
Oligosymptomatické formy myotonické dystrofie typu 2
Oligosymptomatic Forms of Myotonic Dystrophy Type 2
Myotonic dystrophy type 2 (DM2) typically presents in adult life with lower extremity proximal muscle weakness, variously expressed myotonia and cataract being the most common symptoms. On electromyography (EMG) a broad spectrum of spontaneous activity with myotonic discharges can be found and motor unit action potentials may show myopathic pattern. Both clinical and EMG features in DM2 may be greatly variable. We present two cases of DM2 with proximal muscle weakness, spontaneous EMG activity but no myotonic discharges and no characteristic myopathic EMG pattern.
Key words:
myotonic dystrophy type 2 – myopathy – myotonia
Autoři:
Z. Ambler
Působiště autorů:
Neurologická klinika LF UK a FN Plzeň
Vyšlo v časopise:
Cesk Slov Neurol N 2012; 75/108(2): 237-239
Kategorie:
Kazuistika
Souhrn
Myotonická dystrofie typu 2 (DM2) se typicky manifestuje v dospělosti a slabost pletencového svalstva dolních končetin, různě vyjádřená myotonie a katarakta patří k hlavním příznakům. Při elektromyografickém (EMG) vyšetření může být přítomno široké spektrum spontánní aktivity s myotonickými výboji a akční potenciály motorických jednotek vykazují myopatický vzorec. Klinické i EMG projevy DM2 mohou být značně variabilní. Uvádíme kazuistiky dvou nemocných s DM2 s proximální svalovou slabostí, spontánní EMG aktivitou bez myotonických výbojů a bez charakteristických myopatických změn v EMG.
Klíčová slova:
myotonická dystrofie typ 2 – myopatie – myotonie
Úvod
Myotonická dystrofie (DM) je autozomálně dominantní multisystémová porucha, která postihuje kosterní i hladké svaly, ale také oči, srdce, endokrinní a centrální (ale někdy i periferní) nervový systém. Geneticky se rozlišuje DM typu 1 (DM1) a typu 2 (DM2). Lokus pro DM2 byl zmapován na 3. chromozomu (3q21) a byla prokázána mutace v prvním intronu genu pro ZNF9 (zinc finger protein 9), který nese expandovanou tetranukleotidovou repetitivní sekvenci CCTG. Proteiny se zinkovými prsty (zinc finger proteins) jsou jednou z největších proteinových rodin, které se vyskytují u eukaryotních organizmů. Vzhledem k tomu, že většina těchto proteinů funguje jako transkripční faktory regulující genovou expresi (3 % genů lidského genomu), jsou přítomny uvnitř buňky ve velmi malém množství a jejich výzkum nebyl možný před rozvojem technologií molekulární genetiky [1–3].
DM2 se typicky manifestuje v dospělosti proximální slabostí dolních končetin, často s myalgiemi, různě vyjádřenou myotonií a kataraktou. Udává se, že její výskyt je méně častý nežli DM1, ale bude zřejmě výrazně poddiagnostikována [4,5]. Chtěli bychom upozornit na možný výskyt oligo- nebo i monosymptomatických forem DM2 a nutnost častější indikace genetického vyšetření.
Kazuistiky
Kazuistika I
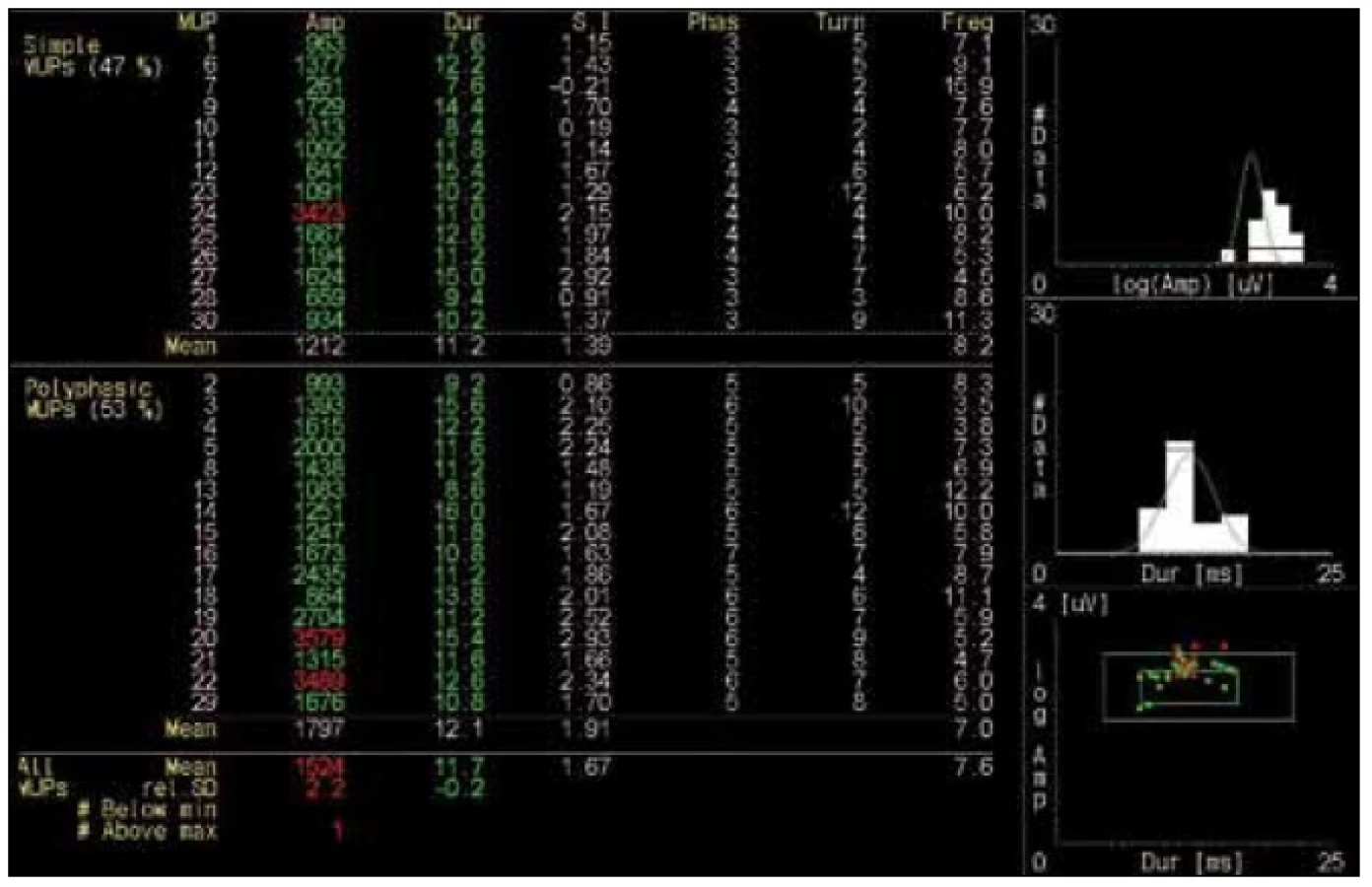
Šestapadesátiletý muž, hypertonik a diabetik léčený perorálními antidiabetiky. Asi šest let se u něho postupně rozvíjela nebolestivá slabost obou dolních končetin (DK) a v poslední době pozoroval mírnou slabost i horních končetin (HK) při práci s rukama nad hlavou. Dosud byl veden na okresním pracovišti podle EMG jako středně těžká polyneuropatie. Při vyšetření jsme zjistili normální nález na hlavových nervech i HK, proximální slabost na DK (flexe v kyčli podle svalového testu 4 st., extenze v koleni 4+ st.) s pouze naznačenou nevýznamnou asymetrií. Šlachookosticové reflexy i čití byly normální. Dřep samostatně nesvedl, chůze byla bez zjevné abnormity, chůze po špičkách i patách normální. Nebyly žádné známky akční ani poklepové myotonie. Kreatinkináza v séru (CK) 4 µkat/l (norma do 3,2). EMG vyšetření prokázalo v m. vastus lateralis nepříliš výraznou spontánní aktivitu (fibrilace) 2+, ojediněle i náznak komplexního repetitivního výboje. Myotonické výboje nebyly přítomny. Při maximální kontrakci byla patrna jen velmi lehká simplifikace (bohaté přechodné stadium). Kondukční studie byly normální. Parametry 30 akčních potenciálů motorických jednotek (MUP): trvání 7,6–16, průměr 11,7 ms (norma), amplituda 0,3–3,5, průměr 1,5 mV (lehce nad normu, 3 MUP s amplitudou nad normu – 3,4–3,5 mV), 53 % MUP bylo polyfázických (obr. 1, 2). Klinický ani EMG nález rozhodně nesvědčil pro polyneuropatii. EMG nález měl charakter mírných reinervačních změn, které se obvykle vyskytují u subakutní až chronické neurogenní léze. Charakteristické myogenní změny nebyly přítomny. Svalová biopsie v korelaci s EMG prokázala pokročilou neurogenní svalovou atrofii se známkami floridní denervace a reinervace, se seskupováním typu svalových vláken a skupinovou atrofií (doc. MUDr. J. Zámečník, FN Motol). Molekulárněgenetické vyšetření prokázalo DM2.

Kazuistika II
Sedmdesátiletý muž, léčí se na hypertenzi, asi jeden rok bere simvastatin. Tři roky se postupně rozvíjela nebolestivá proximální slabost obou DK, ze židle se zvedá s pomocí HK, dřep nesvede. Byla patrna lehká hypotrofie stehen v porovnání s trofikou lýtek. Kromě slabosti DK byl normální neurologický nález a hodnoty CK normální. Při EMG vyšetření byla v m. vastus lateralis zjištěna jen sporadická spontánní aktivita (fibrilace a pozitivní ostré vlny) 2+ a žádné myotonické výboje. Při maximální kontrakci byla interferenční křivka, v některých místech jen naznačena lehká simplifikace. Spektrum MUP: trvání 6,2–16,4; průměr 11,2 ms (norma), amplituda 0,3–3,6; průměr 1,6 mV (lehce nad normu, 3 MUP s amplitudou nad normu – 3–3,6 mV). Opět nález odpovídající mírným reinervačním změnám. Molekulárněgenetické vyšetření prokázalo DM2.
Diskuze
DM2 se nejčastěji manifestuje svalovými příznaky (slabost, bolesti, pocity ztuhlosti nebo myotonie). Slabost bývá nejčastěji vyjádřena v proximálním svalstvu DK, ale časté je i postižení krčních flexorů, hlubokých flexorů prstů na HK nebo i extenzorů lokte. Slabost jiných svalových skupin je méně častá. Svalové bolesti mohou být fluktuující, ale i v některých větších souborech se vyskytují jen u 63 % pacientů [2]. Velmi častá katarakta se obvykle prokáže až cíleným vyšetřením štěrbinovou lampou. EMG nález je charakterizován přítomností spontánní aktivity (fibrilace a pozitivní ostré vlny, někdy komplexní repetitivní výboje) a typicky především myotonickými výboji. MUP bývají kratšího trvání, nižší amplitudy a nález má myogenní charakter. Typické myogenní projevy při hodnocení spektra MUP jsou charakterizovány hlavně zkráceným trváním a redukcí arey. Amplituda je často snížená, ale může být i normální (pokud je registrační elektroda v blízkosti nepostiženého svalového vlákna), ale i zvýšená (pokud se registruje v blízkosti hypertrofického svalového vlákna). Někdy mohou být přítomny i komplexní polyfázické MUP s prodlouženým trvání, ale jsou většinou nízké amplitudy, takže area zůstává nezměněna nebo i snížena. Pro myogenní poruchy je charakteristické, že dochází ke snížení počtu vláken v motorické jednotce. Naopak pro neurogenní změny jsou typické zvětšené MUP, nejprve obvykle zvýšení amplitudy, později i prodloužené trvání (zvětšení arey) a v důsledku kolaterální reinervace se zvětšuje počet svalových vláken v teritoriu motorické jednotky.
Typický myogenní EMG nález ale nemusí být u všech pacientů [7]. U obou našich nemocných se postupně rozvíjela nebolestivá slabost DK, v EMG nebyly zjištěny myotonické projevy a spektrum MUP nasvědčovalo spíše reinervačním neurogenním změnám nežli klasické myogenní lézi, což korelovalo u prvního pacienta i s bioptickým nálezem. Trvání MUP bylo sice v mezích normy, ale byl zřetelný posun k vyšším amplitudám a ve spektru MUP nebyly žádné krátké MUP s nízkou amplitudou.
Ani myotonické výboje nemusí být při EMG vždy přítomné, výskyt se udává u DM2 kolem 90 % [6,7], zejména pokud se systematicky nevyšetřuje větší počet svalů. Někdy může mít výboj i netypický charakter bez klasického kolísání frekvence a amplitudy (waxing-waning), ale je přítomna jen postupně se snižující frekvence i amplituda [8].
Z oligosymptomatických forem DM2 byla popsána i izolovaná fokální slabost jen v m. triceps brachii s hyper-CK-emií [9], DM2 byla zjištěna u dvou pacientů ze skupiny 63 s fibromyalgiemi [10] a dokonce i u asymptomatické hyper-CK-emie [11]. Ani histologické změny nejsou u DM2 zcela typické a diagnostické [12,13] a mohou někdy více imponovat jako změny neurogenní nežli myogenní.
Závěrem lze doporučit u každého dospělého nemocného s nevyjasněnou proximální slabostí – myopatickým syndromem – myslet i na možnost DM2 a indikovat genetické vyšetření. Diagnózu podpoří nález i jen sporadické spontánní aktivity v EMG (fibrilace a pozitivní ostré vlny) bez myotonických výbojů. EMG nález nemusí vykazovat typický myogenní vzorec, ale pouze reinervační změny MUP. Svalová biopsie sice odliší zánětlivou myopatii (která může mít obdobný EMG obraz), ale neprokáže DM2.
Přijato k recenzi: 3. 6. 2011
Přijato do tisku: 18. 8. 2011
prof. MUDr. Zdeněk Ambler, DrSc.
Neurologická klinika LF UK a FN Plzeň
Alej Svobody 80
304 60 Plzeň
e-mail: ambler@fnplzen.cz
Zdroje
1. Finsterer J. Myotonic dystrophy type 2. Eur J Neurol 2002; 9(5): 441–447.
2. Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 2003; 60(4): 657–664.
3. Kukačka J, Kizek R, Průša R. Budoucnost zinkových metaloproteinů v laboratorní medicíně. Klin Biochem Metab 2008; 16(3): 161–170.
4. Suominen T, Bachinski LL, Auvinen S, Hackman P, Baggerly KA, Angelini C et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet 2011; 19(7): 776–782.
5. Voháňka S, Bednařík J, Fajkusová L, Sedláčková J. Myotonická dystrofie typ 2: vzácné nebo časté onemocnění v České republice? Cesk Slov Neurol N 2005; 68/101(6): 390–393.
6. Meola G, Moxley RT jr. Myotonic dystrophy type 2 and related myotonic disorders. J Neurol 2004; 251(10): 1173–1182.
7. Chaudhry V, Johnson NM. Spectrum of clinical presentation in myotonic dystrophy type 2. Clin Neurophysiol 2008; 119: e54.
8. Logigian EL, Ciafaloni E, Quinn C, Dilek N, Pandya S, Moxley RT jr et al. Severity, type, and distribution of myotonic discharges are different in type 1 and type 2 myotonic dystrophy. Muscle Nerve 2007; 35(4): 479–485.
9. Milone M, Batish SD, Daube JR. Myotonic dystrophy type 2 with focal asymmetric muscle weakness and no electrical myotonia. Muscle Nerve 2009; 39(3): 383–385.
10. Auvinen S, Suominen T, Hannonen P, Bachinski LL, Krahe T, Udd B. Myotonic dystrophy type 2 found in two of sixty-three persons diagnosed as having fibromyalgia. Arthritis Rheum 2008; 58(11): 3627–3631.
11. Merlini L, Sabatelli P, Columbaro M, Bonifazi E, Pisani V, Massa R et al. Hyper-CK-emia as the sole manifestation of myotonic dystrophy type 2. Muscle Nerve 2005; 31(6): 764–767.
12. Schoser BG, Schneider-Gold C, Kress W, Goebel HH, Reilich P, Koch MC et al. Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve 2004; 29(2): 275–281.
13. Toth C, Dunham C, Suchowersky O, Parboosingh J, Brownell K. Unusual clinical, laboratory, and muscle histopathological findings in a family with myotonic dystrophy type 2. Muscle Nerve 2007; 35(2): 259–264.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2012 Číslo 2
Nejčtenější v tomto čísle
- Využití perkutánní endoskopické gastrostomie – přehled indikací, popis techniky a současné trendy v neurologii
- Posturálna instabilita, poruchy chôdze a pády pri Parkinsonovej chorobe
- Algoritmus vyšetření likvoru v návaznosti na doporučení Sekce neuroimunologie a likvorologie České neurologické společnosti JEP
- Obstrukční spánková apnoe a CPAP – má význam řešit nosní průchodnost?