
Myoklonická epilepsie a hluchota u sourozenců s mutací 7512T>C v genu pro mitochondriální tRNASer(UCN) – kazuistiky
Myoclonic Epilepsy and Deafness in Siblings with the 7512T>C Mutation in the Mitochondrial Encoded tRNASer(UCN) Gene – Case Reports
Mitochondrial diseases are a very heterogeneous group of disorders affecting the nervous system, heart and skeletal muscle. The mutations are situated in nuclear DNA with Mendelian heredity and in mitochondrial DNA with maternal-type heredity. We describe a natural clinical course, the results of biochemical and molecular genetic measurements from two siblings with mitochondrial disorders due to mutation 7512T>C in the gene for mitochondrial tRNA for serine. The mutation is associated with myoclonic epilepsy, deafness, ataxia, calcification in the basal ganglia and decreased cytochrome c oxidase activity. The data should prove helpful in the exact diagnosis of patients with this phenotype.
Key words:
mitochondrial disease – myoclonic epilepsy – deafness – calcification in basal ganglia – cytochrom c oxidase deficiency – transfer RNA
Autoři:
P. Ješina 1; K. Vinšová 1; O. Brantová 1; M. Tesařová 1; Z. Hájková 1; H. Hansíková 1; L. Wenchich 1; T. Honzík 1; M. Magner 1; J. Zámečník 2; P. Ryška 3; J. Zeman 1
Působiště autorů:
Klinika dětského a dorostového lékařství UK 1. LF a VFN v Praze
1; Ústav patologie a molekulární medicíny UK 2. LF a FN v Motole, Praha
2; Radiologická klinika LF UK a FN Hradec Králové
3
Vyšlo v časopise:
Cesk Slov Neurol N 2010; 73/106(1): 68-72
Kategorie:
Kazuistika
Souhrn
Mitochondriální choroby je různorodá skupina nemocí postihujících především nervovou soustavu, srdce a skeletální svalstvo. Kauzální mutace mitochondriálních onemocnění jsou přítomny jak v jaderné DNA (mendelovská dědičnost), tak v DNA mitochondriální (maternální dědičnost). Prezentujeme klinický průběh onemocnění spolu s výsledky biochemických a molekulárněgenetických vyšetření u sourozenců s mitochondriální chorobou na podkladě 7512T>C mutace v genu pro mitochondriální tRNA pro serin. Mutace se projevuje myoklonickou epilepsií, hluchotou, ataxií, kalcifikacemi v bazálních gangliích a snížením cytochrom c oxidázy. Informace by měly napomoci ke zpřesnění diagnózy u pacientů s tímto fenotypem.
Klíčová slova:
mitochondriální porucha – myoklonická epilepsie – hluchota – kalcifikace v bazálních gangliích – deficit cytochrom c oxidázy – transferová RNA
Úvod
Mitochondriální choroby představují klinicky, biochemicky a geneticky heterogenní skupinu chorob spojených s postižením systému oxidativní fosforylace (OXPHOS) a pyruvátdehydrogenázy, často označované jako mitochondriální encefalomyopatie. Nervová soustava, zejména mozek, dále pak srdce a skeletální svalstvo, jsou tkáně s vysokými energetickými nároky, čímž představují nejpostiženější systémy. Mitochondriální choroby jsou řazeny mezi nejčastější dědičná metabolická onemocnění, jejich frekvence je v současné době udávána přibližně 1 : 3 500–1 : 4 000. K růstu incidence dochází díky zpřesňujícím diagnostickým metodám a v jejich důsledku včasné a exaktní diagnostice. Jedná se o závažná, nevyléčitelná, často fatální onemocnění, postihující jak dětskou, tak i dospělou populaci. Geneticky jsou mitochondriální choroby značně komplikované, protože patogenní mutace mohou být jak v jaderné DNA (nDNA) s mendelovským typem dědičnosti, tak v DNA mitochondriální (mtDNA) s typem dědičnosti maternálním [1].
V buňkách je mtDNA přítomna v tisících kopií. Podle podílu mutovaných a nemutovaných mtDNA určujeme hladinu heteroplazmie. MtDNA kóduje 13 polypeptidových podjednotek komplexů dýchacího řetězce, dvě ribozomální RNA (rRNA) a 22 transferových RNA (tRNA). Více než 60 % všech bodových mutací v mtDNA postihuje právě tRNA, což představuje více než 100 dosud popsaných patologických mutací. Častěji jsou přítomny mutace v tRNA pro leucin (UUR), izoleucin, lysin a serin (UCN). Tyto mutace jsou obvykle spojeny s neurosenzorickým postižením, zahrnujícím myoklonickou epilepsii, senzorineurální hluchotu a/nebo cerebelární symptomy. Mezi nejznámější patří klinické jednotky známé jako myoklonická epilepsie s „ragged red“ vlákny (MERRF) a mitochondriální encefalopatie s laktátovou acidózou a „stroke‑like“ atakami (MELAS), které jsou nejčastěji zapříčiněny mutacemi v tRNA pro lysin (tRNALys), resp. leucin (tRNALeu(UUR)). Podobné klinické symptomy byly však popsány i u dalších bodových mutací v mtDNA, a to jak v genech pro tRNA, tak i pro podjednotky komplexů dýchacího řetězce [2]. Bylo identifikováno i několik dalších mutací v tRNA pro serin (tRNASer(UCN)). V rodinách nesoucích některé z těchto mutací byly pozorovány syndromové nebo nesyndromové typy hluchoty jako hlavní patofyziologické příznaky [3].
V textu je dokumentován případ první rodiny z České republiky, ve které se podařilo prokázat vzácnou mutaci 7512T>C v genu pro tRNASer(UCN) v mtDNA. Tato mu-tace byla publikována u pacientů s myoklonickou epilepsií, hluchotou, ataxií a kalcifikacemi v bazálních gangliích [4]. Předkládáme souhrnný klinický popis průběhu onemocnění a výsledků biochemického vyšetření funkce OXPHOS včetně molekulárněgenetických analýz u dvou pacientů s touto mutací s cílem rozšířit diferenciálně diagnostickou rozvahu u pacientů s obdobným fenotypem.
Kazuistika 1
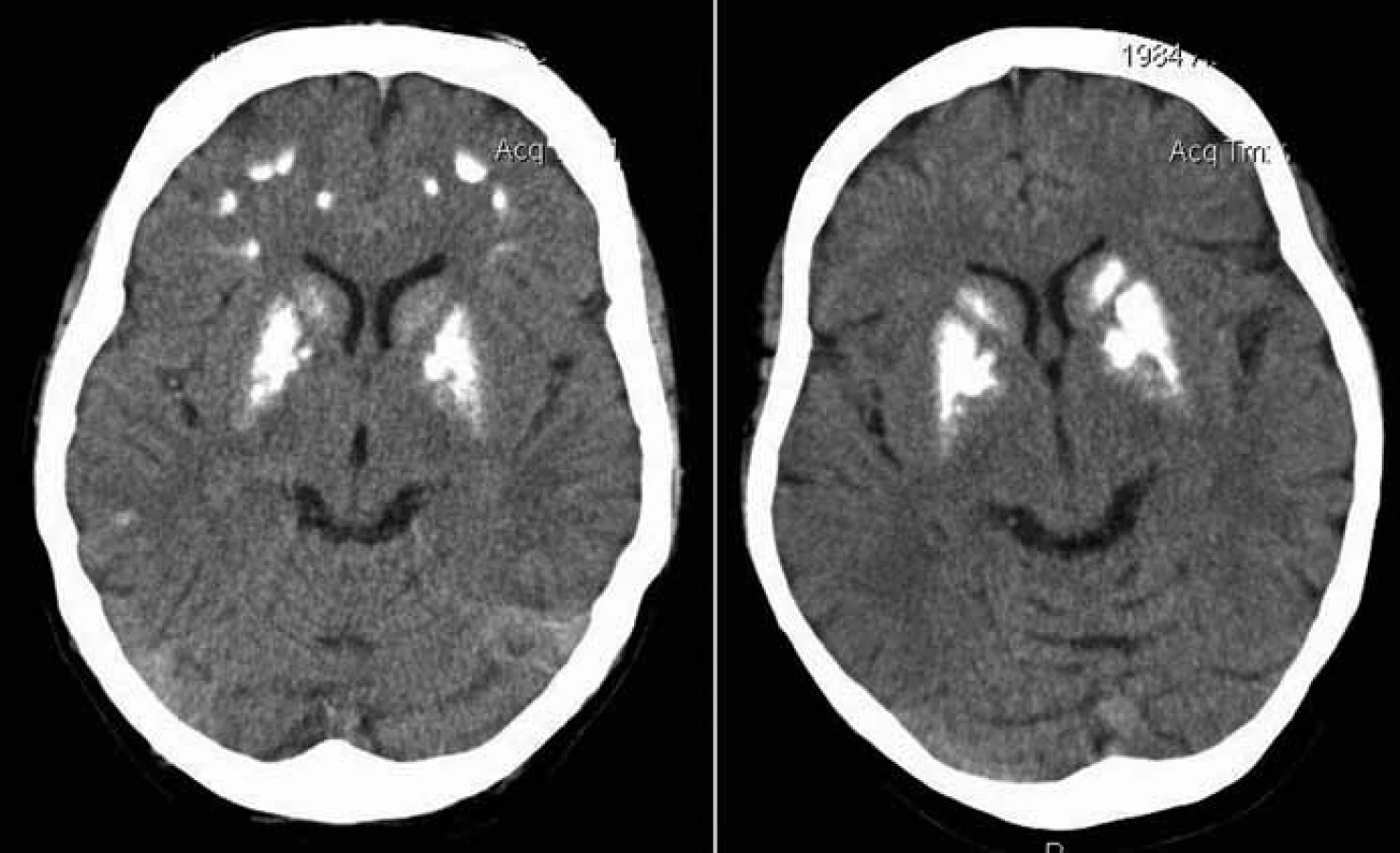
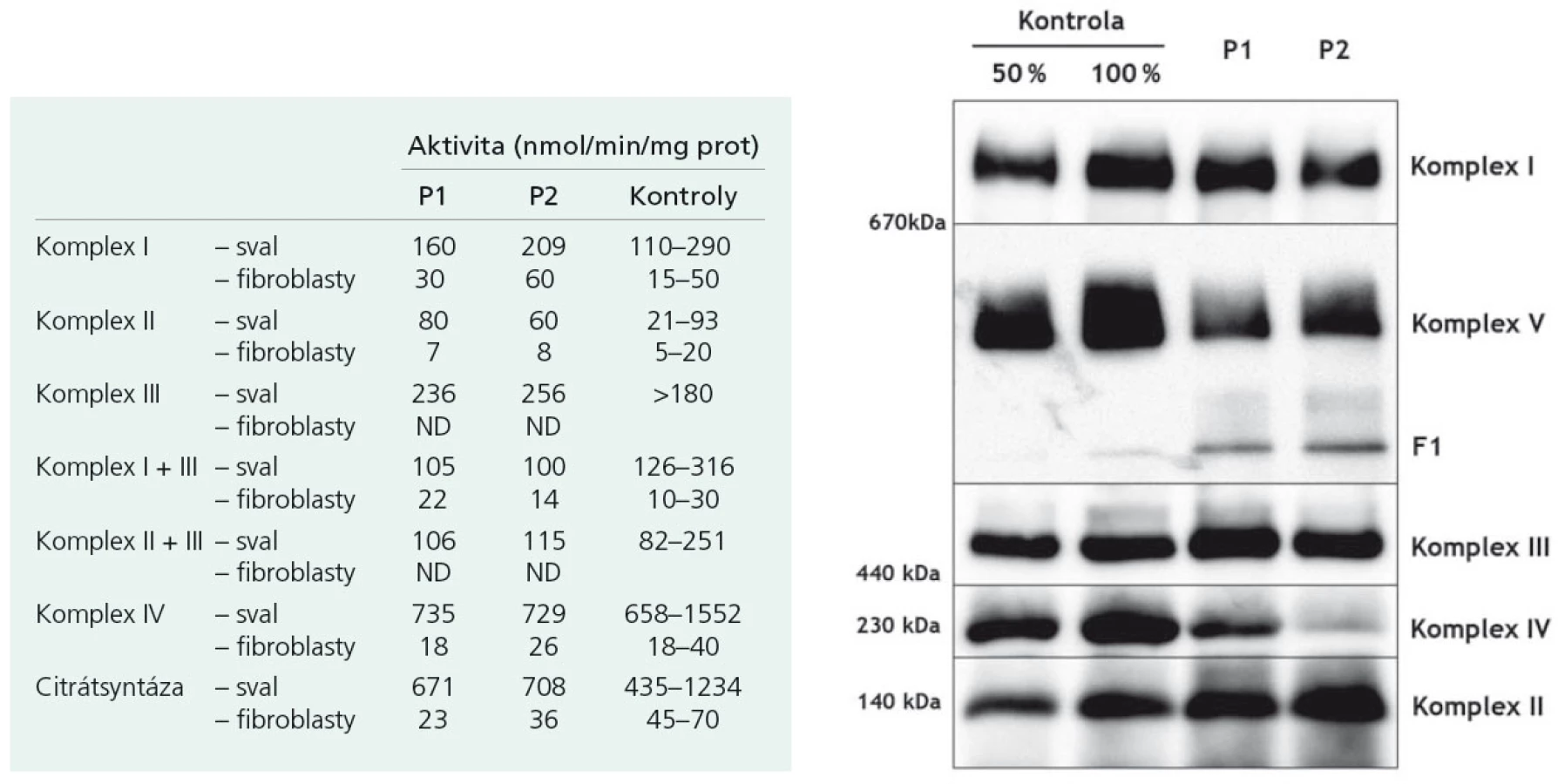
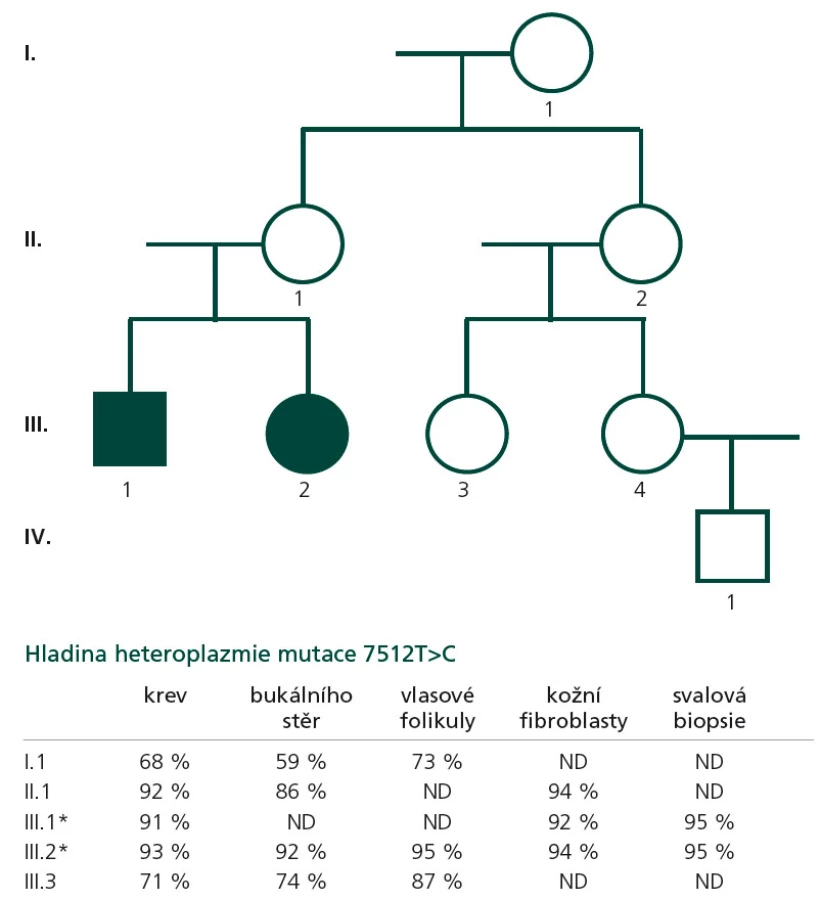
Proband 1 je 29letý muž, starší ze dvou sourozenců s anamnézou myoklonické epilepsie a hluchoty. Rodiče probanda jsou nepříbuzní a zdrávi. Peri- a postnatální data jsou fyziologická. V dětství se projevila porucha sluchu, která je nyní charakterizována jako bilaterální percepční hypakuzie s 84% ztrátou sluchu podle Fowlera. Částečně je korigována pomocí naslouchadel. Ve věku 21 let se objevily první myoklonické záškuby. Generalizované tonicko‑klonické křeče byly pozorovány o dva roky později. Na EEG záznamech byly opakovaně popisovány atypické komplexy hrot-vlna ve fronto-temporo‑parietální oblasti oboustranně. Byla zahájena léčba valproátem, která měla zpočátku dobrý efekt. Pro nakupení záchvatů terapie změněna na lamotrigin (celková denní dávka 150 mg). Nyní jsou myoklonické záškuby končetin pozorovány denně, zejména po ránu. V neurologickém vyšetření je přítomen nález frustního paleocerebelárního syndromu. CT vyšetření mozku prokázalo oboustranné kalcifikace v oblasti nucleus caudatus, putamen a v talamu. Kromě toho byly nalezeny vícečetné hrudkovité kalcifikace v bílé hmotě frontálních a parietálních laloků (obr. 1). Vyšetření očního pozadí ani perimetru neprokázala patologické změny. V základních laboratorních testech dominuje nález elevace CK (max. 49 μkat/l; norma do 3,8), myoglobinu (1900 μg/l; norma do 92) a přechodně zvýšení ALT (max. 1,4 μkat/l; norma do 0,8) a AST (max. 2,1 μkat/l; norma do 0,7). Intermitentně byla zachycena hyperlaktacidurie (max. 157 mM/mol kreatininu; norma do 30) a elevace alaninu v séru (max. 816 μmol/l; norma do 500). Měření enzymové aktivity komplexů dýchacího řetězce ukázalo lehce sníženou aktivitu cytochrom c oxidázy (COX; komplex IV systému OXPHOS) v kožních fibroblastech (obr. 3a). Proto bylo indikováno provedení svalové biopsie, která prokázala snížení jak hladiny aktivity COX (snížení na 20 % kontrolních hodnot), tak i ATP syntázy (30 % kontrolních hodnot). Detekována byla i volná F1-část ATP syntázy. Obsah ostatních komplexů OXPHOS nebyla změněna v porovnání s kontrolou (obr. 3b). Histochemické vyšetření svalové biopsie prokázalo u pacienta přítomnost četných „ragged‑red“ vláken (obr. 2b) a kromě toho byla detekována SDH (sukcinát‑dehydrogenáza, komplex II) pozitivita cév, což jsou nálezy svědčící pro mitochondriální onemocnění. Ve vzorcích byla pozorována vlákna se sníženou aktivitou COX (obr. 2a). Při pátrání po genetické příčině pacientova fenotypu byla provedena molekulárně-genetická analýza v různých tkáních (kožní fibroblasty a sval), která neprokázala větší delece mtDNA, ani prevalentní mutace pro MERRF (8344A>G) a MELAS (3243A>G). Při sekvenování mtDNA byla nalezena mutace 7512T>C v tRNASer(UCN), která byla potvrzena i pomocí PCR-RFLP. V dostupných tkáních (krev, kožní fibroblasty, sval) byla stanovena hladina heteroplazmie (91–95 %) dané mutace (obr. 4).

Kazuistika 2
Proband 2 je 24letá žena, sestra pacienta 1, s normální peri‑ a postnatální anamnézou. Ve věku 5 let podstoupila bez komplikací kardiologický zákrok pro defekt septa síní. Přibližně od 12 let věku začala hůře slyšet a následně byla prokázána oboustranná percepční hypakuzie (ztráta dle Fowlera 66 %), která je nyní korigována naslouchadly. Ve věku 19 let se začaly objevovat první neurologické obtíže – myoklonické záškuby s ranním maximem, pacientce začaly vypadávat předměty z rukou, navíc udávala cefaleu. EEG záznam prokázal četné epizody nepravidelných generalizovaných hrot‑vlna komplexů. Byla zahájena terapie valproátem, po kterém došlo ke zlepšení stavu. V současné době je převáděna na lamotrigin z důvodu negativního účinku valproátu na beta‑oxidaci mastných kyselin v mitochondriích a hladinu karnitinu a pro jeho hepatotoxické účinky. Generalizované záchvaty u této pacientky pozorovány nebyly. V neurologickém nálezu je popisována frustní paleocerebelární symptomatika a divergentní strabizmus. Kardiologické vyšetření (ECHO, EKG) nesvědčí pro kardiomyopatii. Zobrazovací metody (MR a CT) prokázaly výrazné kalcifikace v bazálních gangliích oboustranně v nucleus caudatus a putamen a také v talamu (obr. 1b). Dále byly nalezeny ojedinělé drobné kalcifikace v levé mozečkové hemisféře. V základních laboratorních vyšetřeních je přítomna elevace kreatinkinázy (max. 22 μkat/l; norma do 2,5), myoglobinu (830 μg/l; norma do 76) a transamináz ALT (max. 1,35 μkat/l; norma do 0,8) a AST (max. 2,15 μkat/l; norma do 0,7) v séru. Opakovaně byla nalezena hyperlaktacidurie (max. 2355 mM/mol kreatininu; norma do 30) a elevace alaninu v séru (max. 1 382 μmol/l; norma do 500). Elektroforetická analýza vzorků bioptovaného svalu prokázala obdobný nález jako u bratra. V histologickém nálezu byla potvrzena četná „ragged‑red“ vlákna, přítomnost SDH pozitivity cév a vlákna COX negativní (obr. 2c, d). Obdobně jako v případě jejího staršího bratra byla provedena molekulárněgenetická analýza, která pomocí PCR‑RFLP prokázala mutaci 7512T>C, a byla stanovena hladina heteroplazmie (92–95 %) v dostupných tkáních – krev, kožní fibroblasty a sval (obr. 4).
Diskuze
Představujeme první rodinu v České republice, u které byla jako kauzální příčina onemocnění sekvenací potvrzena mutace (substituce T za C) v mitochondriálním genu pro tRNASer(UCN) na pozici 7512. Ve všech dosud popsaných případech [4] jsou hlavními příznaky tohoto mtDNA defektu myoklonická epilepsie, hluchota, ataxie a kalcifikace v bazálních gangliích.
Mutace 7512T>C narušuje vysoce konzervovaný pár bazí (vazba G-U) v aminoakceptorovém raménku tRNA pro serin. To pravděpodobně vede ke snížení jeho stability a postihuje funkci tRNA [5]. Dochází ke snížení syntézy mitochondriálně kódovaných polypeptidů a následně k defektům respirace v mitochondriích. Podle našich elektroforetických analýz dochází i ke strukturálnímu postižení mitochondriální ATP syntázy. Možným vysvětlením je fakt, že její dvě podjednotky jsou kódovány právě mtDNA. Následkem snížené aktivity COX a strukturálních změn ATP syntázy dochází k poklesu produkce ATP v buňce, a to zejména v energeticky náročných tkáních, mezi které patří mimo jiné i kochleární buňky (vláskové buňky nebo stria vascularis), vedoucí k rozvoji hluchoty [6].
Z genetického hlediska je důležité, že mtDNA je předávána další generaci matkou (maternální dědičnost). Mitochondrie spermie jsou umístěny v bičíku, který neproniká do oocytu, a tím není mtDNA od otce předávána na potomky. Díky mitotické a meiotické segregaci nacházíme v různých tkáních pacientů odlišnou hladinu heteroplazmie. Procesy během segregace mohou vést k situaci, kdy dceřinná buňka může nést více nebo méně mutantních mtDNA než buňka mateřská. Je známo, že ve vertikální linii dochází ke zvyšování heteroplazmie (tedy ke zvyšování počtu buněk s mtDNA nesoucí mutaci). To souvisí s existencí tzv. prahového efektu („thresholdu“), kdy je pro manifestaci dysfunkce nutné, aby byl překročen určitý poměr mitochondrií s mutantní mtDNA (ve většině případů více než 90 %) [7]. Dosud publikované práce zabývající se pacienty s patogenními mutacemi v tRNASer(UCN) uvádějí většinou stupeň heteroplazmie mutované mtDNA vyšší než 95 %. Vyšší „threshold“ se zdá být příčinou fenotypových projevů onemocnění, což u naší rodiny dokumentuje rodokmen s uvedenými stupni heteroplazmie v jednotlivých tkáních jak u probandů, tak u asymptomatických maternálních příbuzných (obr. 4). V průběhu života dochází ke změnám v množství mtDNA molekul v řadě tkání. Tím se také může měnit hladina heteroplazmie mutace. Problémem proto zůstává možnost prenatální diagnostiky mitochondriálních onemocnění způsobenými mutacemi v mtDNA. Zatím je dostupná pouze diagnostika v rodinách s generalizovaným typem onemocnění s mendelovských typem dědičnosti.
Do poruch spojených s „nesyndromovou hluchotou“ je zapojeno více než 50 nukleárních genů. Předchozí studie ukázaly [8], že více než 5 % všech případů hluchoty je zapříčiněno mutacemi v mtDNA (cca > 45 mutací), což představuje jednu z nejvíce frekventovaných genetických příčin hluchoty. Předpokládá se, že funkce buněk kochley jsou více náchylné ke změnám v processingu nebo translaci způsobené mutacemi v mitochondriální tRNA nebo rRNA [9].
Mezi základní projevy mutace 7512T>C patří vedle hluchoty a epilepsie kalcifikace v bazálních gangliích (obr. 1). Řada symptomů spojených s mitochondriálními chorobami (např. Kearns‑Sayerův syndrom; mutace 7480A>G v genu pro tRNASer(UCN)) se obecně projevuje kalcifikacemi v CNS, především v bazálních gangliích [10]. Vedle kalcifikací v bazálních gangliích, které jsou zmiňovány ve všech případech pacientů s touto mutací, jsou uváděny kalcifikace i v jiných částech mozku, zejména v mozečku [8]. V diferenciálně diagnostické rozvaze je třeba vyloučit další příčiny kalcifikací, zejména hypoparatyreózu, případně pseudohypoparatyreózu. Méně pravděpodobně přichází v úvahu renální insuficience, stav po intoxikaci CO, kalcifikace v tumorech nebo po infekcích, manifestující se většinou odlišnými anamnestickými údaji a jiným klinickým průběhem. Mezi další příčiny mohou patřit Cockayneův syndrom, Fahrova choroba (familiární cerebrovaskulární ferrokalcinóza) a Aicardie‑Goutieresův syndrom.
Klinický průběh většinou multisystémového onemocnění s familiárním výskytem a elevací laktátu vede lékaře k podezření na mitochondriální etiologii onemocnění. S rostoucím množstvím nově popsaných mitochondriálních poruch na molekulární bázi se však přesně cílená genetická diagnostika stává komplikovanější. Pacienti s fenotypem myoklonické epilepsie, hluchoty, ataxie, deficience COX a kalcifikacemi v bazálních gangliích, spojené s maternální dědičností, by měli být vyšetřeni na přítomnost mutace v genu pro tRNASer(UCN), především mutaci 7512T>C. Diagnostika mitochondriálních poruch je v České republice dostupná na našem pracovišti, v mitochondriální laboratoři Kliniky dětského a dorostového lékařství 1. LF UK a VFN v Praze.
Seznam zkratek
ATP adenosintrifosfát
COX cytochrom c oxidáza
MELAS mitochondriální encefalopatie s laktátovou acidózou a stroke‑like ataky
MERRF myoklonická epilepsie s „ragged red“ vlákny
mtDNA mitochondriální DNA
nDNA nukleární DNA
OXPHOS systém oxidativní fosforylace
PCR polymerázová řetězová reakce
RFLP polymorfizmus v délce restrikčních fragmentů
rRNA ribozomální RNA
tRNA transferová RNA
Poděkování
Práce vznikla za podpory grantu Interní grantové agentury Ministerstva zdravotnictví ČR IGA MZČR NS 9782-4.
Přijato k recenzi: 18. 5. 2009
Přijato do tisku: 5. 10. 2009
MUDr. RNDr. Pavel Ješina, Ph.D.
Klinika dětského a dorostového lékařství UK 1. LF a VFN v Praze
Ke Karlovu 2
128 08 Praha 2
e-mail: paveljesina@seznam.cz
Zdroje
1. DiMauro S. Mitochondrial medicine. Biochim Biophys Acta 2004; 1659(2–3): 107–114.
2. Schoffner JM, Lott MT, Lezza AMS, Siebel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 1990; 61(6): 931–937.
3. Toompuu M, Levinger LL, Nadal A, Gomez J, Jacobs HT. The 7472insC mtDNA mutation impairs 5’ and 3’ processing of tRNASer(UCN). Biochem Biophys Res Commun 2004; 322 (3): 803–813.
4. Nakanuta M, Nakano S, Goto Y, Ozawa M, Nagahama Y, Fukuyama H et al. A novel point mutation in the mitochondrial tRNASer(UCN) gene detected in a family with MERRF/MELAS overlap syndrome. Biochem Biophys Res Commun 1995; 214(1): 86–93.
5. Chomyn A, Enriguez JA, Micol V, Fernandez‑Silva P,Attardi G. The mitochondrial myopathy, encephalopathy, lactic acidosis and stroke‑like episode syndrome‑associated human mitochondrial tRNALeu(UCN) mutation cause aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J Biol Chem 2000; 275(25): 19198–19209.
6. Guan MX, Enriguez JA, Fischer-Ghodsian N, Puranam R, Lin CP, Marion MA et al. The deafness‑associated mTDNA 7445 mutation, which affects tRNASer(UCN) precursor processing, has long‑range effects on NADH dehydrogenase ND6 subunit gene expression. Mol Cell Biol 1998; 18(10): 5668–5879.
7. Goto YI, Nonaka I, Horai S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalopathies. Nature 1990; 348(6302): 651–653.
8. Jacobs HT, Hutchin TP, Käppi T, Gillies G, Minkkinen K, Walker J et al. Mitochondrial DNA mutations in patients with postlingual, nonsyndromic hearing impairment. Eur J Hum Genet 2005; 13(1): 26–33.
9. Kokotas H, Petersen MB, Willems PJ. Mitochondrial deafness. Clin Genet 2007; 71(5): 379–391.
10. Muñoz A, Mates F, Simon R, Garcia‑Silva MT, Cabello S, Arenas J. Mitochondrial diseases in children: neuroradiological and clinical features in 17 patients. Neuroradiology 1999; 41(12): 920–928.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2010 Číslo 1
Nejčtenější v tomto čísle
- Mitochondriální encefalomyopatie na podkladě deficitu proteinu Sco2 s obrazem SMA‑like neurogenní svalové atrofie – kazuistiky
- Vyšetření čichu u neurologických onemocnění pomocí Testu parfémovaných fixů
- Kongenitální myastenické syndromy – kazuistiky
- Evokované odpovědi a elektromyografie v intraoperační monitoraci v neurochirurgii