
Vrozená myotonie na podkladě mutací v genu pro chloridový kanál ClC-1
Congenital Myotonia Caused by Mutations in the CIC-1 Chloride Channel Gene
Congenital myotonia is caused by mutations in the CLCN1 chloride channel gene. It can be inherited as either an autosomal dominant (Thomsen’s myotonia) or a recessive (Becker’s myotonia) trait. During 2008–2010, mutations in the chloride channel were found in 7 patients with myotonia, making it the third most frequent cause of myotonia in our records (54 patients with type 2 myotonic dystrophy, 18 with type 1 myotonic dystrophy, and 4 suffering from mutations in the sodium channel). All persons revealed hypertrophic muscles, worsening in cold was found in six cases, and no patients have post-exercise weakness. The semi-dominant mutation p.Arg894X was found most frequently: 5 times among a possible 13 mutations. The recessive mutation p.Pro480HisfsX24 was disclosed three times and p.Phe413Cys twice. All the cases but one are sporadic, thus the majority of patients in our population have Becker´s form of myotonia congenita.
Key words:
myotonia congenita – CLCN1 – channelopathy
Autoři:
S. Voháňka 1,2; J. Bednařík 1,2; D. Páclová 3; J. Sedláčková 3; L. Fajkusová 3
Působiště autorů:
Neuromuskulární centrum, Neurologická klinika LF MU a FN Brno
1; CEITEC (Středoevropský technologický institut), MU, Brno
2; Centrum molekulární biologie a genové terapie FN Brno
3
Vyšlo v časopise:
Cesk Slov Neurol N 2011; 74/107(4): 467-470
Kategorie:
Krátké sdělení
Souhrn
Příčinou vrozené myotonie jsou mutace v genu CLCN1, který kóduje chloridový kanál ClC-1. Podle typu dědičnosti se vrozená myotonie tradičně rozděluje na Thomsenovu (dominantní) a Beckerovu (recesivní) variantu. V letech 2008–2010 byla mutace v chloridovém kanálu potvrzena u sedmi nemocných s myotonií. Jde o třetí nejčastější příčinu myotonie v registru (54 nemocných s myotonickou dystrofií 2. typu, 18 nemocných s myotonickou dystrofií 1. typu a čtyři pacienti s mutací v sodíkovém kanálu SCN4A). Klinicky jde ve všech sedmi případech o osoby s dobře vyvinutou muskulaturou. Zřetelná provokace myotonie chladem byla nalezena u šesti z nich. Ani jednou nebyly zaznamenány pozátěžové parézy. Nejčastěji byla prokázána semidominantní mutace p.Arg894X (5 z 13 mutovaných alel), následovala mutace p.Pro480HisfsX24 (tři mutované alely) a p.Phe413Cys (dvě mutované alely). U šesti pacientů by zaznamenán sporadický výskyt onemocnění v rodině, v jednom případě byl dokumentován dominantní přenos. V převážné většině se tedy v naší populaci jedná o tzv. Beckerovu variantu myotonia congenita. Výskyt má sporadický charakter a rodinná anamnéza neposkytuje diagnostické vodítko.
Klíčová slova:
myotonia congenita – CLCN1 – kanálopatie
Rodina genů napěťově řízených chloridových kanálů sestává z devíti příbuzných genů (CLCN1-7, CLCNKA, CLCNKB). Gen CLCN1 kóduje chloridový kanál ClC-1, který je exprimován na sarkolemě kosterních svalů. Chloridový kanál řídí influx negativně nabitých chloridových iontů a je hlavním faktorem klidové vodivosti svalové membrány zajišťujícím její excitabilitu. Při poklesu konduktivity chloridového kanálu pod 40 % depolarizuje kalium akumulované v T tubulech sarkolemu a dochází ke vzniku setrvalého akčního potenciálu, který se projeví jako prolongovaná kontrakce (myotonie). Gen pro chloridový kanál CLCN1 o velikosti 36 kb je umístěn na dlouhém raménku 7. chromozomu (7q35), má 23 exonů, délka transkriptu je 3 093 nukleotidů. Chloridový kanál ClC-1,který je produktem genu CLCN1, je složen z 988 aminokyselin a obsahuje několik transmembránových domén. Je složen pravděpodobně ze čtyř stejných podjednotek. Ultrastrukturální studie ukazují, že kanál tvoří dva póry, každý sestává ze dvou stejných podjednotek [1,2]. Mutace jsou rozprostřeny po celé kódující sekvenci a v přilehlých intronových oblastech. Byly popsány mutace typu nonsense, missense, delece, inzerce i sestřihové mutace. Protože by se pokles chloridové konduktivity do 40 % neměl projevit (jak to vidíme u heterozygotních nosičů recesivních mutací), je vysvětlení dominantních mutací na molekulární úrovni složitější. Předpokládá se tzv. dominantně negativní mechanizmus: dominantní mutace modifikuje otevření obou protoporů (porucha dimerizace) nebo iontovou selektivitu kanálu [3].
Vrozené myotonie na podkladě mutace v CLCN1 jsou vzácné dědičné svalové choroby, které nezkracují délku života ani nevedou většinou k závažné invalidizaci. Dominantním příznakem postižení chloridových kanálů je myotonie, která je definována jako zpomalená relaxace kosterního svalstva po volní kontrakci. Obvykle se zmírňuje opakovanou aktivitou (warm-up fenomén). Dochází-li naopak k zhoršení svalové reakce po opakovaných pohybech, hovoříme o paradoxní myotonii – paramyotonii. Klinicky testujeme akční (intenční) myotonii a méně zřetelnou myotonii perkusní – tedy určitou dobu trvající kontrakci svalu po poklepu. Elektrofyziologickým projevem jsou myotonické výboje, které odpovídají membránové instabilitě jednotlivých svalových vláken. Typické je kolísání frekvence (20–150 Hz) i amplitudy. Všechny formy myotonie se zhoršují chladem.
Porucha chloridových kanálů je buď primární, jako je tomu u chloridové kanálopatie (mutace v genu CLCN1), nebo sekundární, což vidíme u obou typů myotonické dystrofie. Tam je narušena syntéza chloridového kanálu ClC-1 na postranskripční úrovni jeho exprese. Myotonické dystrofie jsou způsobeny expanzí nukleotidů – tripletů CUG u myotonické dystrofie 1. typu a tetrapletů CCUG u myotonické dystrofie 2. typu. Expandované nukleotidy vedou k akumulaci vzniklé RNA v jádře buněk a k vyvázání specifických proteinů potřebných ke správnému sestřihu mRNA genu CLCN1.
Vrozená myotonie – myotonia congenita
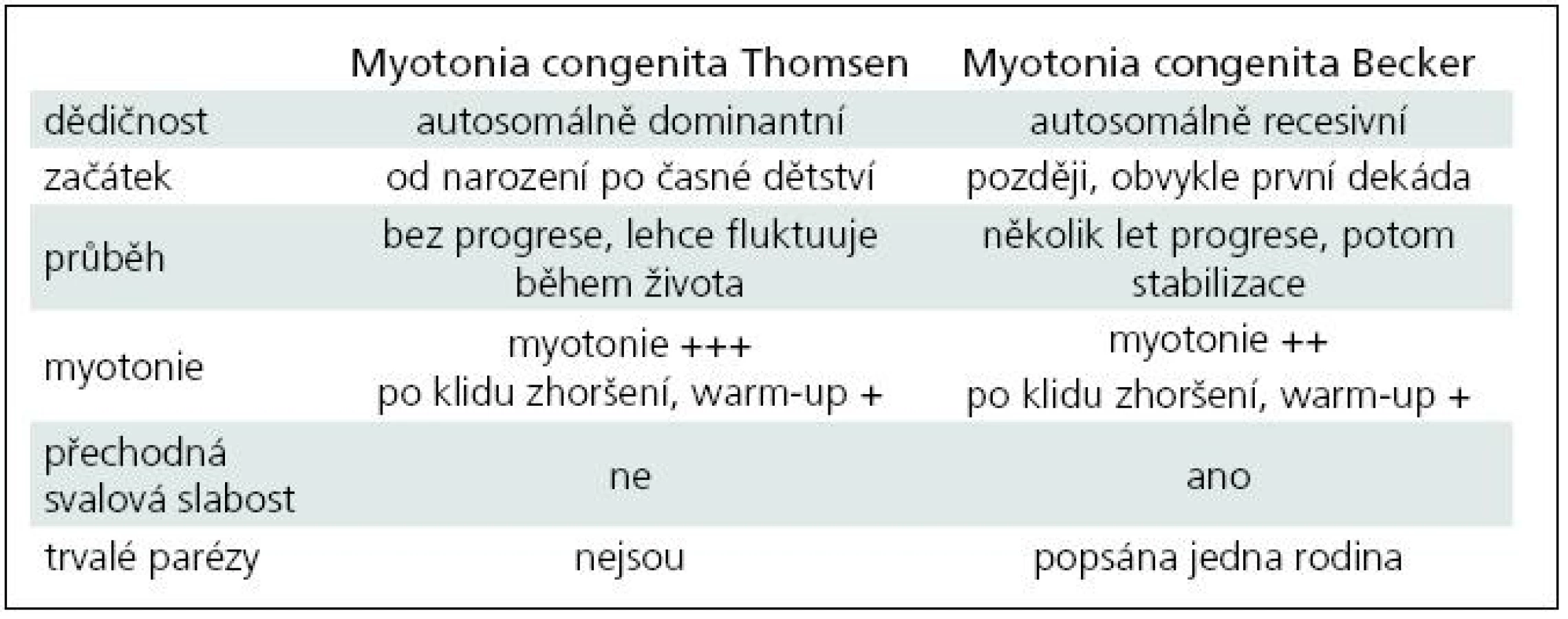
Chorobu popsal poprvé šlesvický lékař Asmus Julius Thomas Thomsen na sobě a u 20 příslušníků vlastní rodiny v roce 1876 [4]. Klasická autosomálně dominantní varianta myotonie nese jeho jméno – Myotonia congenita Thomsen. Čistě dominantní přenos má na svědomí asi 15 definovaných mutací. Podkladem většiny vrozené myotonie (asi 2/3) jsou mutace autosomálně recesivního charakteru, kdy je k manifestaci postižení třeba dvou mutovaných alel. Přenašeči choroby jsou zcela zdraví, někdy lze u nich nalézt elektrofyziologické známky myotonie. Recesivní forma vrozené myotonie byla popsána roku 1957 Peterem Emilem Beckerem, po němž je také pojmenována [5]. Dosud bylo zaznamenáno více než 100 recesivních mutací [6]. Asi 10 mutací sdílí pravděpodobně autosomálně dominantní i recesivní chování, nepochybně to bylo prokázáno u tří typů (p.Gly230Glu, p.Ala531Val a p.Arg894X). Vysvětlení tohoto fenomenu je obtížné: zvažuje se mj. inkompletní dominance, omezená penetrance dominantní mutace či rozdíly v alelické expresi [7–9]. Fenotypická manifestace dominantních a semidominantních mutací může dokonce kolísat i v rámci jedné rodiny. Rozdíly mezi oběma historickými klinickými formami (dominantní Thomsenovou a recesivní Beckerovou) shrnuje tab. 1. V současné době ale stoupá množství informací o korelaci genotypu a fenotypu a variabilitě u různých typů mutací, což tento striktně popisný pohled narušuje a rozvolňuje. Navíc u postižení iontových kanálů lze počítat vždy s významnými modulujícími vlivy vnitřních a vnějších faktorů. Frekvence de novo mutací není známa. Prevalence je udávána hodnotou 1/100 tis. obyvatel, nejvyšší je v severní Skandinávii, kde je 10krát větší [10]. Prevalence je pravděpodobně podhodnocena, protože některé lehké sporadické (AR) formy unikají lékařskému záchytu a diagnostice.
Myotonie je přítomna již od dětství. Pacienti mají potíže hlavně v zimě a zejména s odkrytými částmi těla, jako jsou ruce a obličej. Známý je tzv. lid lag fenomén: opožďování zavření horního víčka při pohledu dolů (myotonie m. levator palpebrae). Výjimečně může myotonie v obličeji imitovat blefarospazmus. Klíčem ke správné diagnóze je EMG vyšetření m. orbicularis oculi, kde najdeme myotonické výboje. Vzhledem k zmírnění myotonie opakovanými pohyby mají postižení především tzv. startovací potíže: nejtěžší je pro ně vystoupit na první schod, druhý je snazší atd. A naopak – myotonie je nejvýraznější po odpočinku, hovoříme o zhoršení – agravaci inaktivitou. U autosomálně recesivní Beckerovy varianty jsou někdy po opakovaných pohybech (zvláště na horních končetinách) známky přechodné slabosti. Nejsou přítomny žádné symptomy systémového postižení (katarakta, kardiomyopatie, arytmie apod.), ani trvalá svalová slabost, jak to vidíme u myotonické dystrofie. Nemocní naopak vynikají většinou dobrou atletickou muskulaturou, jde o pravé svalové hypertrofie. Pacienti ale nedokáží někdy svoje potíže správně formulovat a interpretují problémy spojené s myotonií jako slabost. Jen u jednoho typu autosomálně recesivní mutace byla popsána trvalá progresivní svalová slabost se známkami myopatie ve svalové biopsii [11]. Jde o dva sourozence, kteří mají na jedné alele inzerci v exonu 7 (c.831insG) vedoucí k vzniku stop kodonu a na druhé alele substituci v exonu 23 (p.Pro932Leu).
Laboratorně bývá někdy u pacientů s vrozenou myotonií lehce zvýšená hladina kreatinkinázy. Patrně jako projev poškození některých svalových vláken nadměrnou stimulací, tak jak to vidíme např. po silovém cvičení nebo delším běhu u zdravých osob.
Některé mutace v chloridovém kanálu (p.Phe428Ser) se projevují paradoxní myotonií (paramyotonií), což je tradičně spojováno s mutacemi v sodíkovém kanálu [3].
Terapie myotonie je svízelná, neexistují kvalitní data z randomizovaných studií [12]. Mezi často používané látky patří mexiletin, procainamid (125–1 000 mg/den), chinidin (200–1 200 mg/den), fenytoin (300–400 mg/den) a acetazolamid (250–750 mg/den). Pravděpodobně nejúčinnější je derivát lidokainu mexiletin. Terapie by měla mít charakter postupného zvyšování dávky k optimálnímu účinku. Začíná se na 150 mg dvakrát denně, maximální dávka je 900 mg ve třech denních dávkách. Titrace dávky směrem nahoru by měla proběhnout při použití kteréhokoliv z uvedených léků. Při farmakoterapii je třeba vždy zvážit nežádoucí účinky a potenciální prospěch, protože u myotonie jde o symptomatickou terapii. Potíže jsou často mírné a nemocní jsou na ně od dětství zvyklí.
Soubor a metodika
V Centru molekulárním biologie a genové terapie FN Brno byla v letech 2008––2010 potvrzena mutace v chloridovém kanálu u sedmi nemocných s myotonií, diagnostikovanou v Neuromuskulárním centru Neurologické kliniky FN Brno. Jde o třetí nejčastější příčinu myotonie v registru centra (54 nemocných s myotonickou dystrofií 2. typu, 18 nemocných s myotonickou dystrofií 1. typu a 4 pacienti s mutací v sodíkovém kanálu, graf 1).

Molekulárněgenetická diagnostika vrozené myotonie je založena na amplifikaci a sekvenční analýze všech exonů a přilehlých intronových oblastí genu CLCN1.
Výsledky
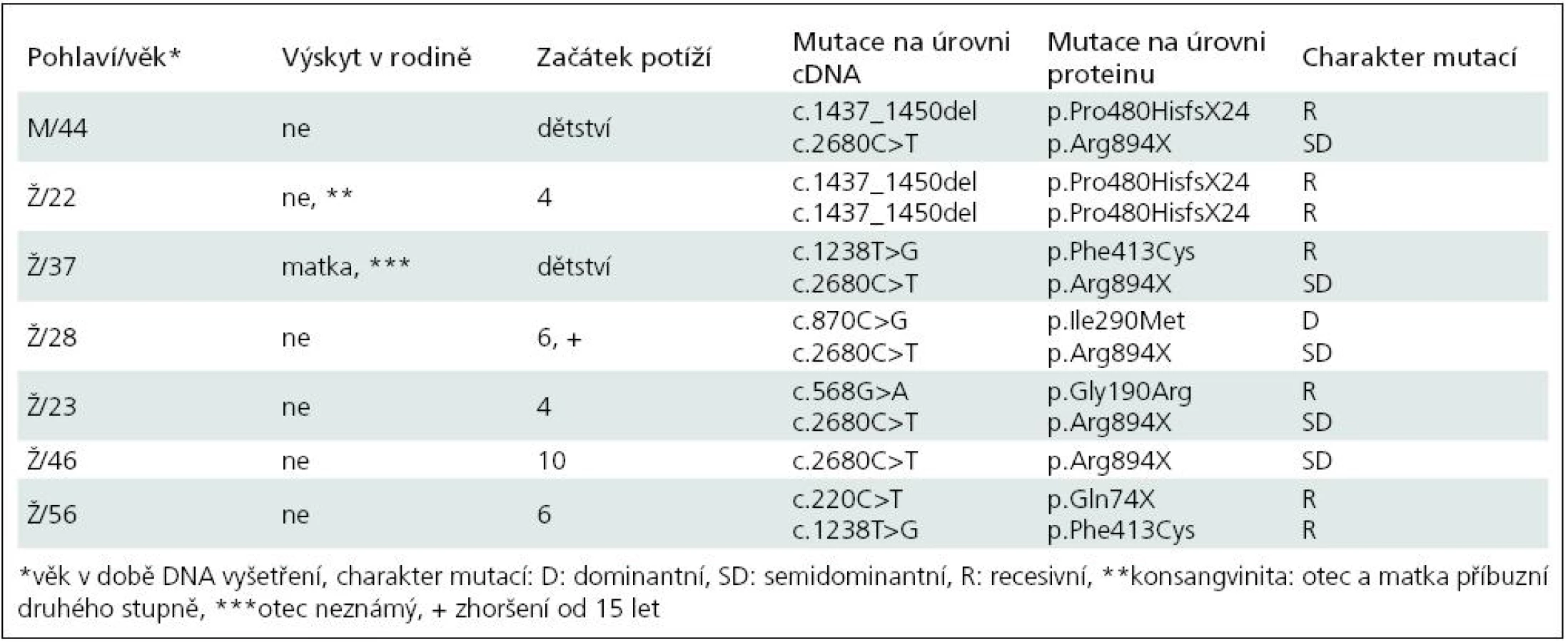
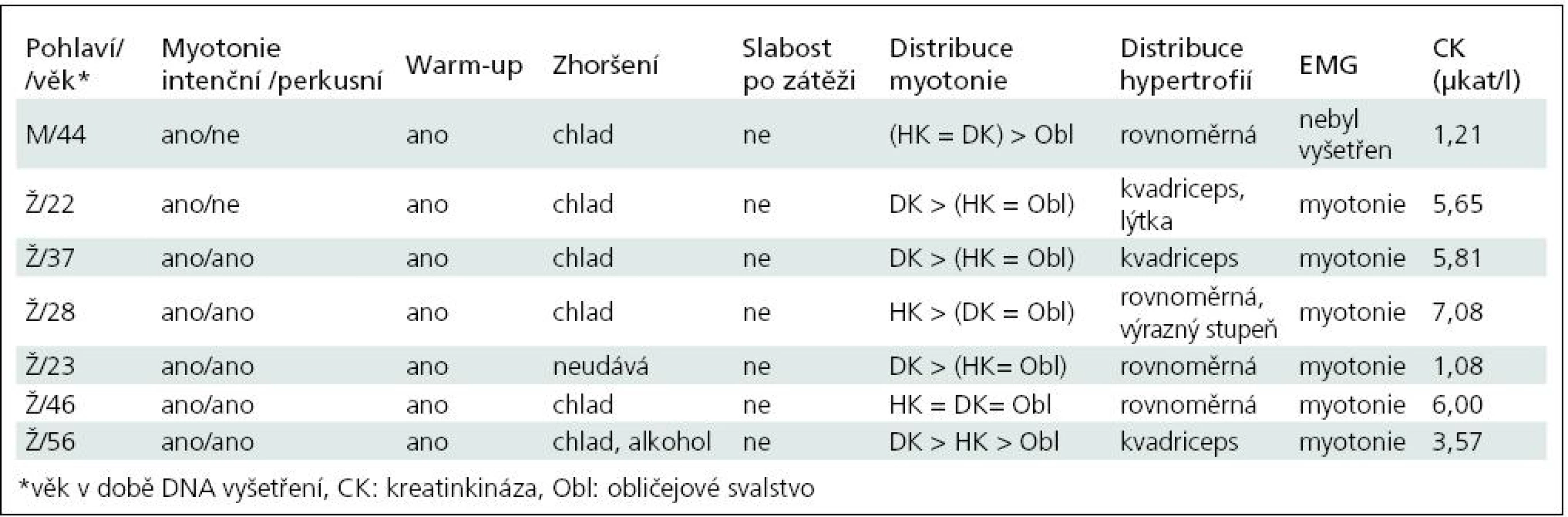
Základní charakteristiku souboru ukazuje tab. 2. Vyplývá z ní, že nejčastěji zachycenou mutací je semidominantní mutace p.Arg894X, která byla nalezena v 5 z 13 mutovaných alel. Třikrát byla zjištěna recesivní mutace p.Pro480HisfsX24 a dvakrát p.Phe413Cys. Klinicky (tab. 3) splňují všichni pacienti představu o kongenitální myotonii: dobře vyvinutá muskulatura, u některých zvláště zřejmá v oblasti stehenního svalstva, myotonie mírnící se opakovanými pohyby. Zřetelná provokace chladem byla nalezena u šesti osob. Ani jednou jsme nezaznamenali pozátěžové parézy. Elektromyograficky byly u všech vyšetřených (6 případů) přítomny myotonické výboje bez změn architektury potenciálů motorických jednotek. Kreatinkináza je lehce zvýšena u pěti osob, nejvíce u pacientky s nejvýraznějším stupněm myotonie.
Diskuze
Dvě osoby (Ž/22, Ž/56) nesou recesivní mutace bez výskytu onemocnění v přímé linii, splňují tedy kritéria typické Beckerovy varianty. Pacientka Ž/22 má na obou alelách genu CLCN1 stejnou recesivní mutaci, která je vysvětlitelná pokrevní příbuzností rodičů. Jsou příbuzní druhého stupně – bratranec a sestřenice. U obou bylo také nosičství mutace prokázáno. V dalších třech případech se jedná o kombinaci recesivní a časté semidominantní mutace p.Arg894X. Ta se v případě pacientů M/44 a Ž/23 chová nepochybně jako recesivní, v posledním případě (Ž/37) jde nejspíše o dominantní přenos. Matka pacientky trpí myotonií. Naopak u pacientky Ž/46 je tato mutace v dominantním postavení, přestože rodiče myotonií netrpí. To ilustruje známou skutečnost, že semidominantní alely mají u příslušníků téže rodiny různé chování [13]. Poslední nemocná (Ž/28) nese pozoruhodnou kombinaci dominantní mutace p.Ile290Met a semidominantní mutace p.Arg894X, přestože nikdo z příbuzných netrpí myotonií. Vhled do tohoto problému by pravděpodobně přineslo vyšetření DNA rodičů, kteří jsou anamnesticky bez potíží (nebyli ale vyšetřeni ani klinicky ani elektrofyziologicky). Sourozence tato mladá žena nemá. Trpí výraznými projevy myotonie (nejvýraznější v celém souboru) a zřetelnými hypertrofiemi. Jde patrně o dávkový efekt heterozygotní dominantní a semidominantní mutace.
Závěr
Vrozená myotonie na podkladě mutace v chloridovém kanálu je třetí nejčastější příčina myotonie v naší populaci. V našem souboru se v převážné většině jedná o recesivní typ dědičnosti, tzv. Beckerovu variantu myotonia congenita. Výskyt má tedy sporadický charakter a rodinná anamnéza nám neposkytuje dostatečné diagnostické vodítko.
Tato práce vznikla díky projektu „CEITEC – Středoevropský technologický institut“ (CZ.1.05/1.1.00/02.0068) z Evropského fondu regionálního rozvoje.
prim.
MUDr. Stanislav Voháňka, CSc.,
MBA
Neurologická
klinika
LF
MU a FN Brno
Jihlavská
20
625
00 Brno
e-mail:
svohanka@fnbrno.cz
Přijato
k recenzi: 31. 1. 2011
Přijato
do tisku: 18. 2. 2011
Zdroje
1. Mindell JA, Maduke M, Miller C, Grigorieff N. Projection structure of a CIC-type chloride channel at 6.5-angstrom resolution. Nature 2001; 409(6817): 219–223.
2. Estevez R, Schroeder BC, Accardi A, Jentsch TJ, Pusch M. Conservation of chloride channel structure revealed by an inhibitor binding site in CIC-1. Neuron 2003; 38(1): 47–59.
3. Wu FF, Ryan A, Devaney J, Warnstedt M, Korade--Mirnics Z, Poser B et al. Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain 2002; 125(11): 2392–2407.
4. Thomsen J. Töniche Krämpfe in willkürlich beweglichen Muskeln in Folge von ererbter psychisscher. Arch Psychiatrie Nervenkranheiten 1876; 6: 702–718.
5. Becker PE. Zür frage der heterogenit der erblichen myotonien. Nervenarzt 1957; 28(10): 455–460.
6. Fialho D, Schorge S, Pucovska U, Davies NP, Labrum R, Haworth A et al. Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain 2007; 130(12): 3265–3274.
7. Mailander V, Heine R, Deymeer F, Lehmann-Horn F. Novel muscle chloride channel mutations and thein effects on heterozygous carriers. Am J Hum Genet 1996; 58(2): 317–324.
8. Plassart-Schiess E, Gervais A, Eymard B, Lagueny A, Pouget J, Warter JM et al. Novel muscle chloride channel (CLCN1) mutations in myotonia congenita with various modes of inheritance including incomplete dominance and penetrance. Neurology 1998; 50(4): 1176–1179.
9. Dunø M, Colding-Jørgensen E, Grunnet M, Jespersen T, Vissing J, Schwartz M. Difference in allelic expression of the CLCN1 gene and the possible influence on the myotonia congenita phenotype. Eur J Hum Genet 2004; 12(9): 738–743.
10. Sun C, Tranebjaerg L, Torbergsen T, Holmgren G, van Ghelue M. Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia. Eur J Hum Genet 2001; 9(12): 903–909.
11. Nagamitsu S, Matsuura T, Khajavi M, Armstrong R, Gooch C, Harati Y et al. A “dystrophic” variant of autosomal recessive myotonia congenita caused by novel mutations in the CLCN1 gene. Neurology 2000; 55(11): 1697–1703.
12. Trip J, Drost G, van Engelen BG, Faber CG. Drug treatment for myotonia. Cochrane Database Syst Rev 2006; 1: CD004762.
13. Colding-Jørgensen E. Phenotypic variability in myotonia congenita. Muscle Nerve 2005; 32(1): 19–34.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie
2011 Číslo 4
Nejčtenější v tomto čísle
- Porucha pozornosti s hyperaktivitou (attention deficit/hyperactivity disorder – ADHD)
- Opožděný akutní subdurální hematom
- Neurologické komplikace při onemocnění herpes zoster – kazuistika
- Vrozená myotonie na podkladě mutací v genu pro chloridový kanál ClC-1