
Mitochondriální encefalomyopatie na podkladě deficitu proteinu Sco2 s obrazem SMA‑like neurogenní svalové atrofie – kazuistiky
Sco2 Protein Deficiency-Based Mitochondrial Encephalomyopathy with the SMA‑like Picture of Neurogenic Muscle Atrophy – Case Reports
Various mitochondrial diseases with clinical, electromyographical and histological signs imitating spinal muscular atrophy (“SMA-like diseases”) with no detected mutation in SMN1 gene have been described in recent years. Respiratory chain disorder was considered due to broad impairment of the central and peripheral nervous systems (encephalopathy, neuropathy) and other tissue and organ involvement (cardiomyopathy, myopathy, hepatopathy). The number of previously-reported mitochondrial syndromes with “SMA-like” sign development is limited. Revealing the phenotype in a detailed clinical context may lead to targeted molecular-genetic analysis and specific diagnostics without calling upon other investigations. A typical representative of this syndrome group is mitochondrial encephalopathy with cardiomyopathy and lactic acidosis arising out of a mutation in gene coding protein Sco2 protein, which is involved in the assembly of mitochondrial cytochrome c oxidase. Two case histories are presented, of patients in whom SMA-like phenotype development led to SCO2 gene investigation and correct diagnosis.
Key words:
mitochondrial encephalopathy – neurogenic muscle atrophy – spinal muscular atrophy – SMA – cytochrome c oxidase – SCO2 gene
Autoři:
M. Magner 1; K. Veselá 1; T. Honzík 1; P. Ješina 1; V. Vobruba 1; B. Petrák 2; J. Zeman 1; P. Klement 1
Působiště autorů:
Klinika dětského a dorostového lékařství UK 1. LF a VFN v Praze
1; Klinika dětské neurologie UK 2. LF a FN v Motole, Praha
2
Vyšlo v časopise:
Cesk Slov Neurol N 2010; 73/106(1): 73-75
Kategorie:
Kazuistika
Souhrn
V posledních letech byly popsány mitochondriální onemocnění s klinickými, elektromyografickými a histologickými projevy napodobujícími spinální svalovou atrofii („SMA‑like onemocnění“), u kterých však nebyla zjištěna mutace SMN 1 genu. Diagnóza mitochondriálního onemocnění byla zvažována vzhledem k širšímu postižení centrální a periferní nervové soustavy (encefalopatie, neuropatie) a dalších orgánů a tkání (kardiomyopatie, myopatie, hepatopatie). Počet známých mitochondriálních syndromů s rozvojem „SMA‑like“ příznaků je omezený. Zjištění tohoto fenotypu v širším klinickém kontextu může vést k cílené molekulárně genetické analýze a specifické diagnóze bez potřeby četných zatěžujících vyšetření. Typickým představitelem této skupiny chorob je mitochondriální encefalomyopatie s kardiomyopatií a laktátovou acidózou, způsobená mutací v genu pro asemblační protein Sco2, která vede k poruše syntézy a funkce mitochondriální cytochrom c oxidázy. Předkládáme dvě kazuistiky pacientů s tímto deficitem, u kterých rozvoj SMA‑like fenotypu vedl k cílenému vyšetření SCO2 genu a správné diagnóze.
Klíčová slova:
mitochondriální encefalopatie – neurogenní svalová atrofie – spinální muskulární atrofie – SMA – porucha cytochrom c oxidázy – SCO2 gen
Úvod
Mitochondriální onemocnění tvoří širokou skupinu chorob, které postihují především orgány s vysokými energetickými nároky (CNS, svaly, myokard) [1]. K nejčastějším onemocněním z této skupiny patří poruchy funkce nebo biogeneze mitochondriální cytochrom c oxidázy (COX; komplex IV dýchacího řetězce). COX je složena ze 13 proteinových podjednotek; tři tvoří katalytické jádro enzymu, ostatní se podílejí na regulaci aktivity a asemblaci enzymu. Biogeneze enzymu je komplexní proces, který probíhá v několika krocích a vyžaduje přes 30 dalších proteinů [2]. Jedním z těchto proteinů je metalochaperon mědi – protein Sco2, který je lokalizován na vnitřní mitochondriální membráně [3].
Onemocnění s deficitem Sco2 se klinicky projevuje většinou jako encefalomyopatie, neuropatie, oftalmoplegie a hypertrofická kardiomyopatie [4,5]. V roce 2004 byly u několika pacientů popsány klinické, EMG i histologické známky typické pro spinální muskulární atrofii [6]. Naše práce přináší klinická a laboratorní data dvou dosud nepublikovaných pacientů s deficitem Sco2, u nichž vedl rozvoj neurogenní svalové atrofie k cílenému molekulárně genetickému vyšetření a diagnóze. Cílem naší práce je zdůraznit potřebu rozšíření diferenciální diagnostiky hypotonie a neurogenní svalové atrofie v kojeneckém věku o toto autozomálně recesivně dědičné mitochondriální onemocnění.
Kazuistika 1
Dívka se narodila ze druhé gravidity zdravých nepříbuzných rodičů. Prenatální a perinatální anamnéza byla bez rizik, vývoj dívky během prvních dvou měsíců probíhal bez pozoruhodností. Od druhého měsíce věku byly pozorovány epizody snížené motorické aktivity až apatie a atypické pohyby očních bulbů charakteru okulogyrní krize. První neurologické vyšetření dítěte proběhlo v 8. měsíci věku pro opoždění vývoje vzpřimování. Byl zjištěn hypertonický syndrom, hyperreflexie, mikrocefalie, mírná kraniofaciální dysmorfie, ptóza. Od 9. měsíce věku došlo během dvou měsíců k regresi psychomotorického vývoje na úroveň patologického novorozence, progredovala ptóza, rozvíjel se bulbární syndrom, objevily se epizody centrální hypoventilace s nutností trvalé umělé plicní ventilace. V 10 měsících věku byly přechodně pozorovány epizody dystonie. Generalizované tonické křeče, zaznamenané v 11. měsíci věku, ustoupily na terapii fenobarbitalem a midazolamem. Od 12. měsíce výrazně progredovala hypotonie kořenového a axiálního svalstva, svalová slabost a atrofie, hyporeflexie; projevy spasticity byly omezeny na akra dolních končetin (tonus, extenční odpověď plantárního reflexu). Od 13 měsíců věku byla dívka bez sociálního kontaktu, těžce hypotonická, bez spontánních motorických projevů, s areflexií a absencí kmenových reflexů.
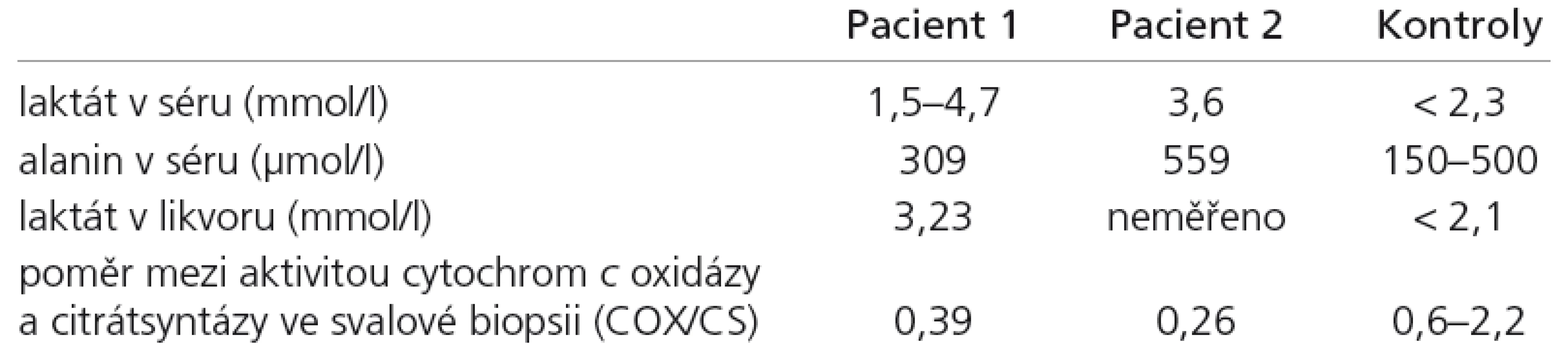
Laboratorní vyšetření ukázala normální nebo zvýšenou hladinu laktátu v krvi, v likvoru a v moči (tab. 1). Intermitentní metabolickou acidózu a laboratorní známky hepatopatie (ALT 4,06 μkat/l; norma < 0,85; AST 0,95 μkat/l; norma < 0,97).
V 10. a 13. měsíci věku nebyly u pacientky patrné klinické ani echokardiografické známky hypertrofické kardiomyopatie.
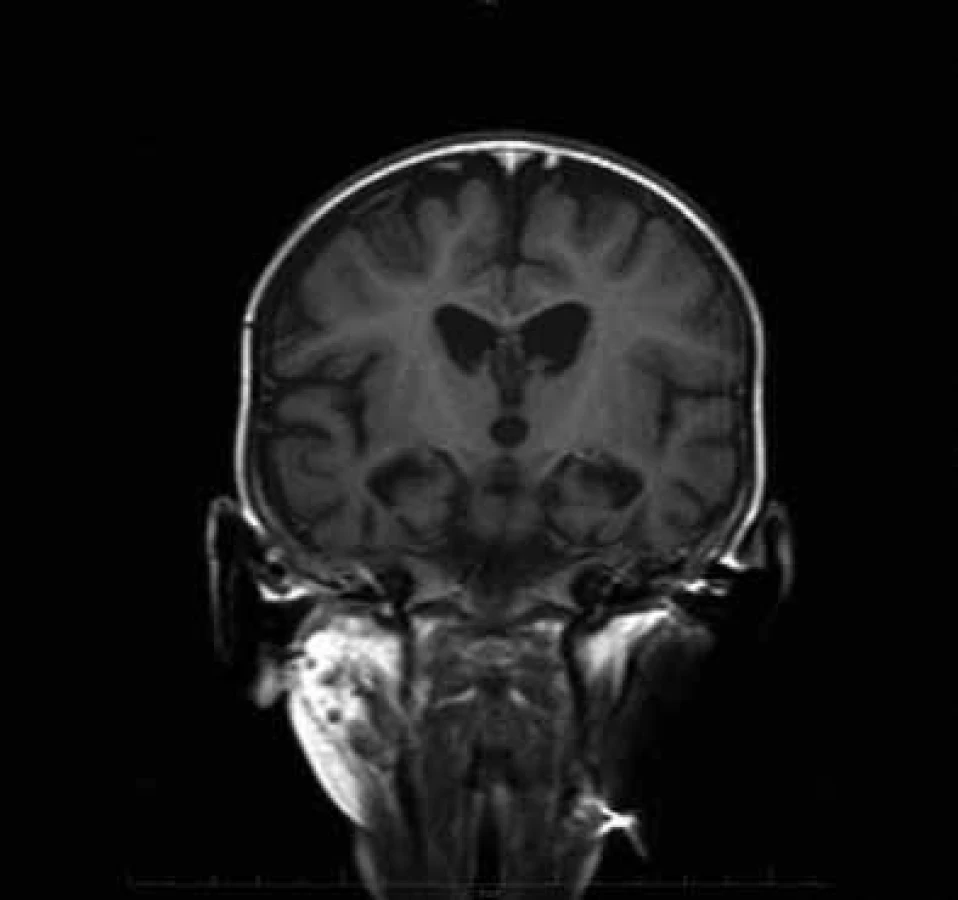
CT a MR vyšetření mozku v 10. měsíci věku ukázala difuzní atrofii CNS. Na kontrolním MR ve 13. měsíci věku byla kromě atrofie i změny odpovídající nálezu u Leighova syndromu (stranově symetrická ložiska zvýšeného signálu v oblasti dorzální části putamen, substancia nigra a v okolí akveduktu, obr. 1). Elektroencefalografické vyšetření ve 12 měsících prokázalo nízkovoltážní nediferencovanou aktivitu. Elektromyografické vyšetření prokázalo výrazně snížený počet činných motorických jednotek a známky myogenní léze. Molekulárně genetické vyšetření spinální muskulární atrofie I. typu, myotonické dystrofie, prevalentních bodových mutací v mtDNA pro syndromy MELAS, MERRF, NARP byly negativní. Enzymologické vyšetření neprokázalo deficit pyruvát dehydrogenázy a Pompeho nemoc. Svalová biopsie ukázala známky neurogenní atrofie svalu s fenoménem mozaikovité atrofie vláken II. typu při hypertrofii vláken I. typu. Histochemické vyšetření SDH a COX nejevilo významné odchylky od kontrol. Enzymologické vyšetření svalu svědčilo o snížené aktivitě komplexů dýchacího řetězce IV s patologickým poměrem mezi cytochrom c oxidázou a kontrolním enzymem citrátsyntázou (COX/CS) (tab. 1). Cílené molekulárně genetické vyšetření SCO2 genu ukázalo mutaci 1541G>A v homozygotním stavu.

Kazuistika 2
Dívka s nezávažnou rodinnou anamnézou se narodila ve 34. gestačním týdnu císařským řezem indikovaným pro nález placenta previa. Bezprostřední poporodní průběh byl nekomplikovaný, ale již ve věku šesti týdnů byla vyšetřována pro chudou motorickou aktivitu, laryngeální stridor a těžký hypotonický syndrom s hyporeflexií. Vzhledem k opakovaným epizodám tonických a klonických křečí byla zahájena léčba fenobarbitalem. Pro hrozící rozvoj ventilační insuficience byla indikována tracheostomie a umělá plicní ventilace. Stav byl komplikován aspirační pneumonií a rozvojem sepse a dívka ve třech měsících věku zemřela.
Byla zjištěna elevace laktátu a alaninu v séru (tab. 1). Sonografie CNS ukázala počínající kortikální atrofii. Echokardiograficky byla diagnostikována progredující hypertrofická kardiomyopatie. Váha srdce při autoptickém vyšetření byla 65g (norma 26g). Elektronmikroskopické vyšetření periferního nervu (n. suralisl. sin. a n. tibialis ant.l. sin) ukázalo axonální degeneraci, demyelinizaci a známky reinervace (axonal sprouting). EMG i histologický nález ve svalu svědčily pro neurogenní svalovou atrofii.
Enzymologické vyšetření biopsie svalu ukázalo snížené aktivity COX, aktivity ostatních komplexů a citrátsyntázy byly v normě. Pompeho nemoc byla enzymologicky vyloučena. Molekulárně genetické studie nepotvrdily delece exonu 6 a 7 genu SMN1. Nebyly zjištěny běžné bodové mutace, delece ani deplece mitochondriální DNA. Molekulárně genetické vyšetření SCO2 genu odhalilo heterozygotní stav pro již výše uvedenou mutaci 1541G>A a další dosud nepopsanou mutaci 1518delA.
Diskuze
Mitochondriální encefalomyopatie představují diagnosticky problematickou skupinu onemocnění s multiorgánovými projevy a variabilním průběhem. Onemocnění způsobená mutací v SCO2 genu jsou typickým příkladem. Klinický průběh je variabilní, typická je různá kombinace příznaků onemocnění CNS, PNS a dalších orgánů, a variabilní věk jejich manifestace. Výsledky běžných laboratorních analýz nejsou specifické, laktátová acidóza nemusí být vždy přítomna, nebo může být jen mírného stupně. Enzymologické analýzy komplexů oxidativní fosforylace ze svalové biopsie nebo z tkáňoavých kultur kožních fibroblastů často prokáží pouze mírné snížení aktivity mitochondriální cytochrom c oxidázy. Specifická diagnostika je proto závislá na cílené indikaci genetického vyšetření SCO2 genu.
Kromě SCO2 genu může být fenotyp „SMA‑like“ onemocnění ještě projevem mitochondriální encefalopatie s deficitem mitochondriální tymidinkinázy 2 a mitochondriální DNA deplece [7]. Ostatní známé mitochondriální choroby s neurogenní svalovou atrofií mají výrazně odlišný klinický průběh (autozomálně dominantní optická atrofie, Mohr-Tranebjaerg syndrom [8] nebo syndrom CPEO (chronická progresivní externí oftalmoplegie).
Průběh onemocnění a laboratorní výsledky u našich dvou pacientek jsou ve shodě s publikovanými případy Sco2 deficitu [4,6,9]. Vodítkem k diagnóze se ukázaly být klinické, EMG a histologické známky progredující neurogenní svalové atrofie. Na jejich základě bylo indikováno molekulárně genetické vyšetření SCO2 genu, které potvrdilo diagnózu. Genetickou analýzou SCO2 genu jsme ukázali, že druhá pacientka je složený heterozygot pro známou mutaci 1541G>A a dosud nepopsanou mutaci 1518delA.
Protože specifická léčba mitochondriálních onemocnění není známa, sehrává molekulárně genetické vyšetření nezastupitelnou roli v genetickém poradenství v postižených rodinách. Zjištění mutace v případě poruch nukleárně kódovaných proteinů umožňuje provedení prenatální diagnostiky. Správná a cíleně volená indikace molekulárně genetického vyšetření mitochondriálních onemocnění je však problematická pro stále se zvětšující počet nově identifikovaných poruch asemblačních, transportních a strukturálních mitochondriálních proteinů při zatím minimálním počtu popsaných pacientů. Proto nalezení kombinace specifických klinických příznaků, jako je spojení encefalopatie, kardiomyopatie a neurogenní svalové atrofie v případě deficitu Sco2 proteinu, může přispět k cílené indikaci molekulárně genetického vyšetření, a tím i k urychlení a zpřesnění diagnózy.
Seznam zkratek
COX cytochrom c oxidáza, komplex IV dýchacího řetězce
CPEO chronická progresivní externí oftalmoplegie
CT počítačová tomografie
EEG elektroencefalografie
EMG elektromyografie
MELAS mitochondriální encefalopatie s laktátovou acidózou a iktu podobnými příhodami
MERRF myoklonická epilepsie s ragged-red vlákny
MR magnetická rezonance
mtDNA mitochondriální DNA
NARP neuropatie, ataxie, retinitis pigmentóza
SCO2 „Synthesis of cytochrome c oxidace“ gen
SMN „Survival motor neurone“ gen
Podpořeno GAČR 303-0707-81 a IGA NS 9782-4.
Přijato k recenzi: 31. 3. 2009
Přijato do tisku: 15. 12. 2009
MUDr. Petr Klement, Ph.D.
Klinika dětské neurologie UK 2. LF a FN v Motole, Praha
Ke Karlovu 2
128 08 Praha 2
e-mail: petrklement@seznam.cz
Zdroje
1. DiMauro S, Schon EA. Mitochondrial respiratory‑chain diseases. N Engl J Med 2003; 348(26): 2656–2568.
2. Barrientos A, Barros MH, Valnot I, Rötig A, Rustin P, Tzagoloff A. Cytochrome oxidase in health and disease. Gene 2002; 286(1): 53–63.
3. Stiburek L, Hansikova H, Tesarova M, Cerna L, Zeman J. Biogenesis of eukaryotic cytochrome c oxidase. Physiol Res 2006; 55 (Suppl 2): 27–41.
4. Jaksch M, Horvath R, Horn N, Auer DP, Macmillan C, Peters J et al. Homozygosity (E140K) in SCO2 causes delayed infantile onset of cardiomyopathy and neuropathy. Neurology 2001; 57(8): 1440–1446.
5. Vesela K, Hansikova H, Tesarova M, Martasek P, Elleder M, Houstek J et al. Clinical, biochemical and molecular analyses of six patients with isolated cytochrome c oxidase deficiency due to mutations in the SCO2 gene. Acta Paediatr 2004; 93(10): 1312–1317.
6. Tarnopolsky MA, Bourgeois JM, Fu MH, Kataeva G,Shah J, Simon DK et al. Novel SCO2 mutation (G1521A) presenting as a spinal muscular atrophy type I phenotype. Am J Med Genet A 2004; 125(3): 310–314.
7. Galbiati S, Bordoni A, Papadimitriou D, Toscano A,Rodolico C, Katsarou E et al. New mutations in TK2 gene associated with mitochondrial DNA depletion. Pediatric Neurol 2006; 34(3): 177–185.
8. Binder J, Hofmann S, Kreisel S, Wöhrle JC, Bäzner H, Krauss JK et al. Clinical and molecular findings in a patient with a novel mutation in the deafness-dystonia peptide (DDP1) gene. Brain 2003; 126(8):1814–1820.
9. Salviati L, Sacconi S, Rasalan MM, Kronn DF, Braun A,Canoll P at al. Cytochrome c oxidase deficiency due to a novel SCO2 mutation mimics Werdnig-Hoffmann disease. Arch Neurol 2003; 60(5): 749.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2010 Číslo 1
Nejčtenější v tomto čísle
- Mitochondriální encefalomyopatie na podkladě deficitu proteinu Sco2 s obrazem SMA‑like neurogenní svalové atrofie – kazuistiky
- Vyšetření čichu u neurologických onemocnění pomocí Testu parfémovaných fixů
- Kongenitální myastenické syndromy – kazuistiky
- Evokované odpovědi a elektromyografie v intraoperační monitoraci v neurochirurgii