
Rettův syndrom
Rett Syndrome
Rett syndrome (RTT) is a severe X‑linked neurodevelopmental disorder affecting almost exclusively girls. It belongs to the family of autistic spectrum disorders, and it is characterized by psychomotor regression, loss of acquired speech, microcephaly, repetitive stereotypic hand movements, and seizures. Most of RTT cases are caused by de novo mutations in the gene for the methyl‑ CpG‑binding protein 2 (MECP2), and familial cases are extremely rare. The MECP2 gene product plays an important role in chromatin remodeling, regulation of gene expression and is also involved in RNA splicing. Some atypical RTT cases are caused by mutations in other genes, such as CDKL5, FOXG1 or NTNG1. In this paper we give an overview of RTT, its clinical aspects, molecular basis, diagnostic criteria, medical management and DNA diagnosis.
Key words:
Rett syndrome – mental retardation – MECP2 gene – CDKL5 gene – FOXG1 gene – NTNG1 gene
Autoři:
D. Záhoráková; P. Martásek
Působiště autorů:
Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
Vyšlo v časopise:
Cesk Slov Neurol N 2009; 72/105(6): 525-533
Kategorie:
Přehledný referát
Souhrn
Rettův syndrom (RTT) je vážné X‑ vázané neurologické onemocnění, postihující především dívky. Patří mezi poruchy autistického spektra a je charakterizován zejména regresem psychomotorického vývoje, ztrátou řeči, mikrocefalií, stereotypními pohyby rukou a záchvaty. Většina případů RTT je způsobena de novo mutacemi v genu pro metyl‑CpG‑vazebný protein 2 (MECP2) a jen velmi ojediněle se jedná o familiární výskyt. Produkt tohoto genu, MeCP2 protein, sehrává důležitou roli v chromatinové remodelaci, regulaci genové exprese a také se účastní modulace RNA sestřihu. Některé případy atypického RTT mohou být způsobeny mutacemi v dalších genech, např. CDKL5, FOXG1 nebo NTNG1. Tento přehledový článek uvádí souhrn současných poznatků o Rettovu syndromu, klinickém obrazu pacientů v jednotlivých stadiích, molekulární podstatě, diagnostických kritériích, terapeutických přístupech a o možnostech DNA diagnostiky.
Klíčová slova:
Rettův syndrom – mentální retardace –MECP2 gen – CDKL5 gen – FOXG1 gen – NTNG1 gen
Práce vznikla za podpory grantů GA UK 257927/154 a IGA NR9215‑3
Úvod
Rettův syndrom (RTT, MIM #312750, MKN‑10 F84.2, DSM‑IV 299.8) postihuje téměř výlučně dívky, u kterých je s prevalencí 1 : 10 000 jednou z nejčastějších příčin mentální retardace a vývojového regresu. Tento syndrom byl v německé literatuře poprvé popsán již v roce 1966 vídeňským pediatrem dr. Andreasem Rettem [1]. Do širšího lékařského povědomí se zapsal až o mnoho let později, po uveřejnění prvního sdělení v anglickém jazyce švédským neurologem dr. Hagbergem a jeho kolegy [2]. Téměř úplná prevalence sporadických případů RTT (více než 99%) značně ztížila vědecké bádání směrující k určení způsobu dědičnosti a k identifikaci odpovědného genu. Teprve v roce 1999 byly u postižených pacientek nalezeny kauzální mutace v MECP2 genu lokalizovaném na chromozomu X, jehož produktem je transkripční represor metyl‑CpG‑vazebný protein 2 (MeCP2) [3]. RTT se tak stal prvním onemocněním ve skupině pervazivních vývojových poruch (neboli poruch autistického spektra), u kterého je již známa genetická příčina. Do této skupiny patří i dětský autizmus, se kterým bývá RTT kvůli podobným příznakům někdy zaměňován. Po stránce klinické i molekulární je RTT považován za prototyp pro genetické, molekulární a neurobiologické studium onemocnění spojených s poruchou neurologického vývoje.
Klinický obraz Rettova syndromu
Klasická forma RTT je charakterizována specifickým vývojovým profilem (obr. 1). Klinická diagnóza je postavena na definovaných kritériích [4], která jsou shrnuta v tab. 1.
![Doba nástupu klinických příznaků a nejčastější projevy RTT [98].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/e942503dc5e71233774b3b2d7cc9023c.jpg)
![Revidována diagnostická kritéria pro klasický Rettův syndrom [4].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/21e8caf48ee507a7f96502a419ae808d.jpg)
Původně se v literatuře uvádělo, že první příznaky RTT se objevují až po několikaměsíčním bezpříznakovém období normálního psychomotorického vývoje. Klinické studie za posledních několik let však prokázaly, že nepatrné abnormality se v mnoha případech vyskytují již v prvních šesti měsících po narození. Dítě může mírně zaostávat v motorickém vývoji (např. přetáčení na bříško, držení hlavičky), i když celkově se jeho zdravotní stav jeví v normě. Rodiče často popisují, že jejich dítě je příliš klidné až flegmatické, nepláče a většinu dne prospí. Pozorným sledováním je také možné již v tomto období pozorovat mírné problémy s jemnou motorikou a nadměrnou aktivitu rukou a prstů, v extrémech až náznaky repetitivních pohybů [5–7]. Další vývoj klinických příznaků RTT lze rozdělit do čtyř stadií.
1. stadium
Po 6–18 měsících normálního nebo zdánlivě normálního psychomotorického vývoje nastává fáze jeho zpomalení až zastavení. Ve většině případů se zpomaluje růst hlavy, což vede k mikrocefalii. Velmi častá je přidružená růstová retardace, ztráta na váze a svalová hypotonie.
2. stadium
Poté nastává fáze vývojového regresu, kdy postižené dítě ztrácí dosažené dovednosti. Jemná motorika rukou se částečně nebo úplně vytrácí a je nahrazena stereotypními pohyby rukou, které patří k typickým klinickým znakům RTT. Připomínají mytí, ždímání, tleskání, tření jedné ruky o druhou, opakované vkládání do úst, olizování a cucání prstů apod. Patrné je i zhoršení sociálních interakcí, vymizení očního kontaktu, nezájem o okolí, zastavení vývoje řeči nebo její úplná ztráta. Dítě může vykazovat přecitlivělost nebo podrážděnost se sklonem k automutilaci [8]. V tomto stadiu mohou pacientky s RTT někdy připomínat případy dětského autizmu. Zhoršování po mentální a kognitivní stránce provázejí další poruchy motoriky; špatná koordinace pohybů, ataxie až apraxie. Chůze je nejistá, o široké bázi, často jen s oporou a v mnohých případech pacientky přestávají chodit úplně. K prvním příznakům poruchy autonomních funkcí patří ataky hyperventilace a jiné dýchací anomálie (zrychlené dýchání, zadržování dechu, apnea, polykání vzduchu, prudké vydechování) vyskytující se u většiny případů. Nástup těchto příznaků nastává kolem druhého roku s přetrváváním do dospělosti. Ve spánku tyto anomálie pozorovány nejsou [9]. Přestože epilepsie není na seznamu diagnostických kritérií, záchvaty, od farmakologicky dobře kontrolovatelných až po těžké a nezvladatelné, se vyskytují u 50–90 % pacientek. Nejčastěji se jedná o generalizované záchvaty tonicko‑klonické nebo simplexní parciální [10]. S postupujícím věkem mají tendenci slábnout a v dospělosti již obyčejně nepředstavují zásadní problém.
3. stadium
Po poměrně dramatickém stadiu psychomotorického regresu nastává stadium stacionární, ve kterém se stav pacientek zdá být stabilizován. Mezi 5. až 10. rokem často dochází ke zlepšení po stránce sociální a komunikační, zejména v neverbální komunikaci, očním a tělesném kontaktu. Záchvaty a motorické postižení však nadále přetrvávají [9].
4. stadium
V poslední fázi, která přetrvává až do dospělosti, se motorické funkce většinou pozvolna opět zhoršují, přidává se hypertonie, rigidita, dystonie a progresivní skolióza. Mnohé pacientky úplně ztrácejí schopnost mobility a jsou připoutány na invalidní vozík. Ve vyšším věku se mohou vyskytnout prvky parkinsonizmu. Přidružují se také další poruchy autonomních funkcí, např. hypotrofické, studené, někdy až namodralé nohy a ruce, zácpa, orofaryngeální dysfunkce a srdeční arytmie (bradykardie, tachykardie, prodloužený QT interval) [11,12]. Pozorovány jsou také behaviorální poruchy, jako noční smích a křik, změny nálad, pocity úzkosti a neklidu, vyvolané např. změnou prostředí apod. [13]. Přestože se pacientky s RTT mohou dožít středního i vyššího věku, mortalita je u nich v průměru vyšší než u zdravých osob stejného věku. Přibližně 25 % úmrtí je náhlých a nevysvětlených. Jejich příčinou mohou být právě již zmíněné kardiologické problémy a jiné poruchy autonomní regulace [14–16].
Bylo popsáno také několik atypických forem, u kterých je průběh mírnější nebo naopak těžší než u klasické formy RTT. U tzv. forme fruste varianty je nástup příznaků pozdější, a to mezi 1. až 3. rokem, jemná motorika rukou může zůstat poměrně dobře zachována, pouze s minimálními stereotypními pohyby. U dalších pacientek zůstává částečně zachována řeč, i když používání jednotlivých slov nemusí být v kontextu, tzv. preserved speech varianta. Tyto pacientky nemívají mikrocefalii a často trpí nadváhou [17]. Těžký průběh má kongenitální forma RTT, u které úplně chybí období zdánlivě normálního psychomotorického vývoje a také varianta, u které záchvaty nastupují již v průběhu prvních šesti měsíců, tzv. early onset seizure varianta [18]. Diagnostická kritéria pro atypické formy RTT jsou shrnuty v tab. 2.
![Revidovaná diagnostická kritéria pro atypické formy Rettova syndromu [4]. Pro stanovení diagnózy musí být splněny nejméně tři základní kritéria a pět podpůrných kritérií.](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/157620b648ec6ce55164f1ea9345f01b.jpg)
Nástup a závažnost příznaků i celkový průběh onemocnění se u jednotlivých pacientek značně liší, což značně ztěžuje stanovení správné diagnózy. Kvůli ztrátě komunikačních dovedností a sociálních interakcí v stadiu regresu mohou být dívky s RTT mylně diagnostikovány jako případy dětského autizmu. S Angelmanovým syndromem má RTT společný výskyt mentální retardace, záchvatů, ataxie a mikrocefalie. U starších pacientek, u kterých dominuje spasticita, těžké postižení motorických funkcí a hluboká mentální retardace, je stanovení diagnózy ještě náročnější. Pokud není znám průběh jejich psychomotorického vývoje v raném dětství, nezřídka bývají tyto případy uzavřeny jako mozková obrna.
Neuropatologie Rettova syndromu
RTT byl původně považován za progresivní neurodegenerativní onemocnění. Tato hypotéza byla brzy vyvrácena, neboť u pacientek nebyla prokázána mozková atrofie, odumírání nervových buněk, zánět ani glióza. Makroskopicky se mozek pacientek jeví bez zřetelných patologických změn [19,20]. Nejvýraznější morfologickou abnormalitou vzhledem ke zdravým věkově odpovídajícím kontrolám je jeho menší hmotnost [21], pravděpodobně v důsledku menších neuronů, především v oblasti kortexu, talamu, substancia nigra, bazálních ganglií, amygdaly a hipokampu [22,23]. Také magnetická rezonance prokazuje snížený objem mozku v některých kortikálních oblastech [20]. Morfologie samotných neuronů ukazuje, že dendrity pyramidálních neuronů ve frontální, temporální a motorické oblasti jsou méně rozvětveny, než je obvyklé, s menším počtem krátkých dendritických výrůstků, avšak bez zjevných abnormalit axonů, což svědčí spíše o porušené neuronální maturaci než o neurodegenerativním onemocnění [24,25].
Definovat jednotné neurochemické změny je u RTT problematické. Abnormality byly pozorovány ve většině systémů, žádnou však nelze považovat za patognomickou, jelikož se nevyskytují u všech pacientek s RTT, a výsledky měření jsou do značné míry ovlivněny jak věkem pacientek, tak závažností jejich klinických příznaků [26]. Podle různých studií byly v mozkomíšním moku naměřeny snížené hladiny acetylcholinu [27], dopaminu [28], substance P [29] a nervového růstového faktoru [30], naopak zvýšené byly hladiny glutamátu [31]. Studium změn v neurotransmiterech a trofických faktorech, ačkoliv dosud bez jednoznačných výsledků, může sehrát důležitou roli při vývoji nové podpůrné terapie RTT.
Neurofyziologické studie ukazují, že k patofyziologii onemocnění přispívá centrální i autonomní nervový systém. Nicméně kvůli různorodosti nálezů u jednotlivých pacientek a absenci jednoznačných patognomických znaků nemají základní zobrazovací a elektrofyziologické metody rozhodující diagnostický význam. Pozitronová emisní tomografie i jednofotonová emisní počítačová tomografie prokazují snížený průtok krve cerebrálním řečištěm, především ve frontálních regionech [32–34]. Poruchy autonomního nervového systému signalizuje zvýšená incidence dlouhých QT intervalů a snížená variabilita tepové frekvence [16,35]. Nálezy evokovaných potenciálů a elektroretinogramů bývají u mladších dívek s RTT (do 15 let) normální [36]. U starších pacientek (10–22 let) s těžkým motorickým postižením byly pozorovány opožděné odpovědi somatosenzorických evokovaných potenciálů, ale elektromyografické a neurografické analýzy u nich neprokázaly závažnou poruchu kořenů míšních nervů ani periferních nervových vláken [37,38]. Vzhledem k častému výskytu záchvatů bývají pacienti s pervazivními vývojovými poruchami běžně podrobováni EEG vyšetření [39–41]. EEG bývá u pacientek s RTT abnormální a jsou pozorovány i jisté charakteristické EEG nálezy. Jedná se především o převažující théta aktivitu v bdělém stavu a opožděný EEG obraz, který neodpovídá věku pacientky. Epileptiformní abnormality zahrnují centrotemporální hroty anebo ostré vlny, později převládají multifokální hroty anebo ostré vlny a generalizované SW komplexy [38]. EEG změny u pacientek s RTT sice nejsou považovány za diagnostický znak, protože se do značné míry liší v jednotlivých případech, i u téže nemocné v různých stadiích onemocnění, nicméně mají význam pro nasazení a úpravu antikonvulzivní terapie [42]. Analýza EEG stejně jako ostatní neurofyziologická vyšetření mají kromě přínosu pro objasnění patofyziologie RTT význam hlavně pro celkové zhodnocení zdravotního stavu nemocných a případné odhalení jiné příčiny neurologických příznaků.
MECP2 gen a MeCP2 protein
Molekulárně genetická podstata RTT byla dlouhá léta předmětem intenzivního výzkumu. Jako nejpravděpodobnější se ukazovala hypotéza, že se jedná o dominantně dědičné X‑vázané onemocnění a kauzální mutace jsou v homozygotní nebo hemizygotní formě, tedy u mužů, letální. Pro odhalení genetické příčiny RTT byly klíčové případy familiárního výskytu onemocnění (postižené sestry nebo dvojice teta a neteř), přestože jsou jinak velmi vzácné. Vazebnými analýzami genetických markerů a systematickým skríningem kandidátních genů se nakonec podařilo na dlouhém raménku chromozomu X identifikovat gen MECP2 jako kauzální gen RTT [3].
MECP2 gen je dlouhý 76 kb a má čtyři exony. Jeho produktem je ubikvitérně exprimovaný jaderný protein, metyl‑CpG‑vazební protein 2 (MeCP2), který se vyskytuje ve dvou izoformách: MeCP2_e1 (kódována exony 1, 3 a 4) a MeCP2_e2 (kódována exony 2, 3 a 4). Obě izoformy mají identické hlavní funkční domény a liší se jen N‑terminální sekvencí (obr. 2). Zajímavé je, že v exonu 2 dosud nebyla popsána žádná mutace a exprese MeCP2_e2 je v mozku asi desetkrát nižší než exprese MeCP2_e1, což svědčí o dominantním postavení izoformy MeCP2_e1 v patogenezi RTT [43,44].
![Struktura MECP2 genu a obě izoformy MeCP2 proteinu [97].
a) struktura MECP2 genu na úrovni genomové DNA.
b) mRNA vznikající alternativním sestřihem. MECP2_e1 mRNA obsahuje exony 1, 3 a 4, MECP2_e2 mRNA obsahuje všechny čtyři exony a exon 1 je součástí 5’ nepřekládané oblasti.
c) srovnání N-terminální části obou izoforem MeCP2 proteinu indikující rozdíly v důsledku translace exonu 1, resp. exonu 2.
Šipky označují začátek translace (iniciační kodon ATG)
MBD: metyl-CpG-vazebná doména, TRD: represorová doména, C-ter: C-terminální doména](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/a45250700886024a4b7614064d225211.jpg)
MeCP2 protein má tři základní funkční domény a dvě signální sekvence pro jadernou lokalizaci proteinu (NLS). Metyl‑CpG‑vazebná doména (MBD) se specificky váže na symetricky metylované CpG dinukleotidy DNA [45,46], ale za jistých podmínek také na nemetylovanou DNA [47]. Druhou funkční doménou je represorová doména (TRD), která odpovídá především za represi transkripce. Asociuje s mnoha proteinovými faktory za vzniku represorového nebo chromatin remodulujícího komplexu [48–51], ale podle nových výzkumů se může uplatnit i při alternativním sestřihu RNA [52,53]. Funkce C‑terminální domény není dosud podrobně prozkoumána, popsané mutace v této doméně nicméně svědčí o jejím významu pro správné fungování MeCP2. Doména napomáhá při vazbě MeCP2 proteinu k nemetylované DNA, při sestřihu RNA a asociuje i s některými neuronálně specifickými faktory [54].
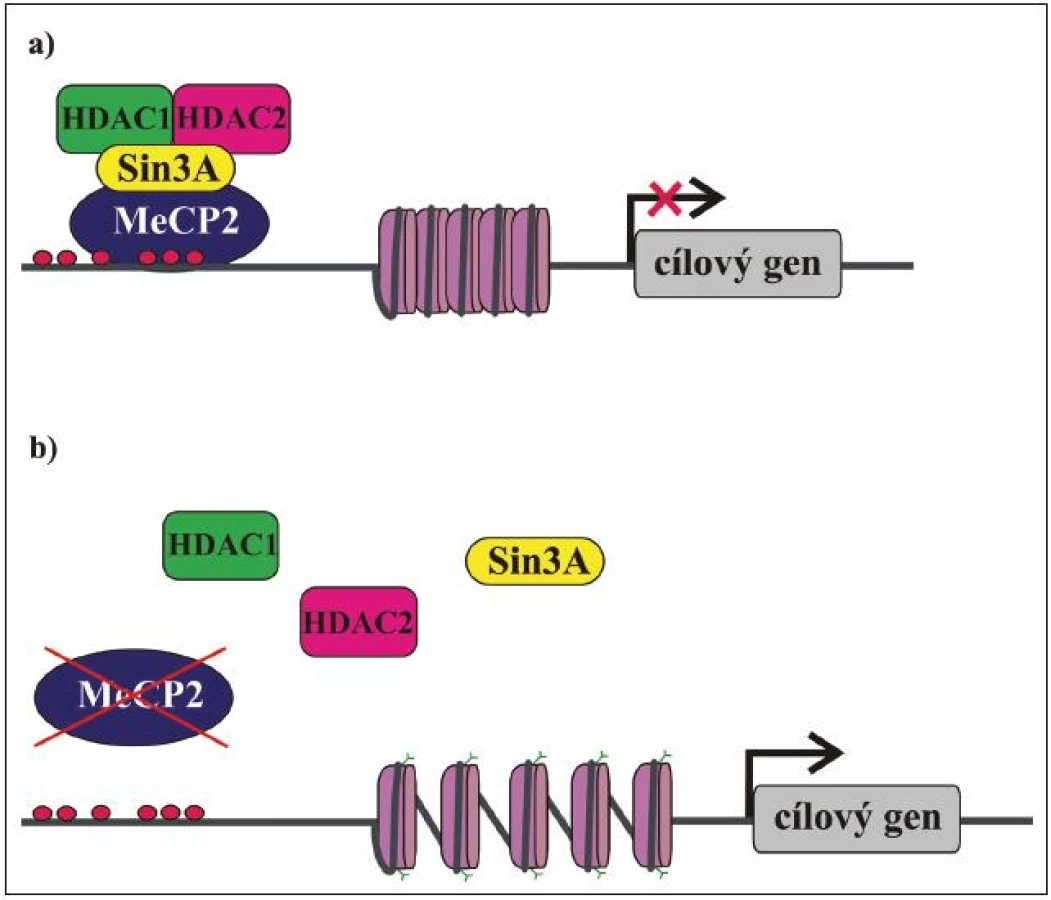
Jak již bylo řečeno, MeCP2 je exprimován ve všech tkáních a uvnitř buněk je lokalizován v jádře. Hladina exprese MeCP2 je relativně nejvyšší v mozku, kde koreluje s maturací neuronů a synaptogenezí. Předpokládá se, že MeCP2 sehrává důležitou roli v modulaci neuronální aktivity anebo plasticity [55,56]. To by mohlo vysvětlit, proč je mozek primárně postiženým orgánem u pacientek s RTT. MeCP2 má klíčovou úlohu v regulaci genové exprese a také funguje jako mediátor komplexní chromatinové remodelace. Původně byl považován za globální transkripční represor [51], který se MBD váže na metylované CpG dinukleotidy v promotorech cílových genů a prostřednictvím své TRD váže korepresor Sin3A a deacetylázy histonů 1 a 2 (HDAC). Deacetylací histonů nukleozomů se mění chromatinová struktura na heterochromatin, který je nepřístupný pro transkripční mašinérii, a dochází tak k transkripční represi (obr. 3) [49,50]. Kromě Sin3A a HDAC bylo později objeveno mnoho dalších faktorů, se kterými MeCP2 asociuje, a účastní se tak nejen modulace genové exprese, ale také RNA sestřihu a pravděpodobně i dalších buněčných procesů. Biologický význam většiny těchto interakcí není zatím zcela objasněn, nicméně jejich existence je důkazem toho, že funkce MeCP2 je mnohem komplexnější, než se původně předpokládalo. Některé cílové geny, jejichž expresi MeCP2 kontroluje, již byly identifikovány a podle očekávání jsou mezi nimi i geny, jejichž produkty se účastní neuronálního vývoje a signalizace (tab. 3). Identifikace dalších cílových genů a funkcí MeCP2 proteinu je klíčová pro pochopení patogeneze RTT a vývoj účinné terapie.
![Některé cílové geny MeCP2 proteinu [9,60].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/0a293419ed389b4e3f660c0bfc232a27.jpg)
Mutace v MECP2 genu
V MECP2 genu bylo dosud popsáno více než 300 různých patogenních mutací (www.hgmd.cf.ac.uk), které jsou identifikovány až u 90–95 % pacientek s klasickým RTT. Frekvence mutací u atypických variant je o mnoho nižší, jen 20–50 % [57]. Spektrum mutací je velmi široké; nejčastěji se jedná o bodové mutace typu missense a nonsense, z nichž osm je výrazně prevalentních (až 70 % případů), dále jsou to menší i rozsáhlejší delece, duplikace, inzerce a komplexní přestavby. Většina popsaných mutací způsobuje částečnou nebo úplnou ztrátu funkce MeCP2 proteinu jako modulátoru genové exprese. Mutace lokalizovány mimo základní funkční domény MBD a TRD mají nejpravděpodobněji negativní vliv na sekundární a terciární strukturu proteinu, jeho stabilitu a transport do buněčného jádra nebo jeho interakce s jinými kofaktory. Jelikož dosud nejsou známy všechny funkce MeCP2 proteinu, je mnohokrát velice těžké posoudit funkční dopad některých mutací (především typu missense).
MECP2 mutace vznikají téměř vždy de novo, většinou v paternální germinální linii [58,59]. Příčinou je pravděpodobně vysoký stupeň CpG metylace v mužské germinální linii a také velký počet buněčných dělení v průběhu spermatogeneze, které mohou vést ke vzniku de novo mutací. Familiární případy RTT bývají téměř výlučně způsobeny mutacemi zděděnými po zdravé nebo velmi mírně postižené matce. Tyto ženy mají buď gonadální mozaicizmus pro MECP2 mutaci (tj. produkují vajíčka s mutací i bez ní), nebo jsou přenašečkami somatické mutace, která se u nich díky příznivé inaktivaci chromozomu X (XCI) na fenotypu neprojevila [60].
V uplynulých letech bylo publikováno velké množství studií zabývajících se závažností klinických příznaků ve vztahu k typu MECP2 mutací u pacientek s RTT. Ačkoliv výsledky těchto studií nejsou zcela konzistentní, všeobecně platí, že:
- mutace postihující NLS jsou mnohem závažnější než ty, které NLS zachovávají;
- delece na konci kódující oblasti (v C‑terminální doméně) jsou nejméně závažné;
- nonsense mutace a delece na začátku kódující sekvence jsou závažnější než missense mutace [61–63].
Jistá asociace mezi genotypem a fenotypem byla pozorována i u některých konkrétních mutací, i když se individuální klinický obraz pacientek se stejnou MECP2 mutací nezřídka podstatně liší [64,65]. Závažnost klinických příznaků může ovlivňovat především rozdílná XCI, která nicméně nevysvětluje všechny rozdíly v manifestaci onemocnění [66,67]. Je proto možné, že průběh onemocnění modulují i další faktory. Možnými kandidáty jsou upstream regulátory exprese samotného MECP2 genu, cílové geny kontrolované MeCP2 proteinem a v úvahu přicházejí také geny, jejichž produkty se účastní vývoje a diferenciace CNS a modulace neuronálních funkcí.
Fenotypové spektrum MECP2 mutací se neomezuje jen na dívky s RTT. Diskutovaným tématem jsou především MECP2 mutace u mužů, které byly původně považovány za prenatálně letální, později však byly nalezeny i u chlapců s těžkou neonatální encefalopatií a mentální retardací. Takto postižení chlapci se však nedožívají více než 1–2 let. Klasický RTT se u mužů celkově manifestuje jen velmi zřídka, a to u pacientů s Klinefelterovým syndromem (47, XXY) nebo se somatickým mozaicizmem pro MECP2 mutaci. Přítomnost nadbytečného X chromozomu v každé jejich buňce, resp. podíl buněk s X chromozomem bez mutovaného MECP2 genu, navozuje situaci podobnou u žen a s tím patrně související i stejný klinický fenotyp [68,69]. Dále byly mutace v MECP2 genu nalezeny i u pacientů s Angelmanovým syndromem, X‑vázanou mentální retardací, autizmem, lehkou mentální retardací nebo jen mírnými psychickými či motorickými problémy. U těchto klinických jednotek je spektrum mutací MECP2 genu častokrát jiné než u RTT a frekvence podstatně nižší [70–73].
Další kauzální geny u Rettova syndromu
Nepřítomnost mutace v MECP2 genu některých pacientek RTT vedla k úvaze, že existují další kauzální geny, jejichž mutace vedou k podobnému fenotypu jako mutace v MECP2.
U některých pacientek s atypickým RTT, u kterých se záchvaty objevují již v prvních šesti měsících po narození, byly nalezeny mutace v genu CDKL5 ležícím na chromozomu X [74–77]. Kromě dívek s atypickým RTT se CDKL5 mutace vyskytují i u některých případů X‑vázaného Westova syndromu (ISSX2, Infantile spasm syndrome X‑linked type 2) [78], X‑vázané retinoschisis s epilepsií [79], autistů a pacientů s neurologickými poruchami a epilepsií [77]. Produktem CDKL5 genu je protein vykazující sekvenční homologii se serin/treonin kinázami (odtud pochází jeho starší název serin/threonin kinase 9, STK9) a pravděpodobně patřící do rodiny cyklin‑dependentních kináz [80]. Ačkoliv funkce CDKL5 (Cyclin‑Dependent Kinase‑Like 5) proteinu zatím není známa, překrývající se fenotyp pacientek s mutacemi v MECP2 a CDKL5 genech vede k teorii, že tyto dva proteiny spolupracují na stejném procesu. Nejenže CDKL5 fosforyluje MeCP2, a tím pravděpodobně moduluje jeho funkci [81], ale také expresní profil CDKL5 je v průběhu maturace neuronů a synaptogeneze velmi podobný profilu exprese MeCP2 [74,82]. Mutace v CDKL5 genu vznikají většinou de novo a jejich frekvence je nízká. Pokud však klinické příznaky jednoznačně svědčí pro atypický RTT s včasnými záchvaty, mělo by se rozhodně uvažovat o analýze tohoto genu.
Kandidátem na pozici dalšího kauzálního genu u RTT je gen FOXG1, který leží na chromozomu 14 a kóduje protein FoxG1 (původně brain factor 1, BF‑1). Nedávno bylo popsáno několik případů RTT s mutacemi v tomto genu [83–85]. FoxG1 protein je transkripční faktor, který je exprimován pouze v mozku a varlatech, a sehrává důležitou roli především v prvních stadiích neuronálního vývoje [86,87].
Dalším kandidátem je gen NTNG1 ležící na chromozomu 1. Jeho produktem je Netrin G1 (též laminet‑1), protein exprimován především v mozku, který má důležitou funkci v průběhu vývoje CNS [88,89]. Dosud byl v literatuře popsán jen jediný případ narušení NTNG1 genu (translokací) u pacientky s RTT [90], proto se nepředpokládá, že by mutace v tomto genu byly častou příčinou onemocnění.
Terapeutické přístupy
Dosud neexistuje žádná účinná terapie, která by zastavila nebo zvrátila postup onemocnění, ačkoliv parciální výsledky získané na myších modelech jsou velmi slibné. Léčba RTT je proto v současnosti pouze symptomatická a podpůrná. Charakter onemocnění a široká variabilita příznaků vyžadují multidisciplinární přístup zaměřený na individuální potřeby každé pacientky. Častým a závažným problémem bývají záchvaty, které jsou však u většiny pacientek s úspěchem kompenzovány antikonvulzivní terapií. Pro rozvoj a zachování svalové hmoty, zlepšení pohyblivosti a rovnováhy se osvědčila rehabilitace a fyzioterapie, případně hydroterapie, hipoterapie apod. Speciální pozornost vyžaduje i skolióza, která se vyskytuje u vysokého procenta pacientek a která má závažný dopad na úroveň jejich mobility. Mnohé dívky s RTT potřebují korzet, u některých je dokonce nutná operace [91]. Důležité je rovněž rozvíjení komunikačních dovedností pacientek. Mnohé dívky jsou i po ztrátě řeči schopny komunikovat pomocí gest, očním kontaktem apod. Při rozvíjení těchto dovedností se osvědčily mnohé techniky, např. muzikoterapie. Pozitivní výsledky na celkovém stavu byly pozorovány po aplikaci L‑karnitinu [92–94], křeče a ataky hyperventilace se zmírnily při ketogenní dietě a dávkách magnezia [95], melatonin se osvědčil při poruchách spánku [96]. Pacientky s RTT mají zvýšené riziko život ohrožujících srdečních arytmií, proto se u nich nedoporučuje aplikace některých léčiv, např. antipsychotik, tricyklických antidepresiv, antiarytmik, některých anestetik (tiopental, sukcynylcholin) a antibiotik (erytromycin, ketokonazol) [97,98].
DNA diagnostika Rettova syndromu
Po objevení kauzálního genu MECP2 se otevřela možnost potvrzení klinicky stanovené diagnózy molekulárně genetickými metodami. Vzhledem k délce nepřekládaných a nekódujících sekvencí (3` nepřekládaná oblast, 3`UTR, MECP2 genu patří k nejdelším v lidském genomu), jsou rutinně analyzovány jen kódující oblasti a přilehlé intronové sekvence potřebné pro správný sestřih mRNA. Základní mutační analýza je prováděna sekvenováním amplifikovaných úseků. Rozsáhlé delece (jednoho nebo více exonů i celého MECP2 genu), které se vyskytují přibližně u 4–8 % pacientek s RTT, nejsou klasickými molekulárně genetickými metodami založenými na PCR odhaleny. Tyto mutace je možné identifikovat hybridizačními metodami, např. MLPA (Multiplex Ligation‑Dependent Probe Amplification). Takovým kombinovaným přístupem je možné odhalit mutaci v MECP2 až u 90–95 % pacientek s klasickou formou RTT a u 20–50 % pacientek s atypickými variantami. Ostatní případy mohou být způsobeny mutacemi v nekódujících, ale funkčně důležitých oblastech MECP2 genu, např. v promotoru nebo konzervovaných úsecích 3`UTR. U atypických variant se vzhledem k variabilnímu fenotypu a nízkému procentu případů s kauzální mutací v MECP2 genu předpokládala a v posledních letech také potvrdila genetická heterogenita, tedy existence dalších odpovědných genů. Dosud byly identifikovány tři takové geny (CDKL5, FOXG1 a NTNG1), ale jejich počet se pravděpodobně ještě rozroste. V současné době je v České republice na Klinice dětského a dorostového lékařství 1. LF UK a VFN v Praze dostupná DNA diagnostika RTT založena na mutační analýze MECP2 genu [99,100] a nově i genu CDKL5. Riziková pacientka, vhodná pro mutační analýzu MECP2 genu, by měla v první řadě splňovat diagnostická kritéria pro klasický nebo atypický Rettův syndrom, jak je uvedeno v tab. 1 a 2. U pacientů a pacientek s dětským autizmem a dalšími výše popsanými fenotypy, jejichž klinický obraz zcela neodpovídá RTT, je možné genetické testování provést také, avšak pozitivní výsledek lze očekávat jen s malou pravděpodobností. Analýza CDKL5 genu je doporučena pouze u pacientek s tzv. early onset seizure variantou RTT, kdy se záchvaty objevují už v průběhu prvních šesti měsíců života.
Nepřítomnost kauzální mutace klinicky stanovenou diagnózu v žádném případně nevylučuje. Molekulárně genetické metody mají své limity a fenotyp podobný RTT může být také způsoben mutacemi v jiných, dosud netestovaných genech. Nicméně DNA diagnostika RTT má velký význam při definitivním potvrzení diagnózy a při diagnostice klinicky nejednoznačných případů, např. u pacientek s atypickým RTT, u velmi mladých pacientek, které ještě neprošly jednotlivými stadii, a není tak možné sledovat typický průběh RTT nebo naopak u dospělých a starších pacientek. Včasné a přesné stanovení diagnózy umožňuje předvídat další vývoj onemocnění a plánovat tak preventivní a podpůrnou terapii. Ačkoliv je riziko rekurence v rodině postižených velmi nízké (méně než 1%), je možné provést skríning na mutaci, která byla nalezena u probanda i u dalších členů rodiny (zejména ženského pohlaví), včetně prenatální diagnostiky.
Zkratky
| 3`UTR | 3`nepřekládaná oblast |
| CDKL5 | cyclin‑dependent kinase‑like 5 protein |
| HDAC | deacetylázy histonů |
| MBD | metyl‑CpG‑vazebná doména |
| MECP2 | gen pro metyl‑CpG‑vazební protein 2 |
| MeCP2 | metyl‑CpG‑vazební protein 2 |
| MLPA | Multiplex Ligation‑dependent Probe Amplification |
| NLS | nuclear localization signal |
| RTT | Rettův syndrom |
| STK9 | serin/threnonin kinase 9 |
| TRD | represorová doména (Transcriptional Repression Domain) |
| XCI | inaktivace chromozomu |
prof. MUDr. Pavel Martásek, DrSc.
Klinika dětského a dorostového lékařství
1. LF UK a VFN v Praze
Laboratoř pro studium mitochondriálních poruch
Ke Karlovu 2
128 08 Praha 2
e-mail: pavel.martasek@gmail.com
Přijato k recenzi: 29. 6. 2009
Přijato do tisku: 21. 8. 2009
Zdroje
1. Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med Wochenschr 1966; 116(37): 723– 726.
2. Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett‘s syndrome: report of 35 cases. Ann Neurol 1983; 14(4): 471– 479.
3. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X‑linked MECP2, encoding methyl- CpG‑binding protein 2. Nat Genet 1999; 23(2): 185– 188.
4. Hagberg B, Hanefeld F, Percy A, Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol 2002; 6(5): 293– 297.
5. Burford B, Kerr AM, Macleod HA. Nurse recognition of early deviation in development in home videos of infants with Rett disorder. J Intellect Disabil Res 2003; 47(Pt 8): 588– 596.
6. Einspieler C, Kerr AM, Prechtl HF. Abnormal general movements in girls with Rett disorder: the first four months of life. Brain Dev 2005; 27 (Suppl 1): S8– S13.
7. Nomura Y, Segawa M. Characteristics of motor disturbances of the Rett syndrome. Brain Dev 1990; 12(1): 27– 30.
8. Nomura Y, Segawa M. Natural history of Rett syndrome. J Child Neurol 2005; 20(9): 764– 768.
9. Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron 2007; 56(3): 422– 437.
10. Jian L, Nagarajan L, de Klerk N, Ravine D, Bower C,Anderson A et al. Predictors of seizure onset in Rett syndrome. J Pediatr 2006; 149(4): 542– 547.
11. Hagberg B. Rett syndrome: long‑term clinical follow‑up experiences over four decades. J Child Neurol 2005; 20(9): 722– 727.
12. Roze E, Cochen V, Sangla S, Bienvenu T, Roubergue A, Leu- Semenescu S et al. Rett syndrome: an overlooked diagnosis in women with stereotypic hand movements, psychomotor retardation, Parkinsonism, and dystonia? Mov Disord 2007; 22(3): 387– 389.
13. Mount RH, Hastings RP, Reilly S, Cass H, Charman T. Behavioural and emotional features in Rett syndrome. Disabil Rehabil 2001; 23(3– 4): 129– 138.
14. Glaze DG, Schultz RJ. Autonomic dysfunction and sudden death in Rett syndrome: Prolonged QTc intervals and diminished heart rate variability. In: Kerr AM, Witt- Engerström I (eds). Rett disorder and the developing brain. Oxford: Oxford University Press 2001: 251– 256.
15. Guideri F, Acampa M, Hayek G, Zappella M, Di Perri T. Reduced heart rate variability in patients affected with Rett syndrome. A possible explanation for sudden death. Neuropediatrics 1999; 30(3): 146– 148.
16. Sekul EA, Moak JP, Schultz RJ, Glaze DG, Dunn JK, Percy AK. Electrocardiographic findings in Rett syndrome: an explanation for sudden death? J Pediatr 1994; 125(1): 80– 82.
17. Zappella M, Meloni I, Longo I, Hayek G, Renieri A. Preserved speech variants of the Rett syndrome: molecular and clinical analysis. Am J Med Genet 2001; 104(1): 14– 22.
18. Hagberg B, Gillberg C. Rett variants – rettoid phenotypes. In: Hagberg B, Anvret M, Wahlstrom J (eds.) Rett syndrome – clinical and biological aspects. London: MacKeith Press 1993: 40– 60.
19. Jellinger K, Armstrong D, Zoghbi HY, Percy AK. Neuropathology of Rett syndrome. Acta Neuropathol 1988; 76(2): 142– 158.
20. Reiss AL, Faruque F, Naidu S, Abrams M, Beaty T, Bryan RN et al. Neuroanatomy of Rett syndrome: a volumetric imaging study. Ann Neurol 1993; 34(2): 227– 234.
21. Jellinger K, Seitelberger F. Neuropathology of Rett syndrome. Am J Med Genet Suppl 1986; 1: 259– 288.
22. Bauman ML, Kemper TL, Arin DM. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett‘s syndrome. Neurology 1995; 45(8): 1581– 1586.
23. Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex 2000; 10(10): 981– 991.
24. Belichenko PV, Oldfors A, Hagberg B, Dahlström A. Rett syndrome: 3- D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport 1994; 5(12): 1509– 1513.
25. Belichenko PV, Dahlström A. Studies on the 3- dimensional architecture of dendritic spines and varicosities in human cortex by confocal laser scanning microscopy and Lucifer yellow microinjections. J Neurosci Methods 1995; 57(1): 55– 61.
26. Percy AK. Neurochemistry of the Rett syndrome. Brain Dev 1992; 14 (Suppl): S57– S62.
27. Wenk GL, Hauss‑ Wegrzyniak B. Altered cholinergic function in the basal forebrain of girls with Rett syndrome. Neuropediatrics 1999; 30(3): 125– 129.
28. Wenk GL. Alterations in dopaminergic function in Rett syndrome. Neuropediatrics 1995; 26(2): 123– 125.
29. Matsuishi T, Nagamitsu S, Yamashita Y, Murakami Y, Kimura A, Sakai T et al. Decreased cerebrospinal fluid levels of substance P in patients with Rett syndrome. Ann Neurol 1997; 42(6): 978– 981.
30. Lappalainen R, Lindholm D, Riikonen R. Low levels of nerve growth factor in cerebrospinal fluid of children with Rett syndrome. J Child Neurol 1996; 11(4): 296– 300.
31. Hamberger A, Gillberg C, Palm A, Hagberg B. Elevated CSF glutamate in Rett syndrome. Neuropediatrics 1992; 23(4): 212– 213.
32. Lappalainen R, Liewendahl K, Sainio K, Nikkinen P, Riikonen RS. Brain perfusion SPECT and EEG findings in Rett syndrome. Acta Neurol Scand 1997; 95(1): 44– 50.
33. Nielsen JB, Toft PB, Reske-Nielsen E, Jensen KE, Christiansen P, Thomsen C et al. Cerebral magnetic resonance spectroscopy in Rett syndrome. Failure to detect mitochondrial disorder. Brain Dev 1993; 15(2): 107– 112.
34. Yoshikawa H, Fueki N, Suzuki H, Sakuragawa N, Iio M. Cerebral blood flow and oxygen metabolism in the Rett syndrome. Brain Dev 1992; 14 (Suppl): S69– S74.
35. Ellaway CJ, Sholler G, Leonard H, Christodoulou J. Prolonged QT interval in Rett syndrome. Arch Dis Child 1999; 80(5): 470– 472.
36. Kálmánchey R. Evoked potentials in the Rett syndrome. Brain Dev 1990; 12(1): 73– 76.
37. Bader GG, Witt- Engerström I, Hagberg B. Neurophysiological findings in the Rett syndrome, I: EMG, conduction velocity, EEG and somatosensory- evoked potential studies. Brain Dev 1989; 11(2): 102– 109.
38. Glaze DG. Neurophysiology of Rett syndrome. J Child Neurol 2005; 20(9): 740– 746.
39. Oslejskova H, Dusek L, Makovska Z, Rektor I. Epilepsia, epileptiform abnormalities, non‑right– handedness, hypotonia and severe decreased IQ are associated with language impairment in autism. Epileptic Disord 2007; 9 (Suppl 1): S9– S18.
40. Ošlejšková H, Dušek L, Makovská Z, Dujíčková E, Austrata R, Šlapák I. Výskyt epileptických záchvatů a/ nebo epileptiformní EEG abnormity u dětí s dětským a atypickým autizmem. Cesk Slov Neurol N 2008; 71/ 104(4): 435– 444.
41. Reinhold JA, Molloy CA, Manning- Courtney P. Electroencephalogram abnormalities in children with autism spectrum disorders. J Neurosci Nurs 2005; 37(3): 136– 138.
42. Moser SJ, Weber P, Lütschg J. Rett syndrome: clinical and electrophysiologic aspects. Pediatr Neurol 2007; 36(2): 95– 100.
43. Kriaucionis S, Bird A. The major form of MeCP2 has a novel N‑terminus generated by alternative splicing. Nucleic Acids Res 2004; 32(5): 1818– 1823.
44. Mnatzakanian GN, Lohi H, Munteanu I, Alfred SE, Yamada T, MacLeod PJ et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet 2004; 36(4): 339– 341.
45. Lewis JD, Meehan RR, Henzel WJ, Maurer- Fogy I, Jeppesen P, Klein F et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992; 69(6): 905– 914.
46. Nan X, Meehan RR, Bird A. Dissection of the methyl- CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res 1993; 21(21): 4886– 4892.
47. Galvão TC, Thomas JO. Structure‑specific binding of MeCP2 to four- way junction DNA through its methyl CpG‑binding domain. Nucleic Acids Res 2005; 33(20): 6603– 6609.
48. Harikrishnan KN, Chow MZ, Baker EK, Pal S, Bassal S,Brasacchio D et al. Brahma links the SWI/ SNF chromatin‑remodeling complex with MeCP2- dependent transcriptional silencing. Nat Genet 2005; 37(3): 254– 264.
49. Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 1998; 19(2): 187– 191.
50. Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN et al. Transcriptional repression by the methyl- CpG‑binding protein MeCP2 involves a histone deacetylase complex. Nature 1998; 393(6683): 386– 389.
51. Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997; 88(4): 471– 481.
52. Jeffery L, Nakielny S. Components of the DNA methylation system of chromatin control are RNA‑binding proteins. J Biol Chem 2004; 279(47): 49479– 49487.
53. Young JI, Hong EP, Castle JC, Crespo‑Barreto J, Bowman AB, Rose MF et al. Regulation of RNA splicing by the methylation– dependent transcriptional repressor methyl- CpG binding protein 2. Proc Natl Acad Sci U S A 2005; 102(49): 17551– 17558.
54. Buschdorf JP, Strätling WH. A WW domain binding region in methyl- CpG‑binding protein MeCP2: impact on Rett syndrome. J Mol Med 2004; 82(2): 135– 143.
55. Jung BP, Jugloff DG, Zhang G, Logan R, Brown S, Eubanks JH. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J Neurobiol 2003; 55(1): 86– 96.
56. Smrt RD, Eaves- Egenes J, Barkho BZ, Santistevan NJ, Zhao C, Aimone JB et al. Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol Dis 2007; 27(1): 77– 89.
57. Amir RE, Fang P, Yu Z, Glaze DG, Percy AK, Zoghbi HY et al. Mutations in exon 1 of MECP2 are a rare cause of Rett syndrome. J Med Genet 2005; 42(2): e15.
58. Trappe R, Laccone F, Cobilanschi J, Meins M, Huppke P, Hanefeld F et al. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am J Hum Genet 2001; 68(5): 1093– 1101.
59. Wan M, Lee SS, Zhang X, Houwink- Manville I, Song HR, Amir RE et al. Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am J Hum Genet 1999; 65(6): 1520– 1529.
60. Matijevic T, Knezevic J, Slavica M, Pavelic J. Rett syndrome: from the gene to the disease. Eur Neurol 2009; 61(1): 3– 10.
61. Huppke P, Held M, Hanefeld F, Engel W, Laccone F.Influence of mutation type and location on phenotype in 123 patients with Rett syndrome. Neuropediatrics 2002; 33(2): 63– 68.
62. Charman T, Neilson TC, Mash V, Archer H, Gardiner MT, Knudsen GP et al. Dimensional phenotypic analysis and functional categorisation of mutations reveal novel genotype- phenotype associations in Rett syndrome. Eur J Hum Genet 2005; 13(10): 1121– 1130.
63. Weaving LS, Williamson SL, Bennetts B, Davis M, Ellaway CJ, Leonard H et al. Effects of MECP2 mutation type, location and X- inactivation in modulating Rett syndrome phenotype. Am J Med Genet A 2003; 118A(2): 103– 114.
64. Bebbington A, Anderson A, Ravine D, Fyfe S, Pineda M, de Klerk N et al. Investigating genotype– phenotype relationships in Rett syndrome using an international data set. Neurology 2008; 70(11): 868– 875.
65. Neul JL, Fang P, Barrish J, Lane J, Caeg EB, Smith EO et al. Specific mutations in methyl- CpG‑binding protein 2 confer different severity in Rett syndrome. Neurology 2008; 70(16): 1313– 1321.
66. Takahashi S, Ohinata J, Makita Y, Suzuki N, Araki A,Sasaki A et al. Skewed X chromosome inactivation failed to explain the normal phenotype of a carrier female with MECP2 mutation resulting in Rett syndrome. Clin Genet 2008; 73(3): 257– 261.
67. Xinhua B, Shengling J, Fuying S, Hong P, Meirong L, Wu XR. X chromosome inactivation in Rett Syndrome and its correlations with MECP2 mutations and phenotype. J Child Neurol 2008; 23(1): 22– 25.
68. Schwartzman JS, Bernardino A, Nishimura A, Gomes RR, Zatz M. Rett syndrome in a boy with a 47,XXY karyotype confirmed by a rare mutation in the MECP2 gene. Neuropediatrics 2001; 32(3): 162– 164.
69. Topçu M, Akyerli C, Sayi A, Törüner GA, Koçoğlu SR, Cimbiş M et al. Somatic mosaicism for a MECP2 mutation associated with classic Rett syndrome in a boy. Eur J Hum Genet 2002; 10(1): 77– 81.
70. Carney RM, Wolpert CM, Ravan SA, Shahbazian M,Ashley-Koch A, Cuccaro ML et al. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol 2003; 28(3): 205– 211.
71. Couvert P, Bienvenu T, Aquaviva C, Poirier K, Moraine C, Gendrot C et al. MECP2 is highly mutated in X‑linked mental retardation. Hum Mol Genet 2001; 10(9): 941– 946.
72. Klauck SM, Lindsay S, Beyer KS, Splitt M, Burn J, Poustka A. A mutation hot spot for nonspecific X‑linked mental retardation in the MECP2 gene causes the PPM- X syndrome. Am J Hum Genet 2002; 70(4): 1034– 1037.
73. Watson P, Black G, Ramsden S, Barrow M, Super M, Kerr B et al. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J Med Genet 2001; 38(4): 224– 228.
74. Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early- onset seizure variant of Rett syndrome. Hum Mol Genet 2005; 14(14): 1935– 1946.
75. Scala E, Ariani F, Mari F, Caselli R, Pescucci C, Longo I et al. CDKL5/ STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet 2005; 42(2): 103– 107.
76. Tao J, Van Esch H, Hagedorn- Greiwe M, Hoffmann K, Moser B, Raynaud M et al. Mutations in the X‑linked cyclin‑dependent kinase‑like 5 (CDKL5/ STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet 2004; 75(6): 1149– 1154.
77. Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 2004; 75(6): 1079– 1093.
78. Kalscheuer VM, Tao J, Donnelly A, Hollway G, Schwinger E, Kübart S et al. Disruption of the serine/ threonine kinase 9 gene causes severe X‑linked infantile spasms and mental retardation. Am J Hum Genet 2003; 72(6): 1401– 1411.
79. Huopaniemi L, Tyynismaa H, Rantala A, Rosenberg T, Alitalo T. Characterization of two unusual RS1 gene deletions segregating in Danish retinoschisis families. Hum Mutat 2000; 16(4): 307– 314.
80. Montini E, Andolfi G, Caruso A, Buchner G, Walpole SM, Mariani M et al. Identification and characterization of a novel serine- threonine kinase gene from the Xp22 region. Genomics 1998; 51(3): 427– 433.
81. Bertani I, Rusconi L, Bolognese F, Forlani G, Conca B, De Monte L et al. Functional consequences of mutations in CDKL5, an X‑linked gene involved in infantile spasms and mental retardation. J Biol Chem 2006; 281(42): 32048– 32056.
82. Lin C, Franco B, Rosner MR. CDKL5/ Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum Mol Genet 2005; 14(24): 3775– 3786.
83. Ariani F, Hayek G, Rondinella D, Artuso R, Mencarelli MA, Spanhol- Rosseto A et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet 2008; 83(1): 89– 93.
84. Mencarelli M, Spanhol- Rosseto A, Artuso R, Rondinella D, De Filippis R, Bahi- Buisson N et al. Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J Med Genet 2009. Epub ahead of print.
85. Papa FT, Mencarelli MA, Caselli R, Katzaki E, Sampieri K, Meloni I et al. A 3 Mb deletion in 14q12 causes severe mental retardation, mild facial dysmorphisms and Rett‑like features. Am J Med Genet A 2008; 146A(15): 1994– 1998.
86. Tan K, Shaw AL, Madsen B, Jensen K, Taylor- Papadimitriou J, Freemont PS. Human PLU- 1 Has transcriptional repression properties and interacts with the developmental transcription factors BF- 1 and PAX9. J Biol Chem 2003; 278(23): 20507– 20513.
87. Yao J, Lai E, Stifani S. The winged- helix protein brain factor 1 interacts with groucho and hes proteins to repress transcription. Mol Cell Biol 2001; 21(6): 1962– 1972.
88. Nakashiba T, Ikeda T, Nishimura S, Tashiro K, Honjo T, Culotti JG et al. Netrin‑G1: a novel glycosyl phosphatidylinositol‑linked mammalian netrin that is functionally divergent from classical netrins. J Neurosci 2000; 20(17): 6540– 6550.
89. Nakashiba T, Nishimura S, Ikeda T, Itohara S. Complementary expression and neurite outgrowth activity of netrin‑G subfamily members. Mech Dev 2002; 111(1– 2): 47– 60.
90. Borg I, Freude K, Kübart S, Hoffmann K, Menzel C, Laccone F et al. Disruption of Netrin G1 by a balanced chromosome translocation in a girl with Rett syndrome. Eur J Hum Genet 2005; 13(8): 921– 927.
91. Budden SS. Management of Rett syndrome: a ten year experience. Neuropediatrics 1995; 26(2): 75– 77.
92. Ellaway C, Williams K, Leonard H, Higgins G, Wilcken B, Christodoulou J. Rett syndrome: randomized controlled trial of L- carnitine. J Child Neurol 1999; 14(3): 162– 167.
93. Ellaway CJ, Peat J, Williams K, Leonard H, Christodoulou J. Medium‑term open label trial of L– carnitine in Rett syndrome. Brain Dev 2001; 23 (Suppl 1): S85– S89.
94. Plioplys AV, Kasnicka I. L- carnitine as a treatment for Rett syndrome. South Med J 1993; 86(12): 1411– 1412.
95. Egger J, Hofacker N, Schiel W, Holthausen H. Magnesium for hyperventilation in Rett‘s syndrome. Lancet 1992; 340(8819): 621– 622.
96. McArthur AJ, Budden SS. Sleep dysfunction in Rett syndrome: a trial of exogenous melatonin treatment. Dev Med Child Neurol 1998; 40(3): 186– 192.
97. Williamson SL, Christodoulou J. Rett syndrome: new clinical and molecular insights. Eur J Hum Genet 2006; 14(8): 896– 903.
98. Weaving LS, Ellaway CJ, Gécz J, Christodoulou J. Rett syndrome: clinical review and genetic update. J Med Genet 2005; 42(1): 1– 7.
99. Rosipal R, Zeman J, Hadac J, Misovicova N, Nevsimalova S, Martasek P. Analysis of the most frequent mutations in girls with Rett syndrome. Cas Lek Cesk 2001; 140(15): 473– 476.
100. Zahorakova D, Rosipal R, Hadac J, Zumrova A, Bzduch V, Misovicova N et al. Mutation analysis of the MECP2 gene in patients of Slavic origin with Rett syndrome: novel mutations and polymorphisms. J Hum Genet 2007; 52(4): 342– 348.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2009 Číslo 6
Nejčtenější v tomto čísle
- Varianty katatonie
- Rettův syndrom
- Neuropatie nervus mentalis jako manifestace systémové malignity
- Syndróm karpálneho tunela