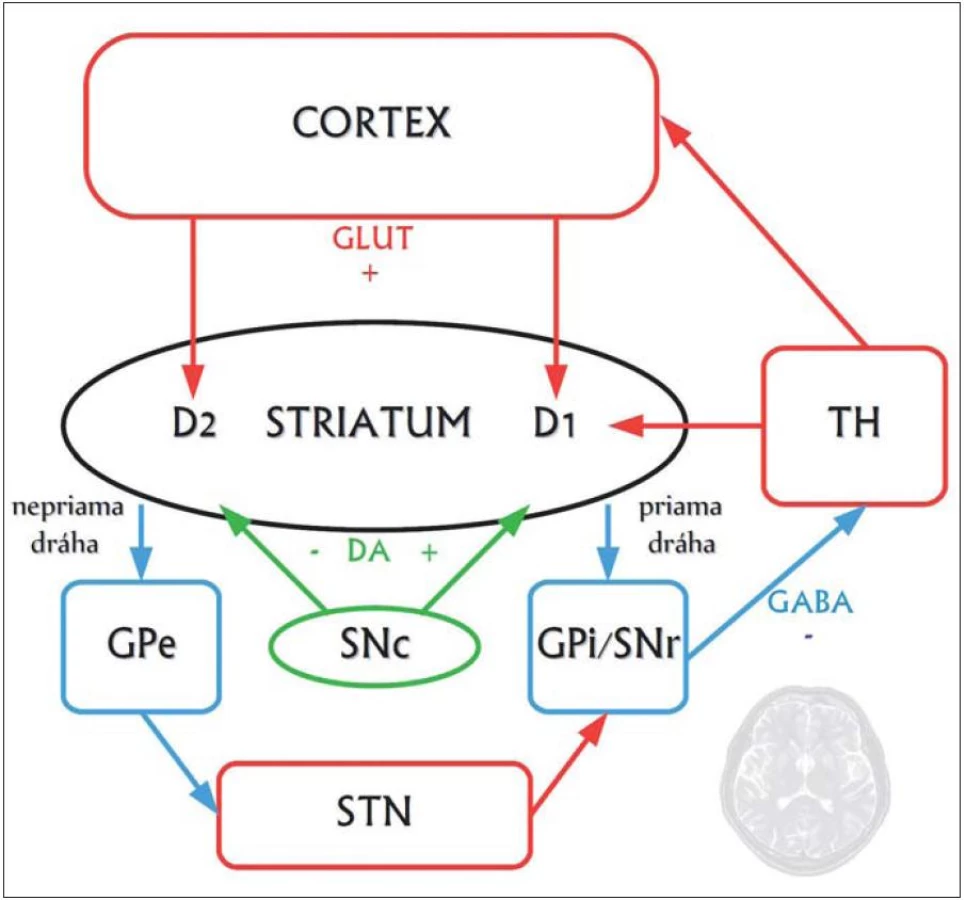

Vzťah huntingtínu k progresívnej neurodegenerácii nie je celkom jasný. Zistilo sa, že v prítomnosti mutovaného huntingtínu, ktorý sa za normálnych okolností špecificky viaže na tzv. huntingtín ‑ asociovaný proteín (HAP1) zabezpečujúci axonálny transport, stráca tento svoju funkciu, čo vedie k degenerácii nervových zakončení. Dlhý polyglutamínový reťazec mutantného huntingtínu oslabuje interakciu s HAP1, ktorý aktivuje proces apoptózy prostredníctvom kaspáz, ktoré štiepia huntingtín, a vytvárajú tak fragmenty formujúce intraneuronálne inklúzie. Okrem toho sa mutovaný huntingtín priamo viaže na domény tzv. CBP proteínu, čím sa redukuje acetyltransferázová aktivita a znižuje acetylácia histónov, čo v konečnom dôsledku vedie k zníženiu génovej transkripcie špecifických génov [3]. K vzniku ochorenia prispieva tiež alterácia transportu proteínov, homeostázy kalcia či mitochondriálneho respiračného reťazca [10].
Klinické charakteristiky
Motorické príznaky
Za počiatok vzniku HCH sa považuje objavenie sa motorických príznakov, jednak extrapyramidálnych, jednak príznakov z poškodenia vôľových pohybov. Hlavným motorickým príznakom je chorea [6]. Involuntárne pohyby, počínajúce v distálnych častiach končatín a malých mimických svaloch, pripomínajú spočiatku nervozitu. Chôdza pacientov sa stáva nestabilnou, často sú považovaní za opitých. Pohyby sa postupne šíria proximálne a axiálne – najnápadnejšie sú extenčné pohyby chrbtového svalstva [2]. Chorea môže postihovať aj respiračné, laryngeálne, orálne a nazálne svalstvo [9]. Má tendenciu zvýrazňovať sa počas prvých približne 10 rokov priebehu HCH, no s jej progresiou ustupuje a je nahradzovaná hypokinézou, rigiditou a dystóniou [6]. Medzi poruchy voluntárnych pohybov patria abnormálne očné pohyby, dyskoordinovaná a spomalená jemná motorika, dysfágia, dyzartria, dysdiadochokinéza, rigidita a poruchy chôdze [9]. Okulomotorické abnormality, patriace medzi prvé príznaky, sa prezentujú hypometriou a spomalením najmä vertikálnych sakád, alteráciou sledovacích pohybov a fixácie pohľadu. V pokročilých štádiách je reč ťažko dyzartrická, vyskytuje sa dysfágia, zvýšený svalový tonus môže viesť ku kontraktúram kĺbov [6,9]. Pacienti nakoniec umierajú v dôsledku komplikácie pádov, inanície, dysfágie alebo aspirácie [11]. Najčastejšou príčinou smrti je pneumónia, nasledovaná suicídiom [2].
Kognitívne poruchy
Napriek klinickej triáde HCH má v praxi primát motorická symptomatológia. Kognitívne a psychiatrické príznaky sú implicitne chápané ako prídavné, hoci motorické prejavy môžu predchádzať aj o viac ako desaťročie [12]. Z časti ide o pochopiteľný prístup k ochoreniu, ktoré má síce jasne definovanú genetickú príčinu, ale ktoré má napriek tomu značne polymorfnú symptomatickú variáciu [12] a ktoré teda nevieme esenciálne uchopiť. Línia vedúca k vystopovaniu patogenézy motorických abnormít je akoby jasnejšia než nitky neuropsychologicko‑neuropsychiatrického poškodenia. To sa zvykne interpretovať ako sekundárny následok štrukturálnych zmien typu atrofie kôry, caudata, prípadne ako dôsledok iných degeneratívnych zmien CNS. Klinická skutočnosť však potvrdzuje kognitívne defekty už v presymptomatickom období HCH [16]. Fakt, že štádium ochorenia pred rozvojom hyperkinéz a po (už) rozvinutých kognitívno ‑ psychiatrických príznakoch sa nazýva presymptomatické, svedčí v prospech primátu motorického/ neurologického chápania, hoci formálne i kognitívne prejavy patria do takzvanej symptomatickej triády [13].
Exekutívna dysfunkcia sa prejavuje v poruche riešenia problémov, koncentrácie, plánovania a pracovnej pamäti [14,15]. Ide pritom o označenie terminologicky správnejšie ako pseudoanatomický termín frontálneho syndrómu, používaný v minulosti [14].
Demencia pri HCH je v začiatkoch charakteristická najmä postihnutím kognitívnej flexibility, schopnosti presúvať a distribuovať pozornosť, psychomotorického tempa, abstrakcie, implicitnej pamäti a učenia sa novým schopnostiam. Na rozdiel od Alzheimerovej demencie jedinci nie sú amnestickí v „klasickom“ zmysle slova a sémantická pamäť, ako aj jazyk sú relatívne zachované [16]. Porucha exekutívnych funkcií koreluje s atrofiou caudata a denzitou D1 a D2 receptorov striata, rezultuje však z narušenia kortiko ‑ subkortikálnych okruhov a nielen z extrastriatálnej patológie, ako sa usudzovalo kedysi [16 – 18]. Podobne ako pri Alzheimerovej demencii, aj tu kognitívnemu poškodeniu charakteru demencie predchádza mierna kognitívna porucha [19]. Znížená schopnosť rozoznávať emócie komunikačného partnera [20,21] patrí k zvláštnym kognitívnym narušeniam, komplikujúcim už aj tak kriticky zaťažené vzťahy pacientov s HCH. Závažnou, no v istom zmysle protektívne pôsobiacou zmenou je progredujúca strata náhľadu na chorobné postihnutie [15]. Niekedy prítomná elevácia nálady je akoby psychologicky zrozumiteľnou obranou voči ťaživému utrpeniu, hoci skôr súvisí s organickými faktormi. Dyskinézy tváre môžu viesť k mimickým grimasám, ktoré majú charakter úsmevu či úškrnu a vzbudzujú pocit dobrej nálady pacienta, ktorá však vôbec nie je prezentná. Praktickým problémom teda nie je len to, že pacient ťažko rozpoznáva a identifikuje emócie druhých ľudí, ale aj to, že v bežnom kontakte ľudia ťažko rozpoznávajú jeho pocity.
Psychiatrické príznaky
Narušené spracovanie emócií naznačené v predchádzajúcom texte, neprimeraná funkcia osi amygdala ‑ mediálna orbitofrontálna kôra (mOFC), ako i porucha serotonínergných modulačných funkcií vedie u pacientov s HCH k často popisovanej zvýšenej iritabilite [22,23]. Tá v kombinácii s nedostatočnou empatickou schopnosťou môže viesť k impulzívnym až agresívnym výbuchom. Apatia sa často vyskytuje paradoxne simultánne s dezinhibíciou (v rámci prefrontality) a nie je vždy sekundárna depresívnej poruche [14,24]. Môžu sa vyskytovať epizódy mánie, schizoformná psychóza, abúzus až závislosť na alkohole, popisuje sa s organickými zmenami spojená porucha osobnosti, ktorá sa obviňuje za rozpad manželstiev, vzťahov a rodinných väzieb [16]. Zvýšený je aj výskyt obsedantno ‑ kompulzívnej symptomatiky [25], ktorá je pravdepodobne viazaná na alteráciu kortiko ‑ striato ‑ thalamo ‑ kortikálnych okruhov [26]. Z psychiatrického hľadiska je veľmi citlivým fenoménom individuálna reakcia pacienta na diagnózu, kde je perspektíva progredujúceho zhoršenia. Reakcie a príslušné obranné mechanizmy osobnosti tu majú pred sebou skutočne náročnejší objekt, v porovnaní s reakciami na diagnózu s trvalým a relatívne stabilným postihnutím [27,28]. Pacientovi a jeho rodine by sa malo dostávať psychosociálnej podpory, hrejivého kontaktu, avšak bez falošných povzbudení a klamných nádejí, čo je i pre odborníka náročnou (ale pacientmi neuveriteľne cenenou!) úlohou. Rovnako uchvacujúce sú kultúrne vplyvy na vyrovnanie sa s HCH. V Latinskej Amerike sa napríklad často chápe ako posadnutosť zlými duchmi, ktorí „nenechajú človeka ticho sedieť“, a trpiaci vyhľadávajú namiesto lekárov ľudových liečiteľov (curanderos) [29].
Spomedzi psychiatrických porúch má najčastejšiu prevalenciu depresia (presné numerické rozmedzia sú ale mätúce, pretože sa v štúdiách väčšinou sleduje depresívna symptomatológia a nie depresia ako syndróm – ide však o problém týkajúci sa všetkých zmienených psychiatrických porúch). Depresia zvyšuje už aj tak vysoké riziko samovraždy [30] a mnohí autori vrátane Huntingtona [31] ju považujú za intrinsický symptóm HCH, nie obyčajný epifenomén. Depresia je v populácii HCH pacientov dvojnásobne častejšia ako vo všeobecnej populácii [32] a údajne viac ako polovica pacientov priznáva suicidálne ideácie [30]. Na tisíc depresívnych pacientov ich pripadá približne 3,3 s abnormálnou CAG expanziou na géne pre huntingtín, pôvodne diagnostikovaných ako depresívna porucha [33]. V pozadí je znížená expresia sérotonínových receptorov v kôre a hipokampe a znížená expresia sérotonínového transportéra v kôre [31], alterácia dorzálnych rafeálnych sérotonínergných jadier [34], narušenie regulácie hypothalamo ‑ pituitárnej‑adrenálnej osi [35], znížená adultná neurogenéza [36 – 38] a narušenie prepojenia caudata s frontálnym lalokom rezultujúci v známu „hypofrontalitu“ [16]. Depresívnu symptomatiku ťažko vyšetriť najmä v pokročilých fázach ochorenia (pre kognitívnu deterioráciu či dyzartriu). Mnohé znaky neuroendokrinnej dysregulácie pri HCH (porucha spánku, strata hmotnosti) imitujú depresiu, a tak môžu viesť nielen k falošne pozitívnemu výsledku, ale ju aj maskovať [13]. Apatia ako symptóm môže súvisieť s depresiou, ale i s organickými zmenami profrontálnej kôry, respektíve príslušných kortiko ‑ striato ‑ thalamo ‑ kortikálnych okruhov, ktoré cez ňu prechádzajú.
Príznaky z dysfunkcie hypothalamu
HCH sa prejavuje i menej známymi metabolickými symptómami, medzi ktoré patrí chudnutie, endokrinná dysfunkcia a poruchy spánku [5]. Okrem klasických neuropatologických zmien najmä v oblasti striata a mozgovej kôry sa dokázalo aj odumieranie buniek a atrofia v oblasti hypothalamu, ktoré nachádzame už v skorých fázach ochorenia [39]. V laterálnej časti hypothalamu pacientov s HCH sa našla 27% atrofia buniek exprimujúcich neuropeptid orexín [40]. Neuróny tejto oblasti ako aj magnocelulárne mamilárne jadrá zadného hypothalamu sú zodpovedné takisto za reguláciu bdelosti. U pacientov sa zistila fragmentácia spánku, redukcia pomalej fázy spánku a narušenie cirkadiánnej rytmicity. Napriek adekvátnej výžive pacienti chudnú, hoci viaceré štúdie poukazujú na ich zvýšenú chuť do jedla a vyšší kalorický príjem oproti zdravým kontrolám. Príčinou napriek tomu sa vyskytujúceho zvýšeného energetického výdaju môže byť zvýšená hladina grelínu, nízka hladina leptínu u pacientov ako i samotná chorea [39]. Okrem toho sa zistilo, že u pacientov s HCH sa vyskytuje diabetes mellitus približne 7 - krát častejšie oproti zdravej populácii [41].
U normoglykemických huntingtonikov sa dokázala porušená inzulínová senzitivita ako i sekrécia inzulínu [42].
Diagnostika
Klinické kritériá pozostávajúce z motorických príznakov s alebo bez prítomnosti psychických či kognitívnych zmien v kombinácii s pozitívnou rodinnou anamnézou podporujú diagnózu HCH [3]. Až 8 – 10 % prípadov ochorenia sa prejaví pred 20. rokom života, kedy hovoríme o juvenilnej forme HCH. Prezentuje sa atypicky: často bez chorey, s dystóniou, stuhnutosťou (najmä dolných končatín), bradykinéziou, epileptickými záchvatmi, poruchou kognitívnych funkcií a zmenami správania (agresivita, sexuálna promiskuita, drogová závislosť a psychické zmeny) [43,44].
Diagnostika HCH sa veľmi zjednodušila pomocou priameho genetického testu, najčastejšie polymerázovou reťazovou reakciou merajúcou dĺžku repetícií na každej alele [10] (tzv. konfirmačné testovanie). Asymptomatické osoby s pozitívnou rodinnou anamnézou môžu podstúpiť prediktívne (presymptomatické) testovanie, ktoré určí, či sú alebo nie sú nosičmi expandovaného génu, a teda či sa v budúcnosti u nich môže vyvinúť HCH [5]. Pri prediktívnom testovaní existujú doporučené postupy, ktoré boli navrhnuté asociáciou IHA (International Huntington Association) a výskumnou skupinou Huntingtonovej choroby WFN (World Federation of Neurology). Test môže byť prevedený iba osobe dospelého veku (v našich podmienkach staršej ako 18 rokov) po jej slobodnom rozhodnutí potvrdenom jej písomným súhlasom. Predchádza mu genetické poradenstvo a psychosociálna podpora [45,46]. Podrobnejšie informácie o pravidlách prediktívneho testovania možno nájsť aj v našej literatúre [47]. V rokoch 1994 – 2005 bolo na území takmer celej Českej republiky prevedených 629 testov, z toho z 567 diagnostických bola diagnóza HCH potvrdená u 72,5 % pacientov [48]. Rozhodnutie podstúpiť takýto test však býva nesmierne ťažké a generuje sériu psychologických dilem. Na jednej strane pomôže zariadiť si život, prácu a rodinu vzhľadom na očakávané postihnutie v budúcnosti, na strane druhej je mnoho ľudí, ktorí dajú prednosť životu v neistote, a teda aj v neistej nádeji, v ktorú dúfajú. Práve po obdŕžaní pozitívnych testov je vysoké riziko samovraždy, takže aj rozhodnutie jedinca pre testovanie má byť sprevádzané a podrobne diskutované s odborníkom.
Najčastejšou zobrazovacou technikou je MR vyšetrenie mozgu. Zachycuje atrofiu nucleus caudatus a putamen, ktoré spolu tvoria striatum. Atrofia striata vedie k hydrocefalu „ex vacuo“ a dilatácii laterálnych mozgových komôr. Možno pozorovať častú celkovú atrofiu mozgu [10].
Patologicko‑anatomicky makroskopicky v pokročilých štádiách ochorenia je hmotnosť mozgu redukovaná o 25 až 30 %. V rámci kortikálnej atrofie existuje laminárna špecificita s najväčším ubúdaním neurónov v VI., III. a V. vrstve. Stratu neurónov sprevádza reaktívna astrocytóza. Degenerácia je pozorovaná aj v iných častiach mozgu, ako je mozgový kmeň, cerebellum, hypothalamus, amygdala alebo thalamus [10]. Vonsatell roku 1985 zistil, že atrofia striata vykazuje v čase dorzoventrálny gradient a vytvoril päťstupňový systém, ktorý sa stal zlatým štandardom neuropatologickej klasifikácie zmien pri HCH. V niektorých prípadoch klinicky zjavného ochorenia patologické abnormality neobjavil (stupeň 0), avšak podľa nových MR štúdií bola u takýchto pacientov dokázaná signifikantná atrofia striata, čo z nej robí skorý marker neurodegeneratívneho procesu [49].
Na monitoring, resp. hodnotenie progresie ochorenia pracovníci z Huntington Study Group vyvinuli užitočný nástroj zvaný UHDRS (Unified Huntington Disease Rating Scale). Pozostáva zo štyroch sekcií vyšetrujúcich motoriku, kogníciu, správanie a funkčné schopnosti pacienta [50].
Liečba
V súčasnosti neexistuje žiadna liečba, ktorá by zastavila alebo spomalila progresiu HCH [4]. Rada symptómov sa dá aspoň prechodne terapeuticky tlmiť. Priebeh ochorenia však vedie nezadržateľne k marazmu, kedy postihnutý jedinec nie je schopný základných životných aktivít [47].
V rámci symptomatickej liečby hyperkinéz sa najčastejšie požívajú antidopaminergné látky, a to tetrabenazín a antipsychotiká (niekde ešte stále nazývané ako neuroleptiká) [3]. Tetrabenazín (predávaný v Kanade a niektorých európskych krajinách pod názvom Xenazine 25 a Nitoman) je reverzibilný dopamínový deplétor, ktorý inhibuje jeho transport do presynaptických vezikúl prostredníctvom vysokej selektivity pre vezikulárny monoamínový transportér typu 2 (VMAT 2) [4,51]. Podľa FDA (U. S. Food and Drug Administration) je jedinou odporúčanou látkou na symptomatickú liečbu HCH. Vo všeobecnosti je pacientmi dobre tolerovaný a nespôsobuje tardívne dyskinézy [52]. Ukázal sa byť účinný, o čom svedčí približne 42 % redukcia skóre chorey v UHDRS s priemerným trvaním účinku 5,4 hod [53]. Môže však exacerbovať alebo spúšťať psychiatrické symptómy, preto by nemal byť používaný u pacientov s anamnézou depresie alebo iného psychiatrického ochorenia [5]. Okrem depresie sa v rámci nežiaducich účinkov môže vyskytovať ospalosť, únava, úzkosť, parkinsonizmus, akatízia či gastrointestinálne problémy [53]. V našich podmienkach tetrabenazín nie je dostupný, preto je prvou voľbu liečba antipsychotikami. Konvenčné antipsychotiká (napr. haloperidol) majú efekt, no kvôli ich nežiaducim účinkom, akými sú tardívna dyskinéza, akútna dyskinéza, parkinsonizmus alebo neuroleptický malígny syndróm, sa dáva prednosť viac atypickým antipsychotikám (napr. risperidon, olanzapín, quetiapín). Majú lepšiu tolerabilitu a navyše menej nežiaducich účinkov v porovnaní s konvenčnými. Pri olanzapíne sa dokázalo signifikantné zlepšenie behaviorálnych príznakov, redukcia chorey a tiež zmiernenie dysfágie [54]. V otázke ovplyvnenia voluntárnych motorických funkcií sa pozornosť sústreďuje na pridopidín (Huntexil), látku patriacu medzi dopamínové stabilizéry. Neskoršie údaje však hovoria len nesignifikantnom zlepšení v tejto oblasti [5,52,55].
Existuje mnoho štúdií s cieľom pozitívneho ovplyvnenia motoriky pri HCH, no bohužiaľ s limitovanými výsledkami (amantadín, kanabidiol, baklofen, lamotrigín, levatiracetam, memantín, minocyklín a i.) [3,52]. V oblasti ochorenie ‑ modifikujúcich stratégií sa popri riluzole, remacemide a kreatíne hovorí najviac o koenzýme Q, ktorého funkcia spočíva hlavne v zapojení do procesu oxidatívnej fosforylácie a takisto v jeho antioxidačných účinkoch. Po roku užívania dennej dávky 600 mg sa zistil trend spomalenia funkčného, ako i kognitívneho postihnutia pacientov s HCH [52,56].
Z hľadiska ovplyvnenia kognitívneho postihnutia sa skúšajú viaceré látky, najmä donepezil a rivastigmín, ktorých účinnosť však nebola signifikantne dokázaná. Behaviorálne prejavy HCH vrátane depresie, apatie, iritability, agresie či porúch sexuality sa môžu ovplyvniť preparátmi zo skupiny SSRI, SNRI, benzodiazepínov alebo antipsychotík [52].
Stereotaktická chirurgia v oblasti vnútorného segmentu globus pallidus je používaná len zriedkavo [57]. V poslednom období sa rýchlo vyvíjajú priamejšie metódy terapeutického ovplyvnenia HCH. V rámci stratégie génovej terapie sa skúšajú metódy ako RNA interferencia alebo antisense olingonukleotidy, ktorých efekt by mal spočívať v utlmovaní tvorby mutantného huntingtínu [52]. Ďalšou možnosťou je neurotransplantácia buniek neurónov v oblasti striata (ostnaté bunky, medial spiny neurons). Traja z piatich pacientov, ktorí podstúpili transplantáciu, vykazovali do dvoch rokov po nej stabilizáciu alebo dokonca zlepšenie v motorickej ako aj kognitívnej sfére, avšak tento efekt nebol trvalý – odoznel 4 až 6 rokov po transplantácii [58]. Symptomatická liečba HCH má svoje mantinely v jej dočasnosti, ako aj účinnosti, preto potreba kauzálnej terapie z hľadiska perspektívy pacienta s týmto ochorením je najdôležitejším cieľom výskumu. Prechod z poznatkov výskumu animálnych modelov do oblasti liečby konkrétneho pacienta je však v tejto dobe stále najťažším „orieškom, ktorý je nutné prelúskať“, a tak naďalej zostáva iba hudbou budúcnosti, dúfajme, že blízkej...
Klinické prípady
V rámci kazuistiky stručne prezentujeme prípad súrodencov s HCH s pozitívnou rodinnou anamnézou, ktorých matka umrela na toto ochorenie a stará matka ukončila život suicídiom pri jeho suspektnom fenotype. Prinášame profily pacientov ako výsledok observácie tejto rodiny so zameraním na primárne neuropsychiatrický pohľad na HCH, vyšetrovacie metódy a niektoré novšie klinické aspekty tejto choroby súvisiace s teoretickou časťou.
Kazuistika 1
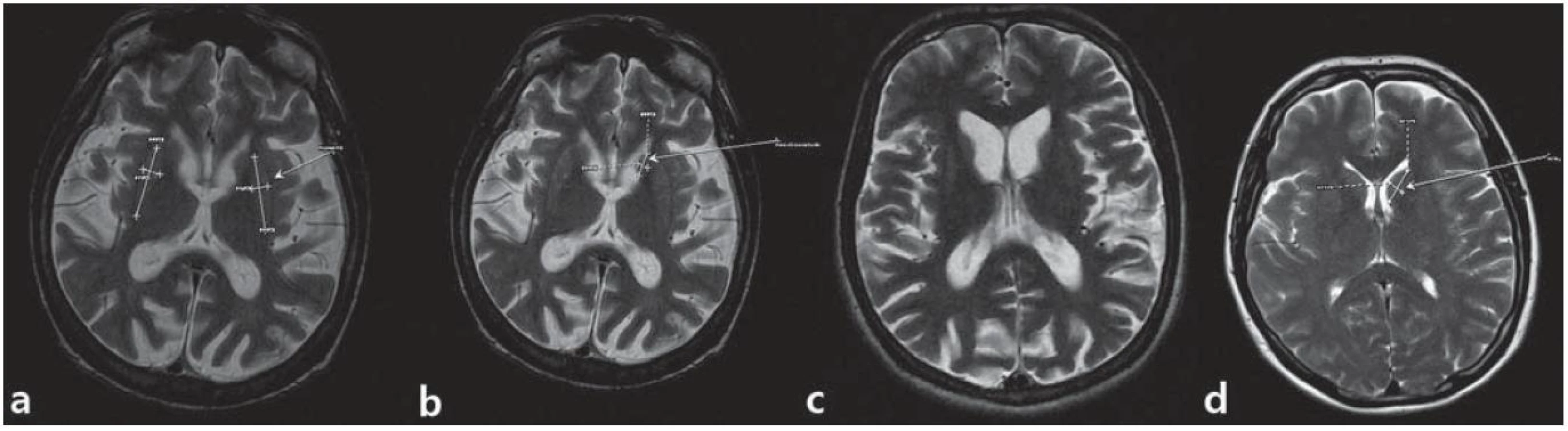
Štyridsať štvorročný muž s anamnézou diskrétnych povahových zmien od mladosti s prejavmi celkovej „zvláštnosti“, hostility, emulácie, ktoré boli príčinou jeho rozvodu; v minulosti mal podmienečný trest pre neúmyselné zabitie. Postupne sa vyvíja u neho akási neposednosť (podľa manželky „tiky“) progredujúceho charakteru do formy mimovoľných choreatických dyskinézií, poruchy chôdze a v poslednom období i reči. Vyšetrený psychológom s nálezom zníženého výkonného tempa, bagatelizácie postihnutia, poruchy krátkodobej pamäti. Ravenov test farebných progresívnych matríc (CMP) odhaľuje rozumové schopnosti v pásme hraničnej subnormy. Psychiatrom vedený pod diagnózou organickej depresívnej poruchy a schizoafektívnej psychózy. V novembri 2010 bola u neho geneticky konfirmovaná diagnóza HCH. Pacient bol v našej starostlivosti sledovaný od opakovaného suicidálneho pokusu stranguláciou (08/ 2011). Neurologicky orientovaný, prítomná dyzartria stredne ťažkého stupňa, porucha sledovacích pohybov, iniciácie sakád, kvadruhyperreflexia, prítomný príznak protrúzie jazyka a stisk dojičky (motorická inperzistencia). Dominujú choreatické pohyby tváre, končatín, trupu, spomalená chôdza o širokej báze s hyperlordózou trupu. Parakinézie a príznak aplauzu neprítomné. Motorické skóre UHDRS 43 bodov. V psychiatrickom náleze bradypsychizmus, myslenie pomalšie, simplexné, bez bludných obsahov, nálada mierne elevovaná, bez prejavov iritability či psychotických symptómov. MMSE test 20 bodov. MR mozgu zobrazuje atrofiu caput nucleus caudatus (caudu nediferencovať), nucleus lentiformis, difúznu atrofiu mozgu a dilatáciu komorového systému (obr. 3). EEG záznam rušený pohybovými artefaktami je prevažne normálny; v predných kvadrantoch prevláda nízkovoltážna b aktivita. Rutinné laboratórne parametre v norme, glykémia 5,3 mmol/ l. V anamnéze údaj o zvýšenej chuti do jedla (napriek tomu hodnota BMI, Body Mass Index 19,8), nadmernom nikotinizme a insomnii (ťažkosti so zaspávaním a fragmentácia spánku). V liečbe tiaprid 300 mg pro die, flunitrazepam 3 × 5 mg denne, koenzým Q10 30 mg denne, neskôr pridaný sertralín a trazodon. Napriek apelu na liečbu v rámci zhoršujúcej sa spolupráce pacienta došlo nakoniec k dokonanému suicídiu.
Kazuistika 2
Štyridsať sedemročná žena s poruchou jemnej motoriky asi od 30. roku života (vypadávanie predmetov z rúk), heteroanamnesticky často nervózna a dysforická. Pred 42. rokom začali byť u nej pozorované choreatické pohyby. Ochorenie relatívne rýchlo progredovalo do terajšieho obrazu pridruženej poruchy chôdze, demencie, urinárnej inkontinencie, vyše roka trvajúcej ťažkej dyzartrie a prehĺtacích ťažkostí. Aktuálne žije v sociálnej izolácii, je nesebestačná a odkázaná na opatrovníctvo. V septembri 2010 bola u nej geneticky verifikovaná HCH. V neurologickom náleze dezorientácia časom, dysfágia, ťažká dyzartria, poruchy sledovacieho systému, iniciácie a rýchlosti reflexných a voluntárnych sakád, kvadruhyperreflexia, choreatické dyskinézie v oblasti tváre, trupu a končatín s dominanciou choreoatetoidných pohybov na ľavej hornej končatine, instabilita stoja a pomalá chôdza o širokej báze, s dystóniou dolných končatín. Príznak aplauzu ani parakinézie nepozorované. Motorické skóre UHDRS 62 bodov. Psychologické vyšetrenie odhaľuje narušenie exekutívnych a intelektuálne ‑ mnestických funkcií, FDT test (test kresby ľudskej postavy) upozorňuje na organicitu ťažkého stupňa. V psychiatrickom náleze spomalené psychomotorické tempo, afektívna labilita, zvýšená pohotovosť k iritabilite, bez suicidálnych ideácií, spomalené myslenie bez bludných obsahov, insomnia (najmä dyskoimesis, tiež fragmentácia spánku), porucha krátkodobej pamäti a koncentrácie, bez prítomnosti psychotických prvkov. MMSE test 20 bodov, o pol roka neskôr MOCA test 10 bodov. MR vyšetrenie mozgu znázorňuje symetrický nález difúznej kortikálnej atrofie s kompenzačne rozšíreným komorovým systémom, atrofie caput nucleus caudatus (cauda nepozorovaná), nucleus lentiformis a corpus callosum. EEG záznam reprezentuje plochý β variant s disperznou prímesou θ a δ komponent. Laboratórne parametre bez hrubších abnormít, glykémia 5,8 mmol/ l. BMI 21,2 bodov – anamnesticky zvýšený apetít. V liečbe okrem koenzýmu Q nastavená na dvojkombináciu tiaprid s olanzapínom v nízkej dávke s dobrou toleranciou a klinickou odpoveďou na motorické symptómy.
Záver
HCH zatiaľ nie je dlho známou diagnózou a za sebou má len pol druha storočia. Napriek intenzívnemu výskumu a námahe odborníkov však doteraz nie je kauzálne liečiteľná a terapia zostáva naskrze symptomatická. Liečba je snahou o čo najväčšie uľahčenie bremena, ktoré si pacienti nesú so sebou a ktoré nikdy nespúšťajú. Neuropsychiatrický prístup má výhodu komplexnejších možností, avšak i ten niekedy bolestne zlyháva, ako to dokladuje suicídium pacienta z prvej kazuistiky. Ostáva nádej, že budúcnosť prinesie lepšie výsledky a na patogenetické mechanizmy viac zaostrenú liečbu.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Ján Necpál
Neurologické oddelenie
Nemocnica Zvolen a.s.
Kuzmányho nábrežie 28
960 01 Zvolen
e-mail: necpal.neuro@gmail.com
Prijato k recenzii: 16. 2. 2012
Prijato do tlače: 26. 2. 2013
Zdroje
1. Heathfield KW. Huntington’s chorea: a centenary review. Postrgrad Med J 1973; 49(567): 32 – 45.
2. Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010; 5(1): 40.
3. Fahn S, Jankovic J. Principles and practice of movement disorders. Philadelphia: Churchill Livingstone Elsevier 2007.
4. Novak MJ, Tabrizi SJ. Huntington’s disease. BMJ 2010; 340: c3109.
5. Margolis RL, Ross CA. Diagnosis of Huntington’s disease. Clinical Chemistry 2003; 49(10): 1726 – 1732.
6. Kosinski CM, Landwehrmeyer B. Huntington’s disease. In: Beal MF, Lang AE, Ludolph A (eds). Neurogenerative Diseases. Neurobiology, Pathogenesis and Therapeutics. New York: Cambridge University Press 2005.
7. Mascalchi M, Lolli F, Della Nave R, Tessa C, Petralli R, Gavazzi C et al. Huntington disease: volumetric, diffusion ‑ weighted, and magnetization transfer MR imaging of brain. Radiology 2004; 232(3): 867 – 873.
8. Montoya A, Price BH, Menear M, Lepage M. Brain imaging and cognitive dysfunctions in Huntington’s disease. J Psychiatry Neurosci 2005; 31(1): 21 – 29.
9. Ross CA, Margolis RL. Huntington disease. In: Davis KL, Chamey D, Coyle JT, Nemeroff C (eds). Neuropsychopharmacology: The Fifth Generation of Progress. 1st ed. Philadelphia: Lippincott Williams Wilkins 2002.
10. Eidelberg D, Surmeier D J. Brain networks in Huntington disease. J Clin Invest 2011; 121(2): 484 – 492.
11. Wolker FO. Huntington’s disease. Lancet 2007; 369(9557): 218 – 228.
12. Waldvogel HJ, Thu D, Hogg V, Tippett L, Faull RLM. Selective neurodegeneration, neuropathology and symptom profiles in Huntington‘s disease. In: Hannan AJ (ed). Tandem Repeat Polymorphisms: Genetic Plasticity, Neural Diversity and Disease. New York: Landes Bioscience and Springer Science+Business Media 2012.
13. Patarák M. Huntingtonova choroba: neuropsychiatrické aspekty [on‑line]. I ‑ med 2012. Available from: http:/ / www.i ‑ med.sk/ moodle/ course/ category.php?id=921&druh_specializacii=1.
14. Rosenblatt A. Neuropsychiatry of Huntington’s disease. Dialogues Clin Neurosci 2007; 9(2): 191 – 197.
15. Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Wang C, Stout JC et al. “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci 2010; 22(2): 196 – 207.
16. Craufurd D, Snowden J. Neuropsychological and neuropsychiatric aspects of Huntington’s disease. In: Bates G, Harper P, Jones L (eds). Huntington’s disease. New York: Oxford University Press 2002 : 62 – 94.
17. Allain P, Gaura V, Fasotti L, Chauviré V, Prundean A,Sherer ‑ Gagou C et al. The neural substrates of script knowledge deficits as revealed by a PET study in Huntington’s Disease. Neuropsychologia 2011; 49(9): 2673 – 2684.
18. Grahn J, Parkinson JA, Owen AM. The role of the basal ganglia in learning and memory: Neuropsychological studies. Behav Brain Res 2009; 199(1): 53 – 60.
19. Duff K, Paulsen J, Mills J, Beglinger LJ, Moser DJ, Smith MM et al. Mild cognitive impairment in prediagnosed Huntington disease. Neurology 2010; 75(6): 500 – 507.
20. Eddy CM, Mitchell IJ, Beck SR, Cavanna AE, Rickards HE. Altered subjective fear responses in Huntington’s disease. Parkinsonism Relat Disord 2011; 17(5): 386 – 389.
21. Illea R, Hollb AK, Kapfhammerb HP, Reisingerb K, Schafera A, Schienlea A. Emotion recognition and experience in Huntington‘s disease: Is there a differential impairment? Psychiatry Res 2011; 188(3): 377 – 382.
22. Klöppel S, Stonnington CS, Petrovic P, Mobbs D, Tuscher O, Craufurd D et al. Irritability in pre‑clinical Huntington‘s disease. Neuropsychologia 2010; 48(2): 549 – 557.
23. Sachdev PS. Huntington’s Disease and related disorders and their association with schizophrenialike psychosis. In: Sachdev PS, Keshavan MS (eds). Secondary Schizophrenia. Cambridge: Cambridge University Press 2010 : 348 – 357.
24. Van Duijn E, Reedeker N, Giltay EJ, Roos RAC, Van der Mast RC. Correlates of apathy in Huntington’s disease. J Neuropsychiatry Clin Neurosci 2010; 22(3): 287 – 294.
25. Eslava JC, Iragorri ‑ Cucalón Á, Ucrós ‑ Rodríguez G,Bonilla ‑ Jácome C, Tovar ‑ Perdomo S, Herin DV et al. Obsessive ‑ Compulsive Disorder Symptoms in Huntington’s Disease: A Case Report. Rev Colomb Psiquiatr 2008; 37(4): 644 – 654.
26. Anderson KE, Louis ED, Stern Y, Marder KS. Cognitive correlates of obsessive and compulsive symptoms in Huntington’s disease. Am J Psychiatry 2001; 158(5): 799 – 801.
27. Uhlmann WR. The Woman Who Walked into the Sea: Huntington’s and the Making of a Genetic Disease. American J Hum Genet 2010; 86(6): 830 – 831.
28. Wexler A. The art of medicine. Stigma, history, and Huntington’s disease. Lancet 2010; 376(9734): 18 – 19.
29. Penarand E, Garcia A, Montgomery L. It wasn’t Witchcraft – It was Huntington Disease! J Am Board Fam Med 2011; 24(1): 115 – 116.
30. Paulsen JS, Hoth KF, Nehl C, Stierman L, The Huntington Study Group. Critical Periods of Suicide Risk in Huntington’s Disease. Am J Psychiatry 2005; 162(4): 725 – 731.
31. Pang TYC, Du X, Zajac MS, Howard M, Hannan AJ. Altered serotonin receptor expression is associated with depression‑related behavior in the R6/ 1 transgenic mouse model of Huntington’s disease. Hum Mol Genet 2009; 18(4): 753 – 766.
32. Gudesblatt M, Tarsy D. Huntington’s Disease: A Clinical Review. Supplement to Neurology Reviews 2011; 1 – 8.
33. Perlis RH, Smolier JW, Mysore J, Sun M, Gillis T, Purcell S et al. Prevalence of incompletely penetrant Huntington’s disease alleles among individuals with major depressive disorder. Am J Psychiatry 2010; 167(5): 574 – 579.
34. Krogias C, Strassburger K, Eyding J, Gold R, Norra C,Juckel G et al. Depression in patients with Huntington disease correlates with alterations of the brain stem raphe depicted by transcranial sonography. Psychiatry Neurosci 2011; 36(3): 187 – 194.
35. Saleh N, Moutereau S, Durr A, Krystkowiak P, Azulay JP, Tranchant C et al. Neuroendocrine Disturbances in Huntington’s Disease. PLoS ONE 2009; 4(3): e4962.
36. Peng Q, Masuda N, Jiang M, Li Q, Zhao M, Ross CA et al. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/ 2 Huntington’s disease mouse model. Exp Neurol 2008; 210(1): 154 – 163.
37. Walker TL, Turnbull GF, Mackay EW, Hannan AJ, Barlett PF. The latent stem cell population is retained in the hippocampus of transgenic Huntington’s disease mice but not wild‑type mice. PLoS One 2011; 6(3): e18153.
38. Zuccato C, Cattaneo E. Role of brain‑derived neurotrophic factor in Huntington’s disease. Prog Neurobiol 2007; 81(5 – 6): 294 – 330.
39. Petersén A, Bjőrkqvist M. Hypothalamic ‑ endocrine aspects in Huntington’s disease. Eur J Neurosci 2006; 24(4): 961 – 967.
40. Petersén A, Gil J, Maat ‑ Schieman ML, Bjőrkqvist M, Tanila H, Araujo IM et al. Orexin loss in Huntington’s disease. Hum Mol Genet 2005; 14(1): 39 – 47.
41. Farrer LA. Diabetes mellitus in Huntington’s disease. Clin Genet 1985; 27(1): 62 – 67.
42. Lalić NM, Marić J, Svetel M, Jotić A, Stefanova E, Lalić K, et al. Glucose homeostasis in Huntington Disease. Abnormalities in Insulin Sensitivity and Early ‑ Phase Insulin Secretion. Arch Neurol 2008; 65(4): 476 – 480.
43. Squitieri F, Frati L, Ciarmiello A, Lastoria S, Quarell O.Juvenile Huntington’s fisease: foes a dosage ‑ effect pathogenic mechanism differ from the classical adult disease? Mech Ageing Dev 2006; 127(2): 208 – 212.
44. Nance M, Jones R, Imbriglio S, Gettig B. The Juvenile Huntington’s Disease Handbook. A Guide for Physicians, Neurologists and Other Professionals. New York: Huntington’s Disease Society of America 2001.
45. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. Neurology 1994; 44(8): 1533 – 1536.
46. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. J Med Genet 1994; 31(7): 555 – 559.
47. Roth J. Huntingtonova nemoc. Cesk Slov Neurol N 2010; 73/ 106(2): 107 – 123.
48. Židovská J, Klempíř J, Kebrdlová V, Uhrová T, Koblihová J, Anders M et al. Huntingtonova nemoc: zkušenosti s genetickým testováním v letech 1994 – 2005. Cesk Slov Neurol N 2007; 70/ 103(1): 72 – 77.
49. Douaud G, Gaura V, Ribeiro M ‑ J, Lethimonnier F,Maroy R, Verny C et al. Distribution of grey mater atrophy in Huntington’s disease patients: a combined ROI‑based and voxel‑based morphometric study. Neuroimage 2006; 32(4): 1562 – 1575.
50. Huntington Study Group. Unified Huntington’s Disease Rating Scale: Reliability and Consistency. Mov Disorders 1996; 11(2): 136 – 142.
51. Frank S. Tetrabenazine as anti‑chorea therapy in Huntington disease: an open label continuation study. Huntington Study Group/ TETRA ‑ HD Investigators. BMC Neurol 2009; 9 : 62.
52. Venuto C S, McGarry A, Ma Q, Kiebutz K. Pharmacologic approaches to the treatment of Huntington’s disease. Mov Disord 2012 27(1): 31 – 41.
53. Kenney C, Hunter C, Davidson A, Jankovic J. Short‑term effects of tetrabenazine on chorea associated with Huntington’s disease. Mov Disord 2007; 22(1): 10 – 13.
54. Paleacu D, Anca M, Giladi N. Olanzapine in Huntington’s disease. Acta Neurol Scand 2002; 105(6): 441 – 444.
55. Lundin A, Dietrichs E, Haghighi S, Göller ML, Heiberg A, Loutfi G et al. Efficacy and safety of the dopaminergic stabilizer Pridopidine (ACR16) in patients with Huntington’s disease. Clin Neuropharmacol 2010; 33(5): 260 – 264.
56. Huntington Study Group. A randomized, placebo ‑ controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology 2001; 57(3): 397 – 404.
57. Kanazawa I. Therapeutic strategies in Huntington’s Disease. J Clin Neurol 2006; 2(4): 213 – 224.
58. Bachoud ‑ Lévi AC, Rémy P, Nguyen JP, Brugières P, Lefaucheur JP, Bourdet C et al. Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet 2000; 356(9246): 1975 – 1979.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2013 Číslo 4
Nejčtenější v tomto čísle
- Úskalí diagnostiky progresivní multifokální leukoencefalopatie u pacientů infikovaných lidským virem imunodeficience – kazuistiky
- Chirurgie baze lební (uvnitř minimonografie video)
- Neuropsychiatrický pohľad na Huntingtonovu chorobu
- Dysembryoplastický neuroepiteliální tumor a jeho atypická varianta u dětí – kazuistiky