
Kongenitální myastenické syndromy – kazuistiky
Congenital Myasthenic Syndromes – Case Reports
Congenital myasthenic syndromes started to be recognised and categorised in the 1970s, when they were first distinguished from myasthenia gravis. They are based on a genetic disorders affecting any of the proteins taking part in neuromuscular transmission. They comprise a clinically and genetically heterogeneous group of disorders, largely of autosomal recessive mode of inheritance. Fatigable weakness of skeletal muscles, also involving ocular and bulbar muscles, often manifests as early as in neonates and infants in the form of feeding difficulties with poor suck and swallowing, weak cry, ptosis, ophthalmoplegia, hypotonia, weakness and delayed motor development. The symptoms may fluctuate: crises with life-threatening acute respiratory failure are typical of some forms. Other forms may manifest as arthrogryposis or myopathy. Rarer are mild cases with onset later in childhood or in adolescence. Patients are often misdiagnosed with congenital or metabolic myopathy, muscle dystrophy or classical myasthenia. What is usually a good response to symptomatic therapy significantly changes the prognosis of the disease. In many instances, molecular genetic diagnosis is possible and prenatal testing is available. Most common in the Czech Republic is a mutation of the epsilon subunit of the acetylcholine receptor in patients of Roma ethnic origin. However, the range of congenital myasthenic syndromes is much wider. We present an overview, illustrated by case reports of the most frequent types.
Key words:
congenital myasthenic syndromes – respiratory failure – neuromuscular transmission disorders – muscle hypotonia – muscle weakness – fatigue – ptosis – ophthalmoplegia
Autoři:
M. Adamovičova 1,2; D. Šišková 2
Působiště autorů:
Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
1; Oddělení dětské neurologie, pracoviště IPVZ, Fakultní Thomayerova nemocnice s poliklinikou, Praha
2
Vyšlo v časopise:
Cesk Slov Neurol N 2010; 73/106(1): 62-67
Kategorie:
Kazuistika
Souhrn
Kongenitální myastenické syndromy jsou postupně rozpoznávány od 70. let 20. století, kdy byly poprvé odlišeny od myasthenia gravis. Jejich podkladem je vrozená porucha některé z bílkovin podílejících se na nervosvalovém přenosu. Jedná se o klinicky i geneticky heterogenní skupinu onemocnění s převážně autozomálně recesivní dědičností. Únavová slabost kosterního svalstva postihující i okohybné a bulbární svalstvo se projevuje většinou již v novorozeneckém či kojeneckém věku jako porucha sání a polykání, slabý pláč, ptóza, oftalmoplegie, hypotonie, svalová slabost a opoždění motorického vývoje. Příznaky mohou kolísat, pro některé jednotky jsou typické krize s život ohrožujícím akutním respiračním selháním. Jiné jednotky se mohou projevit artrogrypózou či myopatií. Vzácnější jsou případy s mírnými projevy a začátkem až později v dětství či v adolescenci. Pacienti často správné diagnóze unikají, mohou být mylně vedeni jako kongenitální či metabolická myopatie, svalová dystrofie nebo klasická myastenie. Obvykle dobrá odpověď na symptomatickou léčbu výrazně mění prognózu onemocnění. Často je možná diagnóza na molekulárně genetické úrovni a prenatální diagnostika. V ČR se nejčastěji vyskytuje mutace epsilon podjednotky acetylcholinového receptoru u pacientů romského etnika, spektrum kongenitálních myastenických syndromů je však mnohem širší. Uvádíme jejich přehled, ilustrovaný kazuistikami nejčastějších typů.
Klíčová slova:
kongenitální myastenické syndromy – respirační selhání – nervosvalový přenos – hypotonický kojenec – svalová slabost – únavnost – ptóza – oftalmoplegie
Úvod
Kongenitální myastenické syndromy (CMS) jsou známy od 70. let 20. století, kdy byly odlišeny od klasického autoimunního mya-stenického syndromu. Liší se výskytem příznaků od raného dětství, rodinná anamnéza může být pozitivní. Postižena je okulomotorika, bulbární a mimické svalstvo a v různé míře i další svalové skupiny. Klinické projevy jsou variabilní, pro některé CMS je typický výskyt život ohrožujících krizí s respiračním selháním.
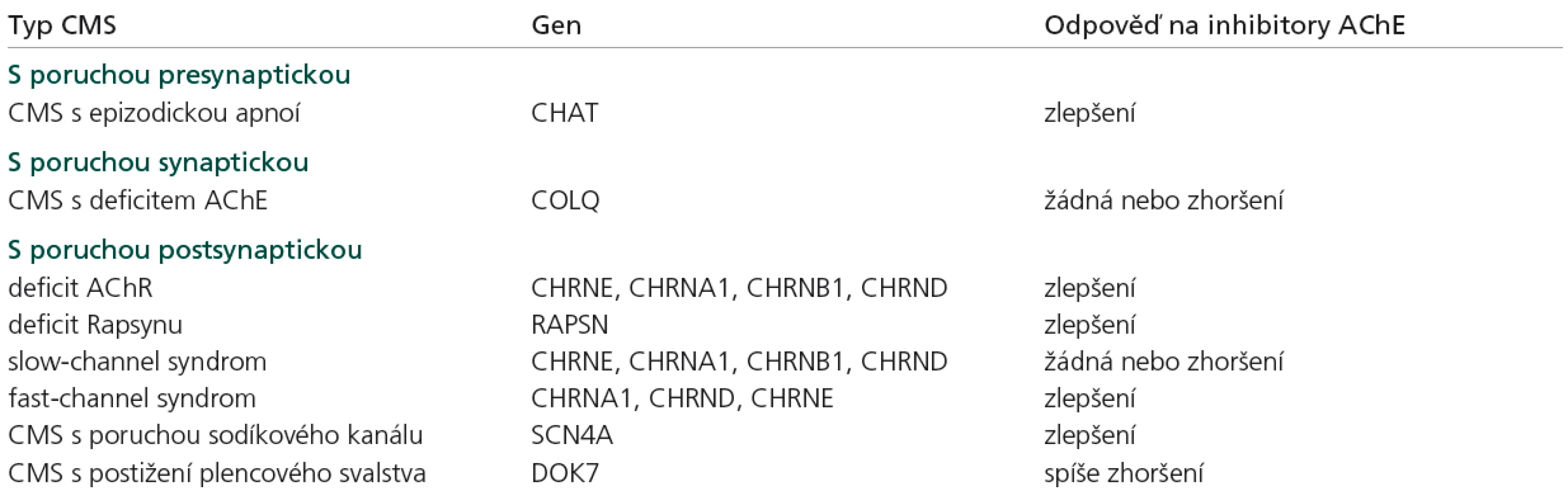
Zatímco klasická myasthenia gravis má autoimunní podklad, u kongenitálních myastenických syndromů se jedná o vrozenou poruchu některé z bílkovin, které se na nervosvalovém přenosu podílejí, ať už na úrovni presynaptické, synaptické či postsynaptické. Postupné odlišení různých jednotek umožnily nejprve klinické, ultrastrukturální a in vivo mikroelektrodové studie a po rozpoznání struktury jednotlivých bílkovin byly postupně objevovány mutace jejich kódujících genů. Dosud byly odhaleny mutace v 12 genech, které způsobují CMS (CHAT, COLQ, CHRNA1, CHRNB1, CHRND, CHRNE, RAPSN, MUSK, DOK7, SCN4A, AGRN, LAMB2) [1–3,19,24] (tab. 1), u řady dalších CMS není dosud molekulární příčina objasněna. Dědičnost je převážně AR, pouze některé „slow-channel“ syndromy jsou dědičné AD. Klinické projevy jsou velmi variabilní, u jednotlivých genů je známa řada mutací, většinou není korelace genotyp‑fenotyp. Na EMG lze při repetitivní stimulaci vybavit dekrement, neplatí to ale ve všech případech. Syntostigminový test je obvykle pozitivní, jsou však i typy CMS, u kterých podání syntostigminu svalovou slabost paradoxně zhorší (COLQ, slow-channel syndrom). Protilátky proti AchR, MuSK a P/Q typu Ca kanálů jsou negativní, hodnoty CK jsou v normě či lehce zvýšené. Postižení je omezeno na kosterní svalstvo. Hladké ani srdeční svalstvo postiženo není. Intelekt, koordinace, čití a šlachosvalové reflexy jsou v normě.
Manifestace CMS je obvykle časná, většinou do dvou let věku, výjimečně později v dětství či adolescenci. Mohou být chudé pohyby plodu, u novorozence slabý pláč, problémy se sáním a polykáním, mající za následek i opakované aspirace, dále ptóza, zevní oftalmoplegie, hypotonie, únavová slabost zejména proximálního svalstva a opoždění motorického vývoje. U některých jednotek se vyskytují epizody apnoe s respiračním selháním – bezprostředně po narození či později, kdy jsou obvykle provokované infektem. Mohou být příčinou náhlého úmrtí kojence. Mezi další možné projevy patří artrogrypóza, syndrom mnohočetných pterygií z akinezie plodu, kraniofaciální dysmorfie, skolióza, neprospívání, kolísající či progresivní respirační insuficience, myastenický či myopatický syndrom od velmi lehkého až po těžký s upoutáním na vozík a výraznou hypotrofií svalstva.
Patofyziologie
V presynaptickém nervovém zakončení jsou připraveny vezikuly obsahující ACh. Na jejich přípravě se podílí cholinacetyltransferáza (ChAT). V místě synapse, tedy na nervosvalové ploténce, je postsynaptická membrána výrazně nařasena, přičemž na vrcholu záhybů jsou nahromaděny AChR (toto nahromadění zajišťuje protein rapsyn) a v hloubce záhybů jsou umístěny napěťově řízené Nav1,4 kanály. Vlastní receptor pro acetylcholin (AChR) tvoří pět podjednotek uspořádaných kolem iontového kanálu. AChR dospělého člověka má dvě podjednotky alfa, dále podjednotky beta, delta a epsilon. Fetální sval má místo epsilon podjednotky podjednotku gama. Při aktivaci AChR dojde k otevření iontového kanálu, do buňky vstupují kationty a vzniká akční potenciál. Pokud je tento potenciál dostatečně vysoký, aktivuje napěťově řízené Nav1,4 kanály v hloubce záhybů, tím je spuštěn šířící se akční potenciál a dojde ke svalovému stahu. Enzym AChE, umístěný v synaptické štěrbině, zajišťuje odbourávání ACh. Tak se podílí na ukončení odpovědi svalové buňky a její přípravě na další podnět. Ukotvení AChE v prostoru synapse zajišťuje její podjednotka ColQ, tzv. „kolagenní ocas“. Komplex agrin, MuSK, Dok7 (geny AGRN, MUSK, DOK7) se ve spolupráci s rapsynem podílí na organizaci uspořádání nervosvalové ploténky, zejména nakupení AChR při tvorbě synapse, a je nezbytný k udržení struktury synapse.
To, zda vznikne akční potenciál, ovlivňuje: množství molekul ACh v presynaptických vezikulech, jejich uvolnění a účinnost uvolněného ACh. Účinnost uvolněného ACh pak závisí na geometrii nervosvalové ploténky, na hustotě AChE v synaptické štěrbině, na hustotě a rozmístění AChR na postsynaptické membráně, dále pak na kinetických vlastnostech (tedy na funkci) jednotlivých AChR a jednotlivých Nav1,4 kanálů.
Léčba
Podle typu CMS se v léčbě používají inhibitory AChE (zvyšují odpověď na uvolněný ACh) 3,4-diaminopyridin (zvyšuje množství ACh uvolněného po podnětu), u poruchy AChE efedrin a u slow-channel syndromu chinidin či fluoxetin. Onemocnění nemá autoimunní podklad, imunosuprese tedy není indikována. U jednotek s apnoí se doporučuje apnea monitor a zacvičení rodičů do kardiopulmonární resuscitace.
Kazuistiky
Mutace 1267delG v CHRNE genu
Dívka narozená v srpnu 1994
Dívka romského etnika, rodiče a asi sedm sourozenců zdrávo. U sestřenice byla zjištěna stejná diagnóza. Dívka je z fyziologické gravidity i porodu v termínu, vážila 2 400 g. Do sedmi měsíců byla opakovaně hospitalizována pro respirační infekty. Vývoj byl lehce opožděn, v 1,5 roce chodila a začala mluvit. Od malička pozorována ptóza, ráno je stav lepší, během dne se zhoršuje. Údaj o oftalmoplegii v časném věku chybí. Od sedmi let byla postupně vyšetřována na více pracovištích pro zhoršování slabosti – nevyšla schody, nedošla do školy, při zpěvu hlas postupně slábl. Běžná biochemie včetně CK a laktátu byla v normě, protilátky proti AChR byly negativní. Pro podezření na mitochondriální onemocnění byly vyšetřeny komplexy dýchacího řetězce a nejčastější mutace mtDNA – vše s normálními výsledky. MR mozku bylo v normě. V osmi letech pak byla provedena i svalová biopsie, jež diagnózu rovněž neobjasnila.
Při našem vyšetření byla dívka drobná a menšího vzrůstu, měla hypomimii s ptózou a poruchou okulomotoriky. Reflexy bylydobře výbavné, nebyla zachycena významná svalová slabost. V EMG byly normální kondukční studie a signifikantní dekrement při repetitivní stimulaci. Při syntostigminovém testu jednoznačně ustoupila ptóza. Léčba inhibitory AchE byla s příznivým efektem, zmenšila se ptóza a zlepšila se celková kondice. V jejích devíti letech jsme odeslali DNA k vyšetření na zahraniční pracoviště (Univerzita Ludvíka Maximiliána, Mnichov) s podezřením na mutaci 1267delG v CHRNE genu, která byla potvrzena. Vzhledem k množství pacientů s touto diagnózou je nyní vyšetření této mutace dostupné i v ČR.
Mutace COLQ genu způsobující deficit AChE
Chlapec narozený v dubnu 1994
V rodině chlapce kavkazského etnika se žádné nervosvalové onemocnění nevyskytuje. Je ze druhé gravidity, těhotenský diabetes byl léčen pouze dietou. Porod byl v termínu, fyziologický, PH byla 3 290 g. Od počátku špatně sál z prsu, byl krmen odstříkaným mateřským mlékem. Nadále měl problémy s příjmem potravy. Sociální kontakt byl včasný, mentálně postupoval v pásmu podprůměru. Po pátém měsíci byl hodnocen jako hypotonický, rehabilitoval, milníky hrubé motoriky byly lehce opožděné, až ve 12 měsících lezl po čtyřech a ve dvou letech samostatně chodil. V roce života byl drobnější, měl symetrické oční štěrbiny bez ptózy, strabizmus convergens a povšechnou výraznou hypotonii s výbavnými reflexy, špatně kousal. Vyšetření biochemie včetně laktátu a CK bylo v normě, dále i metabolické vady, potní test, hormony štítné žlázy, protilátky proti kravskému mléku a gliadinu. Karyotyp byl mužský s nevýznamnou variantou na 10. chromozomu. Rychlosti vedení i nález v jehlové EMG byly v normě, repetitivní stimulace vyšetřovana nebyla. Na CT mozku bylo popsáno cavum septi pellucidi et Vergae. Ve dvou letech nedotahoval levý bulbus temporálně. Byla provedena svalová biopsie, jež diagnózu neobjasnila. Až v sedmi letech začíná být patrná kolísavá semiptóza a lehká porucha okulomotoriky, odpoledne býval unavenější. Vyšetření protilátek proti AchRbylo negativní, při repetitivní stimulaci byl zachycen jednoznačný dekrement, neměl však typický tvar písmene „U“. Byla vylou-čena mutace 1267delG v CHRNE genu (doc. Pavel Seeman). Ve 12 letech jsme doplnili test se syntostigminem, po kterém se akcentovala svalová slabost, chlapec se neudržel na nohou, měl i pocit ztížené ventilace. Tento výsledek je typický pro CMS synaptického typu s deficitem AchE. Ve spolupráci se zahraničním pracovištěm (Univerzita Ludvíka Maximiliána, Mnichov) byly přímým sekvenováním všech 17 exonů COLQ genu prokázány dvě heterozygotní mutace (složený heterozygot): Missense v exonu 11: c.710G>A, p.G237DA frameshift v exonu 12: 797insC. Na základě literárních údajů [24] byla zahájena léčba efedrinem 25-25-0 mg s poměrně příznivým efektem. Je nadále astenický, ale zvládá běžnou denní činnost, školu, jako brankář hraje florbal.
Mutace RAPSN genu pro rapsyn
Dívka narozená v květnu 2005
Rodinná anamnéza je negativní, etnikum kavkazské. Gravidita fyziologická, porod v termínu, plánovanou sekcí v celkové anestezii pro polohu koncem pánevním. PH 2980g, AS 8-9-9, pH pupečníkové krve 7,329. Po vybavení byla dívka nápadně hypotonická, nesála ani nepolykala, vyžadovala sondování, později byla krmena stříkačkou a lžičkou. Druhý den po porodu se při manipulaci objevil výrazný třes a stáčení bulbů. Vyšetření: základní biochemie, adnátní infekce, likvor, oční pozadí, sono a MR mozku, molekulární genetika na SMA byly s normálním nálezem, ve skríningu metabolických vad byla mírná laktacidurie. EEG v měsíci věku s nálezem supression‑burst vzorce, kontrolní EEG již v normě. Dívka přechodně užívala Phenaemaletten, na který reagovala výraznějším útlumem již po 1/4 tbl. Byla sledována neurologem, pro mírné opoždění hrubé motoriky a pro hypotonii rehabilitovala, psychický vývoj byl v normě. Trpěla na zahlenění a záněty HCD, při infektech hůře jedla a zvýrazňovala se hypotonie. Rodiče nepozorovali žádné kolísání výkonnosti během dne, ani ptózu.
V 10 a v 11 měsících došlo vždy při infektu k aspiraci, respiračnímu selhání s hypoventilací s nutností intubace a umělé plicní ventilace. Nápadný byl dlouhý zotavovací čas po sedativech. Až v rámci pobytu na JIP po druhé příhodě v 11 měsících byla pozorována kolísající ptóza, bez oftalmoplegie. Dívka měla poruchu kousání a polykání, nižší svalový tonus, normálně výbavné šlachosvalové reflexy. Hrubá motorika byla na úrovni počátku IV. trimenonu (lezla po čtyřech a stoupala si s oporou), jemná motorika a psychika byly přiměřené věku. CK i kardiologické vyšetření bylo v normě, protilátky proti AChR negativní. EMG repetitivní stimulace bez dekrementu. Kongenitální myastenii potvrdil až syntostigminový test, který byl jednoznačně pozitivní (obr. 1 a 2) a také dobrá klinická odpověď na léčbu inhibitory AChE (Mytelase). Slabost a ptóza se objevují již jen při infektech, ale respirační selhání se nezopakovalo ani při pneumonii. Postupně se upravilo kousání a polykání. Ve 14 měsících obcházela, sama chodila od 16 měsíců. Ve čtyřech letech je při léčbě bez potíží, stačí vrstevníkům.


Byla vyloučena mutace 1267delG v CHRNE genu (doc. Pavel Seeman). Vespolupráci se zahraničním pracovištěm (Univerzita Ludvíka Maximiliána, Mnichov) byly prokázány dvě heterozygotní mutace (složený heterozygot)RAPSN genu (RAPSN N88K and V165M), a tím byla na molekulárně genetické úrovni potvrzena diagnóza kongenitální myastenie s poruchou rapsynu.
Literární přehled jednotlivých CMS a diskuze
CMS s poruchou presynaptickou
CMS s epizodickou apnoí – deficit ChAT (cholin acetyl transferázy)
Při deficitu ChAT je porušena resyntéza ACh v presynaptickém zakončení. Typické jsou náhlé krize s výraznou slabostí, respiračním selháním a apnoí v novorozeneckém či časném kojeneckém věku. Mohou navodit sekundární hypoxicko‑ischemické poškození mozku. V období mezi epizodami je patrný různě vyjádřený myastenický syndrom. Pokud pacienti toto období přežijí, mohou se v adolescenci a v dospělosti příznaky postupně zmírňovat. Inhibitory AChE mohou epizody zmírnit, nebo jim zabránit. V období mezi krizemi je při repetitivní stimulaci vybaven dekrement pouze při únavě po svalové zátěži či po několikaminutové „přípravné“ stimulaci [5–8].
CMS s poruchou na úrovni synaptické štěrbiny
CMS s poruchou AChE, podmíněný mutací COLQ genu
Většina pacientů má závažné potíže od kojeneckého věku, s celkovou slabostí a hypotrofií svalstva, častá je respirační insuficience a postižení axiálního svalstva s rozvojem skoliózy. Popisuje se obleněná fotoreakce, běžná je ptóza, oftalmoplegie, dysfagie. Léčba není známa. Méně časté jsou mírnější formy s pozdější manifestací a postupnou progresí pro rozvoj myopatie. Tito pacienti mohou profitovat z podávání efedrinu [10].
Ukotvení AChE tak, aby byla přítomna prostoru synapse, zajišťuje její podjednotka ColQ, tzv. kolagenní ocas. Při mutaci COLQ genu AChE v synaptické štěrbině chybí a uvolněný ACh není rozkládán. Po jednorázovém podnětu se uvolněný ACh opakovaně váže na AChR. Prodloužená synaptická odpověď tak překročí absolutní refrakterní periodu svalového vlákna a vyvolá opakované akční potenciály svalu. Synapse je přetížena a dochází k řadě strukturálních i funkčních změn včetně rozvoje myopatie, způsobené přetížením postynaptické oblasti kationty. Při syntostigminovém testu dojde k paradoxní reakci – ke zhoršení svalové slabosti. EMG – repetitivní stimulace může, ale nemusí prokázat dekrement [9].
CMS s poruchou postsynaptickou
Byly nalezeny mutace genů pro všechny podjednotky AChR a na různých jejich doménách. Způsobují buď poruchu exprese AChR, nebo poruchu jejich funkce neboli kinetiky.
Primární deficit AChR s mírnou nebo žádnou poruchou kinetiky AChR
Snížená exprese AChR způsobená mutací AChR
Úbytek AChR mohou způsobovat mutace všech podjednotek, nejčastěji nacházíme změnu podjednotky epsilon. Pravděpodobně proto, že její funkci může kompenzovat alespoň částečná exprese fetální podjednotky gama, zatímco pacienti s mutací jiné podjednotky, kde kompenzace není možná, zřejmě nepřežijí.
V našich podmínkách je nejčastější porucha podjednotky epsilon, podmíněná tzv. romskou mutací genu ChRNE v exonu 12 epsilon podjednotky (e1267del G) [4,22,23]. Již od časného kojeneckého věku je pozorována ptóza, zevní oftalmoplegie, hypomimie, slabý pláč a problémy s krmením, mohou být i opakované aspirace (z postižení bulbárního svalstva). Respirační infekty mají těžší průběh, respirační selhání je však vzácné. Je patrná mírná až střední únavová pletencová slabost, většinou je opožděn vývoj hrubé motoriky. Onemocnění bývá neprogresivní a dobře reaguje na inhibitory AChE.
Snížená exprese AChR způsobená mutací RAPSN genu kódujícího protein rapsyn
Protein rapsyn je zodpovědný za seskupování AChR na nervosvalové ploténce. Při jeho deficitu tedy není na postsynaptické membráně dostatečný počet AChR. Typické jsou krize s rychlým rozvojem život ohrožujícího respiračního selhání vyvolané např. stresem, horečkou či infekcí. Pohyby plodu jsou slabé, po narození bývá hypotonie, ptóza, chabý pláč a sání. Oftalmoplegie je popisována zhruba u poloviny pacientů. Bývá opožděný vývoj motoriky.
Příznaky jsou výrazně variabilní. Někdy je postižení tak závažné, že je nutné provést gastrostomii a tracheostomii. Malá hybnost plodu in utero může být příčinou artrogrypózy, mohou být i kraniofaciální malformace (specifická mutace u Židů ze Středního východu [15]). Jen výjimečně se první příznaky objeví později, např. až ve třetí dekádě. Dekrement při vyšetření repetitivní stimulací se popisuje u 50–80 % případů. Dobrá je reakce na inhibitory AChE, používá se i 3,4-diaminopyridin [11–14].
Primární porucha kinetiky AChR s deficitem nebo bez deficitu AChR
Poruchu kinetiky dělíme na dva typy: „slow-channel syndrom“, u kterého jsou po podnětu dlouho otevřeny iontové kanály a synaptická odpověď na ACh je zvýšena, a „fast-channel syndrom“, kdy jsou naopak po podnětu otevřeny iontové kanály velmi krátce a synaptická odpověď na ACh je snížena. Jedná se tedy o fyziologicky protikladné reakce.
Slow-channel syndrom
Jediný CMS s AD dědičností, možná je i AR dědičnost. Typická je selektivní slabost svalstva šíje, lopatek, extenzorů prstů, mírná oftalmoplegie a variabilní postižení dalších svalových skupin. Svaly ruky a extenzorů prstů jsou atrofické. První příznaky se mohou objevit již v kojeneckém věku ale i později, třeba až v sedmé dekádě. Onemocnění je pomalu progresivní – podobně jako u mutace COLQ genu vede přetížení synapse k rozvoji myopatie. Inhibitory AChE příznaky zhoršují. V léčbě jsou úspěšně používány chinidin či fluoxetin.
Fast-channel syndrom
Onemocnění je vzácnější než slow-channel syndrom. Typicky se projevuje v kojeneckém věku či v časném dětství ptózou, oftalmoplegií, dysartrií, dysfagií, poruchou kousání a námahovou slabostí trupového a končetinového svalstva. Může být postiženo i respirační svalstvo, vážnější postižení se projeví artrogrypózou. Postižení je statické či mírně progresivní. Při repetitivní stimulaci pomalou frekvencí se objeví dekrement, který při stimulaci rychlou frekvencí mizí. V léčbě se používá kombinace 3,4-diaminopyridinu s inhibitory AchE.
Porucha funkce sodíkového kanálu způsobená mutací genu SCN4A
V roce 2003 byla popsána jediná pacientka: od narození měla opakované krize s respiračním selháním, opožděný motorický vývoj, ptózu, mírnou oftalmoplegii, bulbární slabost a celkovou slabost zhoršující se po námaze. Při repetitivní stimulaci byl prokázán dekrement. Léčba inhibitory AChE zlepšila výkonnost, acetazolamid zabránil dalším krizím s respiračním selháním. Podkladem je porucha funkce Nav1,4 sodíkových kanálů, kdy akční potenciál vzniklý při aktivaci AChR nevyvolá otevření sodíkových kanálů v hloubce záhybů postsynaptické membrány [16].
CMS s postižení pletencového svalstva při mutaci DOK 7 („downstream-of-kinase“) genu
Mutace popsaná v roce 2006 byla prokázána u mnoha dosud nezařazených pacientů. Komplex agrin, MuSK a Dok7 se ve spolupráci s rapsynem podílí na organizaci uspořádání nervosvalové ploténky, zejména nakupení AChR při tvorbě synapse, a je nezbytný k udržení struktury synapse.
Typická je pletencová slabost, klinický obraz je velmi variabilní. Příznaky se nejčastěji objevují v době nástupu chůze, která je kolébavá, někdy s vnitřní rotací kolen, mohou se ale rozvinout od narození po třetí dekádu. Typická je pletencová slabost, únavnost, ptóza obvykle bez oftalmoplegie, častá je slabost obličejového a bulbárního svalstva, dále respirační problémy, někdy od novorozeneckého období, většinou však později a s progresí, i s epizodami respiračního selhání. Potíže obvykle nekolísají během dne, ale v delších periodách, kdy mohou být horší a lepší týdny. U většiny pacientů se rozvine skolióza či hyperlordóza, u některých i svalová atrofie. Onemocnění bývá progresivní, někdy se ztrátou schopnosti chůze a zhoršováním respiračních funkcí.
V léčbě se používá efedrin a 3,4-diaminopyridin s variabilním efektem, inhibitory AChE potíže spíše zhoršují, i když syntostigminový test může být pozitivní. Repetitivní stimulace trapézového svalu obvykle navodí dekrement [17–21].
CMS s mutací MuSK genu kódujícím protein MuSK; s mutací AGRN genu kódujícím protein agrin a s mutací LAMB2 genu kódujícím b2 podjednotku lamininu.
Byly popsány v roce 2004 (MUSK) a v roce 2009 (AGRN a LAMB2). Jde o ojedinělé případy, proto podrobnosti neuvádíme [25–27].
Diskuze
Poznávání CMS trvá již takřka 40 let. Za tu dobu významně pokročily vyšetřovací možnosti zobrazovací, elektrofyziologické i možnosti genetické diagnostiky. Odhalování nových jednotek přináší i stále podrobnější poznání procesu nervosvalového přenosu. Přesto dosud není známa molekulárně genetická podstata onemocnění zhruba u poloviny pacientů.
Obecné povědomí o kongenitálních myastenických syndromech je zatím poměrně nízké a pacienti diagnóze často unikají. Diagnostiku znesnadňuje výrazná klinická variabilita, kdy u mnoha pacientů nejsou typické příznaky plně vyjádřeny. Ptóza se často rozvine až později (kazuistika 2 a 3), ofatalmoplegie není vždy pravidlem (kazuistika 3). U CMS s apnoickými krizemi může být nález mezi krizemi nenápadný, a CMS se tak může projevit až náhlým respiračním selháním, které může být příčinou syndromu náhlého úmrtí novorozence. Ani dekrement při EMG není vždy přítomen (kazuistika 3). A pokud při syntostigminovém testu dojde k paradoxnímu zhoršení svalové slabosti (slow‑channel syndrom, COLQ), (kazuistika 2), je stanovení správné diagnózy možné pouze při dostatečných teoretických znalostech o problematice CMS.
Pro diagnózu jsou důležité údaje o consanquinitě, familiárním výskytu, náhlém úmrtí sourozence v kojeneckém věku, pohybech plodu, o časném poporodním období (porucha sání, polykání, chabý křik, hypotonie), o epizodickém zhoršování (např. při infektu) či o kolísání příznaků během dne. Pátráme po ptóze, oftalmoplegii (přítomna u většiny pacientů s CHRNE, ale jen u 50 % pacientů s jiným CMS), postižení bulbárního svalstva, hypotonii, skolióze, kontrakturách, případně artrogrypóze či dysmorfických rysech. Akutní respirační selhání může být hlavním či jediným projevem CMS, dále je možné pozorovat nápadný útlum či slabost po podání některých léků (např. sedativa, některá antibiotika).
Prognóza je variabilní. V mnoha případech je choroba neprogresivní, někdy i s tendencí ke zlepšování.
Závěr
Kongenitální myastenické syndromy mají široké spektrum příznaků i závažnosti potíží. Pro možnost účinné léčby a prenatální diagnostiky je třeba po CMS pátrat – zejména u novorozenců a kojenců s hypotonií, svalovou slabostí, únavností, poruchou sání, polykání, hypomimií či epizodami respiračního selhání, ale i u starších dětí s neobjasněným myastenickým či myopatickým syndromem. Na kongenitání myastenii je třeba myslet i u pacientů bez oftalmoplegie či zjevné ptózy, u pacientů, u kterých repetitivní stimulace při EMG vyšetření nevyvolá dekrement a dokonce i u pacientů s paradoxní reakcí na syntostigminový test.
Seznam zkratek
CMS kongenitální myastenický syndrom
RA rodinná anamnéza
ChAT cholinacetyltransferáza
AChR acetylcholinový receptor
AChE acetylcholinesteráza
ACh acetylcholin
PH porodní hmotnost
COLQ gen kódující ColQ podjednotku AChE, tzv. kolagenní ocas
CHRNA1 gen kódující alfa podjednotku AChR
CHRNB1 gen kódující beta podjednotku AChR
CHRND gen kódující delta podjednotku AChR
CHRNE gen kódující epsilon podjednotku AChR
RAPSN gen kódující rapsyn
MUSK gen kódující pro sval specifický receptor tyrozin kinázy
MuSK pro sval specifický receptor tyrozin kinázy
SCN4A gen kódující napěťově řízený sodíkový kanál kosterního svalu
DOK7 “downstream‑of‑kinase” gen
AGRN gen kódující protein agrin
LAMB2 gen kódující b2 podjednotku lamininu
Poděkování:Prof. Dr. med. Hannsi Lochmüllerovi, Univerzita Ludvíka Maximiliána, Mnichov, a doc. MUDr. Pavlu Seemanovi, Gennet, Praha – za molekulárně genetickou diagnostiku.
Výzkumný záměr MSM0021620849.
MUDr. Miriam Adamovičová
Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
Ke Karlovu 2
121 09 Praha 2
e-mail: adamovicova.m@seznam.cz
Přijato k recenzi: 1. 7. 2009
Přijato do tisku: 21. 11. 2009
Zdroje
1. Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: a diverse array of molecular targets. J Neurocytol 2003; 32(5–8): 1017–1037.
2. Harper CM. Congenital myasthenic syndromes. Semin Neurol 2004; 24(1): 111–123.
3. Engel AG, Sine SM. Current understanding of congenital myasthenic syndromes. Curr Opin Pharmacol 2005; 5(3): 308–321.
4. Abicht A, Stucka R, Karcagi V, Herczegfalvi A, Horváth R, Mortier W et al. A common mutation (epsilon1267delG) in congenital myasthenic patients of Gypsy ethnic origin. Neurology 1999; 53(7): 1564–1569.
5. Ohno K, Tsujino A, Brengman JM, Harper CM, Bajzer Z, Udd B et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci U S A 2001; 98(4): 2017–2022.
6. Byring RF, Pihko H, Tsujino A, Shen XM, Gustafsson B,Hackman P et al. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul Disord 2002; 12(6): 548–553.
7. Barisic N, Müller JS, Paucic‑Kirincic E, Gazdik M, Lah‑Tomulic K, Pertl A et al. Clinical variability of CMS‑EA (congenital myasthenic syndrome with episodic apnea) due to identical CHAT mutations in two infants. Eur J Paediatr Neurol 2005; 9(1): 7–12.
8. Kraner S, Laufenberg I, Strassburg HM, Sieb JP, Steinlein OK. Congenital myasthenic syndrome with episodic apnea in patients homozygous for a CHAT missense mutation. Arch Neurol 2003; 60(5): 761–763.
9. Mihaylova V, Müller JS, Vilchez JJ, Salih MA, Kabiraj MM, D’Amico A et al. Clinical and molecular genetic findings in COLQ‑mutant congenital myasthenic syndromes. Brain 2008; 131(3): 747–759.
10. Bestue‑Cardiel M, Sáenz de Cabezón‑Alvarez A, Capablo‑Liesa JL, López‑Pisón J, Peña‑Segura JL, Martin‑Martinez J et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrin. Neurology 2005; 65(1): 144–146.
11. Ohno K, Engel AG, Shen XM, Selcen D, Brengman J, Harper CM et al. Rapsyn mutations in humans cause endplate acetylcholine‑receptor deficiency and myasthenic syndrome. Am J Hum Genet 2002; 70(4): 875–885.
12. Burke G, Cossins J, Maxwell S, Owens G, Vincent A,Robb S et al. Rapsyn mutations in hereditary myasthenia: distinct early‑ and late‑onset phenotypes. Neurology 2003; 61(6): 826–828.
13. Ioos C, Barois A, Richard P, Eymard B, Hantaï D, Estournet‑Mathiaud B. Congenital myasthenic syndrome due to rapsyn deficiency: three cases with arthrogryposis and bulbar symptoms. Neuropediatrics 2004; 35(4): 246–249.
14. Müller JS, Mildner G, Müller‑Felber W, Schara U, Krampfl K, Petersen B et al. Rapsyn N88K is a frequent cause of congenital myasthenic syndromes in European patients. Neurology 2003; 60(11): 1805–1810.
15. Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG. E‑box mutations in the RAPSN promotor region in eight cases with congenital myasthenic syndrome. Hum Mol Genet 2003; 12(7): 739–748.
16. Tsujino A, Maertens C, Ohno K, Shen XM, Fukuda T,Harper CM et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci U S A 2003; 100(12): 7377–7382.
17. Selcen D, Milone M, Shen XM, Harper CM, Stans AA, Wieben ED et al. Dok‑7 Myasthenia: Phenotypic and Molecular Genetic Studies in 16 Patients. Ann Neurol 2008; 64(1): 71–87.
18. Okada K, Inoue A, Okada M, Murata Y, Kakuta S,Jigami T et al. The muscle protein Dok‑7 is essential for neuromuscular synaptogenesis. Science 2006; 312(5781): 1802–1805.
19. Beeson D, Higuchi O, Palace J, Cossins J, Spearman H, Maxwell S et al. Dok‑7 mutations underlie a neuromuscular junction synaptopathy. Science 2006; 313(5795): 1975–1978.
20. Palace J, Lashley D, Newsom‑Davis J, Cossins J, Maxwell S, Kennett R et al. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain 2007; 130: 1507–1515.
21. Müller JS, Herczegfalvi A, Vilchez JJ, Colomer J,Bachinski LL, Mihaylova V et al. Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromes. Brain 2007; 130(6): 1497–1506.
22. Morar B, Gresham D, Angelicheva D, Tournev I, Gooding R, Guergueltcheva V et al. Mutation history of the roma/gypsies. Am J Hum Genet 2004; 75(4): 596–609.
23. Seeman P, Sisková D. Autosomal recessive ethnic diseases of Czech Gypsies. Čas Lék Česk 2006; 145(7): 557–560.
24. Bestue‑Cardiel M, Sáenz de Cabezón‑Alvarez A, Capablo‑Liesa JL, López‑Pisón J, Peña‑Segura JL, Martin‑Martinez J et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrin. Neurology 2005; 65(1): 144–146.
25. Chevessier F, Faraut B, Ravel‑Chapuis A, Richard P,Gaudon K, Bauché S et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Human Molecular Genetics 2004 13(24): 3229–3240.
26. Chevessier F, Faraut B, Ravel‑Chapuis A, Richard P,Gaudon K, Bauché S et al. Identification of an Agrin Mutation that Causes Congenital Myasthenia and Affects Synapse Function. Am J Hum Genet 2009; 85(2):155–167.
27. Maselli RA, Ng JJ, Anderson JA, Cagney O, Arredondo J, Williams C et al. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J Med Genet 2009; 46(3): 203–208.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2010 Číslo 1
Nejčtenější v tomto čísle
- Mitochondriální encefalomyopatie na podkladě deficitu proteinu Sco2 s obrazem SMA‑like neurogenní svalové atrofie – kazuistiky
- Vyšetření čichu u neurologických onemocnění pomocí Testu parfémovaných fixů
- Kongenitální myastenické syndromy – kazuistiky
- Evokované odpovědi a elektromyografie v intraoperační monitoraci v neurochirurgii