
Genetika Parkinsonovy nemoci
The Genetics of Parkinson’s Disease
Parkinson’s dise ase (PD) is the second most common ne urodegenerative disorder affecting more than 1% of the populati on over the age of 60 ye ars. PD is ca used by loss of dopaminergic ne urons in the substanti a nigra pars compacta; however the exact eti ology of the cell damage is unknown. Multiple risk factors are supposed in the eti ology of PD, such as harmful environmental influences, genetic alterati ons and bi ological ageing. Altho ugh PD is mainly sporadic dise ase, abo ut 5– 10% of cases are ca used by a gene mutati on (monogenic form of PD). To date, 12 genetic loci and eight genes associ ated with monogenic PD were identifi ed. In this revi ew, we present the basic overvi ew of the genetics of PD. Individu al genes are introduced, including the molecular pathology of their protein products.
Key words:
Parkinson’s disease – genetics – mutation – molecular pathology
Autoři:
O. Fiala; E. Růžička
Působiště autorů:
Neurologická klinika 1. LF UK a VFN v Praze
Vyšlo v časopise:
Cesk Slov Neurol N 2009; 72/105(5): 419-428
Kategorie:
Přehledný referát
Souhrn
Parkinsonova nemoc (PN) je druhé nejčastější ne urodegenerativní onemocnění postihující více než 1 % osob starších 60 let. Nemoc vzniká na podkladě ztráty dopaminergních ne uronů v substanti a nigra pars compacta, přesná eti ologi e buněčného poškození však není známa. Předpokládá se, že na vzniku PN se podílí více faktorů, mezi které patří negativní vlivy prostředí, genetické změny a bi ologické stárnutí. Ačkoli je PN převážně sporadické onemocnění, asi v 5– 10 % případů je způsobena geneticko u mutací (monogenní forma PN). Doposud bylo identifikováno 12 lokusů a osm genů zodpovědných za monogenní PN. Tento so uhrnný článek přináší základní přehled genetiky PN. Uvedeny jso u jednotlivé geny včetně molekulární patologi e jejich bílkovinných produktů.
Klíčová slova:
Parkinsonova nemoc – genetika – mutace – molekulární patologie

Úvod
Parkinsonova nemoc (PN) je druhým nejčastějším neurodegenerativním onemocněním postihujícím celosvětově více než 1 % osob starších 60 let [1]. Klinický obraz PN je charakterizován přítomností klidového třesu, bradykineze, rigidity a posturální instability. Hybné postižení bývá stranově asymetrické a je doprovázeno non‑motorickými projevy, k nimž patří vegetativní dysfunkce, kognitivní deficit nebo psychiatrické komplikace [2]. Důležitým rysem onemocnění je odpovídavost příznaků na dopaminergní terapii. PN má progresivní charakter, kauzální terapie neexistuje.
V neuropatologickém nálezu nacházíme masivní úbytek dopaminergních neuronů v substantia nigra pars compacta (SNc), makroskopicky se manifestující depigmentací této oblasti (ztráta neuromelaninu) [3]. Typická je přítomnost eozinofilních nitrobuněčných inkluzí, tzv. Lewyho tělísek, která obsahují agregáty alfa‑synukleinu a dalších substancí [4]. Degenerativní změny a Lewyho tělíska bývají kromě SNc přítomny i v jiných částech mozku, což odpovídá novému pohledu na PN jako na onemocnění, které postihuje celý nervový systém [5].
Molekulární patogeneze PN je složitou mozaikou různě významných a vzájemně interagujících patogenních mechanizmů. K nejvýznamnějším patří agregace alfa‑synukleinu [6], poruchy odbourávání proteinů v ubikvitin‑proteazomovém systému (rámeček 1) [7], mitochondriální dysfunkce a oxidativní stres , zánětlivá reakce spojená s gliální aktivací [10], aberantní reaktivace buněčného cyklu [11] či dysregulace apoptózy [12]. Tyto patologické změny vznikají na základě několika primárních příčin. Jsou jimi negativní vlivy vnějšího prostředí [13], biologické stárnutí [14] a genetické změny (mutace a polymorfizmy) [15], o kterých budeme dále hovořit.
Fenotypové varianty PN
PN začíná nejčastěji mezi 60.–70. rokem věku. V anglosaské literatuře bývá pro tuto formu užíváno označení late-onset (PN s pozdním začátkem) a její projevy odpovídají „klasickému“ obrazu PN. Asi 5–10 % nemocných má první příznaky nemoci již před 40. rokem věku [16]. Tato podoba PN, zvaná early-onset (PN s časným začátkem), se do určité míry odlišuje. Pacienti s časným začátkem mají obvykle pomalejší progresi onemocnění, dobrou odpovídavost na dopaminergní terapii a brzký rozvoj polékových dyskinezí. Častá je přítomnost dystonie a zlepšení motorických příznaků po vyspání (tzv. sleep benefit). Kognitivní funkce zůstávají u většiny nemocných dlouhou dobu normální. V následujícím textu bude popis fenotypu jednotlivých mutací zjednodušen na dělení early-onset/late-onset s tím, že v některých případech budou uvedeny další klinické rysy.
Genetika PN
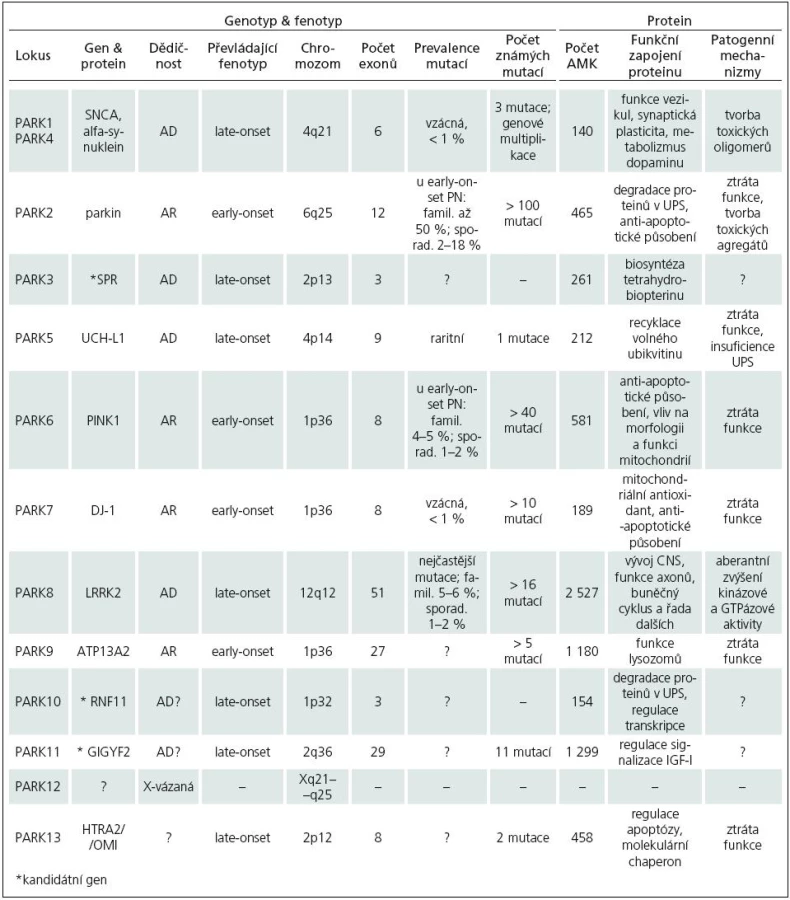
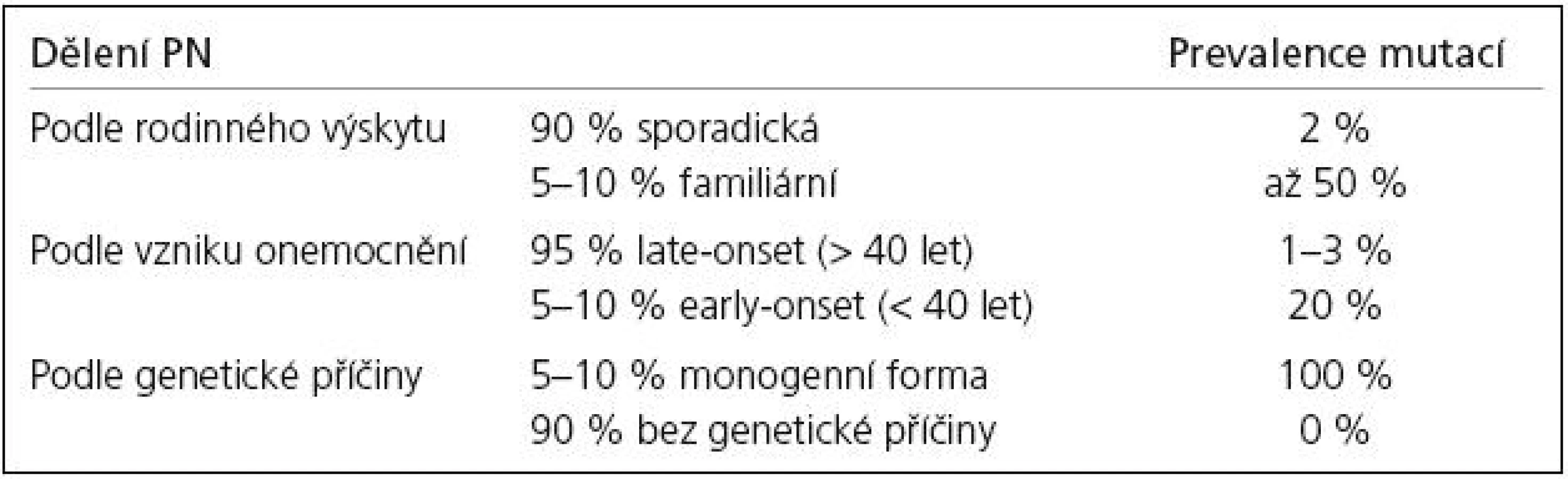
Zmínky o hereditárních aspektech PN jsou staré více než 100 let. Již Leroux [17] a Gowers [18] upozorňují na relativně vysoké procento familiárního výskytu PN. Přesto byla PN dlouho považována za výhradně sporadické onemocnění. Přelom ve výzkumu genetiky PN nastal až v 90. letech 20. století. V současné době je známo 12 lokusů a 8 genů (tab. 1), v nichž byly identifikovány mutace zodpovědné za monogenní PN. Tato forma PN je přenášena vždy jen jedním genem a tvoří asi 5–10 % z celkového počtu onemocnění.
Výskyt PN u několika členů rodiny (5–10 %) svědčí pro možný genetický přenos, nicméně svoji roli mohou sehrát i sdílené vlivy prostředí. Pravděpodobnost záchytu mutace u familiární formy PN je značně vysoká (v některých případech až 50 %), nelze ji však vyloučit ani u sporadických případů (vznik mutace de novo).
Přítomnost mutace ještě neznamená, že dojde k rozvoji onemocnění. O jejím fenotypovém projevu – penetranci, rozhoduje celá řada proměnných. Některé mutace zasahují do patogeneze v takové míře, že nemoc propukne vždy bez ohledu na okolní vlivy (kompletní penetrance). V jiných případech genetická změna indukuje pouze částečnou odchylku od normy a pro její uplatnění je třeba spolupůsobení dalších faktorů (inkompletní penetrance). Takovým příkladem je věkově vázaná penetrance, kdy se zděděná vloha manifestuje až v přítomnosti patologických změn hromadících se v průběhu stárnutí.
Genetické odchylky nenalézáme jen v exonovém úseku genu (části kódující protein), ale mohou se vyskytovat i v jiných oblastech, např. promotoru, jehož změny ovlivňují genovou expresi. Varianty genů, které zvyšují riziko rozvoje PN, ale samy o sobě ji nevyvolávají, nazýváme polymorfizmy. Na rozdíl od mutací, jež způsobují poškození proteinu, polymorfizmy pouze pozměňují funkci bílkoviny nebo zasahují do exprese genu.
U monogenní PN se předpokládají mendelovská pravidla autozomálně dominantní (AD) a autozomálně recesivní (AR) dědičnosti. Prevalence mutací a typ jejich přenosu se u jednotlivých forem monogenní PN liší. U late-onset fenotypu, který je obvykle spojen s AD přenosem, mutace nacházíme v 1–3 %. Early-onset forma PN se dědí převážně AR a mutace jsou přítomny až ve 20 % případů. Souhrn jednotlivých forem PN a četnost výskytu mutací uvádí tab. 2.
Geny asociované s monogenní PN
PARK1/4 – SNCA
Genotyp a fenotyp
V roce 1996 byl objeven první lokus pro PN – PARK1 [19]. V tomto lokusu byla u rodiny s AD PN nalezena bodová mutace A53T genu SNCA, kódující alfa‑synuklein [20] a později identifikovány další dvě mutace, A30P [21] a E46K [22]. Projevy onemocnění se blíží late-onset fenotypu, mají ale časnější začátek (40–50 let), rychlejší progresi, častá je také přítomnost demence. Fenotyp mutace E46K odpovídá spíše demenci s Lewyho tělísky. Kromě bodových mutací jsou známy také duplikace a triplikace celého genu (nesou označení PARK4) [23]. Nemocní s triplikací genu mají časný začátek, rychlejší progresi a těžší projevy PN (demence, vegetativní dysfunkce) než pacienti s duplikacemi, jejichž fenotyp se více podobá obrazu late-onset. U sporadické PN byly zachyceny též polymorfizmy SNCA, které jsou spojovány s vyšším rizikem vzniku onemocnění. Mutace SNCA jsou vzácné (méně než 1 % nemocných), polymorfizmy bývají ale přítomny poměrně často.
Protein
Alfa‑synuklein je za fyziologických podmínek nesbalený presynaptický protein s nízkou tendencí zaujímat specifickou sekundární strukturu [26]. Tvoří asi 1 % všech bílkovin CNS a jeho agregáty jsou hlavní součástí Lewyho tělísek [4]. Depozita alfa‑synukleinu nacházíme také u dalších neurodegenerativních onemocnění, souhrnně označovaných synukleinopatie (např. multisystémová atrofie, demence s Lewyho tělísky a další). Přesná funkce alfa‑synukleinu není známa. Existují doklady o jeho vlivu na funkci vezikul, synaptickou plasticitu či metabolizmus dopaminu [27,28].
Konformační chování alfa‑synukleinu reaguje na vnější vlivy a může být modifikováno jejich změnami [29]. V monomerním stavu dokáže původně nesbalený protein zaujímat různé sekundární struktury a má schopnost polymerizovat. Polymerizace postupuje od dimerů k morfologicky odlišným formám oligomerů a protofibril a je zakončena tvorbou nerozpustných agregátů. Agregaci indukuje zvýšená koncentrace alfa‑synukleinu, změna pH či přítomnost některých kovů [30]. Tvorba inkluzí byla prokázána také při působení organických rozpouštědel [31] a látek blízkých herbicidům (např. paraquat) [32]. Úlohu v jejich formování má zřejmě i tkáňová transglutamináza [33]. Fosforylace a další posttranslační modifikace alfa‑synukleinu mohou rovněž ovlivnit jeho solubilitu [34]. Mutace mají za následek snadnější tvorbu fibril [35], která je přítomna také u poruch odbourávání alfa‑synukleinu v UPS a lysozomech [36].
Která z forem alfa‑synukleinu je toxická a jakým způsobem indukuje buněčnou smrt, jsou dvě klíčové otázky, na které stále neznáme detailní odpověď. Obecné mechanizmy neurotoxického působení alfa‑synukleinu lze shrnout do tří skupin: mechanické poškození buněčných kompartmentů, ztráta fyziologických funkcí a získání nových toxických vlastností. Tyto procesy se vzájemně nevylučují a mohou působit synergicky [37]. Stále více dokladů nasvědčuje hypotéze, že za toxicitu alfa‑synukleinu jsou zodpovědné rozpustné oligomery (protofibrily) spíše než maturované fibrily a agregáty [38]. Jsou‑li oligomery skutečně toxické, pak by formování agregátů mohlo mít ochranný efekt. Tuto úvahu podporuje studie, ve které farmakologicky indukovaná tvorba inkluzí působila neuroprotektivně [39]. Lze tedy spekulovat, že Lewyho tělíska vznikají jako výsledek snahy neuronu eliminovat toxické oligomery a nejsou příčinou, nýbrž důsledkem neurotoxicity. Kromě nitrobuněčné patologie byla toxická aktivita alfa‑synukleinu prokázána také v extracelulárním prostoru, kde agregáty vyvolávají mikrogliální aktivaci [40].
PARK2 – parkin
Genotyp a fenotyp
Roku 1998 byla v oblasti lokusu PARK2 nalezena mutace genu kódujícího protein parkin [41]. Je známo více než 100 mutací parkinu, zahrnující bodové mutace či exonové delece a duplikace [42]. Jejich výskyt je asociován s AR PN a early-onset fenotypem, ve kterém se může navíc objevit hyperreflexie či symetrický rozvoj onemocnění. V sekčním nálezu často chybějí Lewyho tělíska. Mutace jsou přítomny až u 50 % familiární a 2–18 % sporadické early‑onset PN. Frekvence záchytu závisí na věku při prvních projevech nemoci (< 20 let – 60 %; > 30 let – 10 %). Riziko vzniku onemocnění mohou zvyšovat také genetické varianty promotoru [43]. Řada nemocných (až 50 %) má pouze jednu mutaci v heterozygotní konstituci [44]. Předpokládá se, že přítomnost heterozygotní mutace potencuje riziko rozvoje PN a snižuje věk vzniku onemocnění [45]. Studie využívající 18F-dopa pozitronovou emisní tomografii (PET) ukázaly u asymptomatických přenašečů heterozygotní mutace presynaptickou dopaminergní dysfunkci ve striatu [46].
Protein
Parkin hraje úlohu v odbourávání proteinů pomocí UPS. Má aktivitu E3 ubikvitin ligázy, enzymu, který váže ubikvitin na bílkoviny určené k degradaci v proteazomu [47]. Mezi jeho substráty patří např. Pael-R (Parkin‑associated endothelin Receptor‑like receptor) [48], alfa‑synuklein [49] a další. Anti‑apoptotický efekt parkinu byl prokázán jak in vitro, tak in vivo [50,51]. Mechanizmem, jímž parkin přispívá k inhibici apoptózy, může být např. ubikvitinace substrátu Pael-R, který má schopnost indukovat buněčnou smrt [52]. Parkin také zvyšuje transkripční aktivitu NK- κB (Nuclear factor Kappa B), významného induktoru anti‑apoptotických genů [53], a podílí na regulaci metabolizmu mitochondriální DNA (mtDNA) [54]. Rozšířená hypotéza předpokládá, že nedostatečná degradace substrátů parkinu (na základě jeho dysfunkce) způsobuje buněčné poškození. Tím lze však vysvětlit podíl parkinu na patogenezi PN jen zčásti, neboť řada mutací s ligázovou aktivitou parkinu neinterferuje [55], ale např. zhoršují jeho rozpustnost [56]. Agregovaný parkin poškozuje buněčný cytoskelet, narušuje funkci UPS a přispívá k zániku dopaminergních neuronů [57].
PARK3
Genotyp a fenotyp
PARK 3 je lokus asociovaný s AD PN a late--onset fenotypem blízkým obrazu sporadického onemocnění [58]. Z kandidátních genů je nejsuspektnější gen kódující sepiapterin reduktázu (SPR) [59].
Protein
SPR katalyzuje biosyntézu tetrahydrobiopterinu (BH4), důležitého kofaktoru hydroláz aromatických aminokyselin, mezi které patří také tyrosin hydroxyláza (TH), esenciální enzym syntézy dopaminu. Analýza mozkové tkáně pacientů s PN doložila zvýšenou expresi SPR v porovnání s kontrolami [60]. Detailnější popis úlohy SPR v patogenezi PN je předmětem dalšího výzkumu.
PARK5 – UCH-L1
Genotyp a fenotyp
Mutace I93M v genu kódujícím UCH-L1(Ubiquitin Carboxy‑terminal Hydrolase L1) byla identifikována v roce 1998 u sourozenců s AD PN, fenotypově blízkou sporadické late‑onset formě [61]. Tento nález však nebyl nikdy zopakován. Byla ale objevena častá genetická varianta S18Y, která je asociována s nižším rizikem rozvoje PN [62].
Protein
Neuron-specifický enzym UCH-L1 představuje 1–2 % všech bílkovin mozkové tkáně a byl detekován také v Lewyho tělíscích [63]. UCH-L1 se podílí na degradaci proteinů skrze UPS (rámeček 1). V závislosti na konformaci vykazuje dvojí enzymatickou aktivitu. V monomerní konstituci hydrolyzuje polyubikvitinové řetězce, a umožňuje tak recyklaci ubikvitinu, dimer má naopak funkci ubikvitin ligázy (katalyzuje vazbu volného ubikvitinu na cílové proteiny). Monomerní forma přispívá k proteazomové degradaci alfa‑synukleinu (díky zvýšenému poolu volného ubikvitinu), dimer váže ubikvitin na alfa‑synuklein pomocí vazby Lys-63, která však nemá za následek odstranění v proteazomu, ale podporuje jeho agregaci [64]. Mutace I93M přispívá k tvorbě dimerů a potlačuje hydrolázovou aktivitu UCH-L1. Oproti tomu varianta S18Y kóduje protein, který nedimerizuje, akceleruje degradaci alfa‑synukleinu a má protektivní antioxidační účinky [65].

PARK6 – PINK1
Genotyp a fenotyp
Lokus PARK6 byl popsán v roce 2001 u rodiny s výskytem AR formy PN [66]. Později byla v tomto lokusu nalezena mutace G309D genu kódujícího PINK1 (PTEN-Induced Kinase 1) [67] a dále identifikováno přes 40 dalších genetických variant. Fenotyp zpravidla odpovídá early‑onset PN, jsou však známy i případy s late‑onset fenotypem. Mutace jsou přítomny u 4–5 % pacientů s familiárním výskytem early‑onset PN a v 1–2 % u sporadické formy onemocnění. Význam heterozygotních mutací je předmětem diskuze [68]. PET ukázala u asymptomatických heterozygotů s PINK1 mutací presynaptickou dopaminergní dysfunkci ve striatu [69].
Protein
PINK1 je mitochondriální kináza s anti‑apoptotickým vlivem, jejíž aktivita byla prokázána též v Lewyho tělíscích [70]. PINK1 snižuje uvolňování cytochtomu c (mitochondriálního mediátoru apoptózy) a aktivaci kaspázy 3 [71]. PINK1 interaguje také s parkinem (PARK2), oba jsou pravděpodobně součástí jedné anti‑apoptotické kaskády [72] a mají vliv na morfologii a funkci mitochondrií [73]. Řada mutací se nachází v oblasti kinázové domény PINK1 [74] a předpokládá se, že ztráta kinázové aktivity je hlavním patogenním mechanizmem.
PARK7 – DJ-1
Genotyp a fenotyp
V roce 2001 byl v rodině s AR dědičnou PN a early‑onset fenotypem popsán lokus PARK7 [75] a později byla v tomto lokusu identifikována bodová mutace L166P genu DJ-1 [76]. V současnosti je známo více než 10 mutací, jejichž výskyt u pacientů s early‑onset PN nepřesahuje 1 %. Role heterozygotních mutací DJ-1 v patogenezi PN není zcela jednoznačná. U přenašečů této formy nebyla na PET zachycena patologie dopaminergního metabolizmu ve striatu [77] a zdá se, že její přítomnost nemá významnější klinický dopad.
Protein
DJ-1 byl poprvé identifikován jako onkogen [78]. Tato homodimerní bílkovina má funkci mitochondriálního antioxidantu, jehož exprese je indukována přítomností volných radikálů (ROS, Reactive Oxygen Species) [79]. K eliminaci ROS přispívá DJ-1 více mechanizmy. Je schopen vlastní autooxidace [80], stabilizuje regulační faktor transkripce antioxidantů Nrf2 (Nuclear factor erythroid 2‑related factor) [81] a zvyšuje buněčnou koncentraci glutationu [82]. DJ-1 má také anti‑apoptotické vlastnosti [83]. Mutace narušují funkci DJ-1 zejména snížením jeho stability [84]. Např. mutace L166P mění strukturu proteinu a zabraňuje jeho dimerizaci, což vede k zvýšené degradaci DJ-1 v UPS [85].
PARK8 – LRRK2
Genotyp a fenotyp
Lokus PARK8 byl popsán v roce 2002 u rodiny s AD dědičnou late‑onset PN [86]. Později se podařilo v místě lokusu identifikovat mutace genu kódujícího protein dardarin (název odvozen z baskického slova „dardara“, znamenající třes). Nyní je označován jako LRRK2 – Leucine Rich Repeat Kinase 2 [87]. Dodnes bylo popsáno více než 50 genetických variant, nejméně 16 patogenních. Fenotyp obvykle odpovídá projevům late‑onset sporadické PN, onemocnění však může mít i časný začátek. Substituce G2019S, s penetrancí závislou na věku (50 let – 17 %; 70 let – 85 %), představuje vůbec nejčastější mutaci u PN. Vyskytuje se u 5–6 % familiární a 1–2 % sporadické formy onemocnění [88]. Její prevalence však značně kolísá v závislosti na studovaném etniku. Mezi severoafrickými Araby [89] a aškenázskými Židy [90] ji nacházíme u pacientů v řádu desítek procent, naopak u Asiatů je záchyt mutace vzácný [91]. V této populaci je ale častá přítomnost polymorfizmů G2385R a R1628P, které jsou asociovány se zvýšeným rizikem vzniku PN [92,93].
Protein
LRRK2 je multifunkční protein s kinázovou a GTPázovou aktivitou [94]. Široké spektrum funkčního působení LRRK2 není stále objasněno. Studie využívající DNA čipy (microarray) nalezla po vyřazení LRRK2 změnu v expresi 187 genů důležitých pro vývoj CNS, funkci axonů, buněčný cyklus apod. [95]. LRRK2 ovlivňuje endocytózu a dynamiku vezikul [84] a má schopnost vázat α- a β-tubulin [96]. Exprese mutantní LRRK2 vyvolává oxidativní stres a indukuje mitochondriální apoptotickou dráhu [97]. LRRK2 může vytvářet komplexy s chaperonem Hsp90 (Heat shock protein 90), který ji stabilizuje a potencuje tak toxický vliv [98]. Pro patogenní působení LRRK2 je klíčová její aberantní kinázová aktivita. Nejčastější mutace G2019S tuto aktivitu zvyšuje [99]. Rovněž modifikace GTPázové aktivity, způsobená mutacemi, má význam pro toxicitu LRRK2 [100].
PARK9 – ATP13A2
Genotyp a fenotyp
V roce 2001 byla u rodiny s výskytem AR dědičného Kuforova-Rakebova syndromu (KRS) objevena asociace s lokusem PARK9 [101]. KRS je charakterizován časným rozvojem rychle progredujícího parkinsonského syndromu se zachovanou odpovídavostí na dopaminergní terapii, pyramidovu symptomatikou, supranukleární obrnou pohledu a demencí [102]. Později se podařilo prokázat v lokusu PARK9 mutace genu ATP13A2 (ATPáza typ 13A2) [103]. Další studie doložila výskyt homozygotní mutace G504R u pacienta s juvenilní PN (= vznik onemocnění do 20 let věku) a přítomnost dvou mutací v heterozygotní konstituci u nemocných s early‑onset PN [104].
Protein
Gen ATP13A2 kóduje lysozomální ATPázu. U pacientů s PN byla doložena její zvýšená aktivita v SNc [103]. Detailní funkce ATP13A2 jsou stále předmětem výzkumu.
PARK10
Genotyp a fenotyp
Lokus PARK10 je asociován s late‑onset fenotypem PN [105]. Nejsuspektnějším kandidátním genem se zdá RNF11 (RING-finger protein 11), jehož změny exprese byly popsány v SNc u pacientů s PN [106].
Protein
RNF11 byl prokázán v různých částech mozku a je přítomen i v Lewyho tělíscích [107]. Podobně jako parkin (PARK2) se podílí na ubikvitinaci a degradaci proteinů [108]. RNF11 má také úlohu v signalizačních drahách růstových faktorů a regulaci transkripce [109].
PARK11
Genotyp a fenotyp
V roce 2008 byly v lokusu PARK11 nalezeny mutace genu GIGYF2 (Grb10-Interacting GYF protein‑2, zvaný též TNRC15 – Trinucleotide Repeat Containing 15;) u rodin s late‑onset fenotypem PN, AD přenosem a inkompletní, věkově vázanou penetrancí [110].
Protein
Protein GIGYF2 interaguje s Grb10 (Growth factor receptor-bound protein) a společně s ním kooperuje na regulaci signalizace zprostředkované IGF‑I (Insulin‑like Growth Factor I) a jeho receptorem [111]. Vzhledem k tomu, že IGF‑I a inzulin mají důležitou úlohu v CNS [112] a zřejmě i v patogenezi PN [113], lze předpokládat, že také GIGYF2 bude zasahovat do patogenetických mechanizmů tohoto onemocnění.
PARK12
Dalším lokusem pro PN je X-vázaný PARK12. Kromě asociace tohoto lokusu s PN však zatím nebyly identifikovány žádné kandidátní geny.
PARK13 – HRTA2/OMI
Genotyp a fenotyp
V roce 2005 byla v oblasti lokusu PARK13 objevena mutace genu HTRA2/OMI (High Temperature Requirement protein A2, nebo též Omi stress-regulated endoprotease) u pacientů se sporadickou PN a late‑onset fenotypem [115]. V rozporu s tímto nálezem je však jiná studie, která asociaci mutací HTRA2/OMI s PN neprokázala [116].
Protein
HTRA2/OMI je mitochondriální proteáza, která se vyskytuje také v Lewyho tělíscích [115]. Má částečný pro‑apoptotický vliv [117], avšak hlavní funkce HTRA2/OMI (podobně jako u její bakteriální varianty) spočívá v podpoře buněčného přežití skrze stabilizaci důležitých bílkovin a degradaci defektních proteinů [118]. Mutace HTRA2/OMI mají za následek snížení proteolytické aktivity.
Mitochondriální DNA
Mitochondriální dysfunkce je významný patogenetický mechanizmus nejen u PN, ale i u dalších neurodegenerativních onemocnění [8]. Maternálně dědičná PN (resp. parkinsonský syndrom), způsobená mutacemi mitochondriálních genů, je raritním nálezem, obvykle spojeným s širší symptomatikou (např. neuropatií a myopatií) [119]. Řada prací poukazuje na vztah PN k specifickým haplotypům a polymorfizmům mtDNA. Polymorfizmus 10398G genu ND3 (NADH dehydrogenáza 3, podjednotka komplexu I) signifikantně snižuje riziko vzniku PN v evropské populaci [120], naopak vyšší pravděpodobnost rozvoje PN byla zaznamenána u jedinců s haplotypy ze skupiny JTIWX (finská populace) [121].
Jelikož jsou mitochondrie významným producentem ROS, dochází k poškození mtDNA poměrně často. U pacientů s PN byl doložen zvýšený počet získaných delecí mtDNA v SNc [122,123]. Opravu poškozené mtDNA zajišťuje mitochondriální DNA polymeráza gama POLG1. Mutace POLG1 se vyskytují u nemocných s progresivní externí oftalmoplegií, jež je doprovázena L-dopa responzivním parkinsonizmem, a existují doklady o tom, že polymorfizmy POLG1 jsou rizikovým faktorem sporadické PN [124].
Kandidátní geny
Polymorfizmy a mutace v řadě kandidátních genů mohou ovlivnit riziko vzniku PN. Produkty těchto genů hrají roli v rozličných molekulárních mechanizmech, jako je metabolizmus dopaminu (např. MAO-B – monoaminooxidáza B) či detoxikace cizorodých látek (např. cytochrom P450) [125]. Stále narůstající seznam kandidátních genů, včetně přehledu asociačních studií, je pravidelně doplňován na internetových stránkách PDGene database (www.pdgene.org).
NR4A2 (Nuclear Receptor subfamily 4,group A, member 2; označovaný též Nurr1 – Nuclear receptor‑related 1), je transkripční faktor, který se podílí na regulaci exprese alfa‑synukleinu [126]. Nálezy polymorfizmů a mutací NR4A2 u pacientů s PN svědčí pro jeho možný význam v patogenezi onemocnění [127].
U Gaucherovy nemoci, střádavého AR dědičného onemocnění způsobeného deficitem lysozomálního enzymu glukocerebrosidázy (GBA), byl opakovaně popsán výskyt parkinsonského syndromu (ojediněle dokonce i PN) [128] a u PN zase zachyceny mutace GBA [129]. Vzhledem k tomu, že lysozomální metabolizmus se podílí na degradaci alfa‑synukleinu [130], lze předpokládat, že mutace GBA narušují lysozomální funkce rezultující v jeho snížené odbourávání.
Gen MAPT (Microtubule-Associated Protein Tau) kóduje protein tau, který tvoří intracelulární neurofibrilární klubka (tangles) u tautopatií (např. kortikobazální degenerace či progresivní supranukleární obrna). Agregáty proteinu tau byly zachyceny také u sporadické [131] i familiární PN [132]. Genetické analýzy definovaly dva haplotypy genu MAPT, H1 a H2. Bylo prokázáno, že osoby s haplotypem H1 mají zvýšené riziko rozvoje PN [133].
Synphilin‑1 (též SNCAIP – Synuclein‑Alpha-Interacting Protein) je součást Lewyho tělísek, má schopnost vázat alfa‑synuklein a přispívá k tvorbě jeho agregátů [134]. Substituce R621C v genu kódujícím synphilin‑1 byla popsána u dvou pacientů se sporadickou formou PN [135]. Signifikantnost vazby polymorfizmů synphilinu-1 s PN je však sporná [136].
Závěr
Výzkum genů a jejich proteinových produktů významně napomáhá odhalovat molekulární patogenezi PN. Její detailní poznání je klíčovým předpokladem pro vývoj nové neuroprotektivní léčby a preklinické diagnostiky. Vzhledem k tomu, že PN začíná převážně v pokročilém věku, je pravděpodobné, že se na jejím vzniku výrazně podílí biologické stárnutí [14]. Stárnoucí organizmus má sníženou schopnost čelit negativním vlivům prostředí, které spolu s genetickou dispozicí rozhodují o rozvoji nemoci. Podíl genetické složky však není konstantní. Vedoucí úlohu má u monogenní formy, kde je hlavní příčinou PN mutace. Vysoký záchyt mutací u pacientů s časným začátkem (až ve 20 %) ukazuje, že v těchto případech je genetický defekt silným patogenním podnětem, který se dokáže uplatnit do značné míry nezávisle na ostatních faktorech. Monogenní forma představuje ale jen malý zlomek (5–10 %) všech případů PN. U sporadického onemocnění, kde není jednoznačná genetická příčina známa, mohou k rozvoji nemoci přispět genové polymorfizmy [137]. Hledání nových polymorfizmů, stejně tak jako kandidátních genů, však vyžaduje kritický přístup. Rozsáhlý seznam genů v PDGene databázi vyvolává otázku, zda má skutečně každý registrovaný gen reálný podíl na vzniku PN. Pro výběr kandidátních genů by bylo vhodné stanovit spíše přísnější kritéria, která zohlední pouze geny, u nichž byla opakovaně prokázána signifikantní asociace s PN a jejichž proteinové produkty hrají významnou úlohu v patogenezi onemocnění. Podobné podmínky by měly platit i pro geny zodpovědné za monogenní PN. Sporným zůstává zařazení PARK5 mezi lokusy pro PN, neboť popis mutace UCH-L1 (PARK5) je stále ojedinělým nálezem [61]. Podobnou kontroverzi přináší i lokus PARK13 (gen HTRA2/OMI), kde není doložena jednoznačná asociace mutací s PN [116].
V genetice PN lze najít určitá pravidla, která ale mají řadu výjimek. Obecně platí, že nejvyšší pravděpodobnost záchytu mutace je u mladého pacienta s familiární PN, nejnižší pak u nemocného s pozdním začátkem sporadické formy. Z klinického pohledu by bylo ideální znát pro každý typ mutace specifický obraz onemocnění. Uvedené klinické rysy mutací jsou však jen orientační a neplatí vždy. Díky řadě epigenetických faktorů, které ovlivňují vznik PN, není možné přesně stanovit korelaci genotyp-fenotyp. Navíc rozdílné mutace stejného genu mohou vyvolat různou funkční poruchu proteinu, a tím i odlišný fenotyp. Klinická podoba mutací může být v některých případech také shodná s projevy sporadické PN (např. LRRK2, ale i část PINK1) [138].
Dědičnost mutací nemusí vždy odpovídat dogmatu mendelovské genetiky. U AR nemocí je mutace jedné alely kompenzována alelou druhou a nemělo by dojít k rozvoji patologie. Přesto až 50 % pacientů s mutací parkinu (PARK2), představitele AR formy PN, má jen jednu heterozygotní mutaci [44]. Zdá se tedy, že za určitých podmínek je tato mutace schopna imitovat AD typ dědičnosti a vyvolat příznaky onemocnění. Pro tento jev lze najít více vysvětlení. Množství proteinu produkovaného zdravou alelou nemusí stačit k zajištění fyziologických funkcí nebo mutantní protein negativně ovlivňuje expresi své normální varianty. Mutace může také indukovat novou patogenní funkci bílkoviny. Tento mechanizmus je častý u AD přenosu, který je typický pro LRRK2 (PARK8). Přestože je dědičnost LRRK2 považována za AD, překvapivě mnoho přenašečů heterozygotní mutace (až 70 %) nejeví známky onemocnění [44]. Navíc byl registrován značný počet pacientů s mutací v homozygotní konstituci, což je u AD onemocnění neobvyklé. Pozoruhodný je také záchyt asymptomatických homozygotů [139].
V histopatologickém nálezu pacientů s mutací parkinu často chybí Lewyho tělíska, nicméně i zde existují výjimky [141]. Variabilita přítomnosti Lewyho tělísek nasvědčuje hypotéze, že PN může vznikat na základě odlišných patogenních mechanizmů rezultujících ve ztrátu dopaminergních neuronů. Je‑li toto tvrzení pravdivé, pak lze PN ve skutečnosti považovat za etiologicky heterogenní nozologickou jednotku s více či méně podobným fenotypem [141].
Přehled užitých zkratek
| AD | autozomálně dominantní |
| AR | autozomálně recesivní |
| ATP13A2 | ATPáza typ 13A2 |
| ATPáza | enzym, který hydrolyzuje ATP – adenosintrifosfát |
| BH4 | tetrahydrobiopterin |
| CNS | centrální nervová soustava |
| GBA | glukocerebrosidáza |
| GIGYF2 | grb10-interacting GYF protein‑2 |
| Grb10 | Growth factor receptor-bound protein |
| GTPáza | enzym, který hydrolyzuje GTP – guanosintrifosfát |
| Hsp90 | Heat shock protein 90 |
| HTRA2/OMI | High Temperature Requirement protein A2 |
| IGF‑I | Insulin‑like Growth Factor I |
| KRS | Kuforův-Rakebův syndrom |
| LRRK2 | Leucine Rich Repeat Kinase 2 |
| MAO-B | monoaminooxidáza B |
| MAPT | Microtubule-Associated Protein Tau |
| mtDNA | mitochondriální DNA |
| ND3 | NADH dehydrogenáza 3 |
| NK- κB | Nuclear factor kappa B |
| NR4A2 | Nuclear Receptor subfamily 4, group A, member 2 |
| Nrf2 | Nuclear factor erythroid 2‑related factor |
| Pael-R | Parkin‑associated endothelin Receptor‑like receptor |
| PET | pozitronová emisní tomografie |
| PINK1 | PTEN (Phosphatase and Tensin homolog) – induced kinase 1 |
| PN | Parkinsonova nemoc |
| POLG1 | mitochondriální DNA polymeráza gama |
| RNF11 | RING-finger protein 11 |
| ROS | Reactive Oxygen Species; volné radikály |
| SNc | Substantia Nigra pars compacta |
| SNCA | alfa‑synuklein |
| SPR | sepiapterin reduktáza |
| TH | tyrosin hydroxyláza |
| UCH-L1 | Ubiquitin Carboxy‑terminal Hydrolase L1 |
| UPS | ubikvitin‑proteazomový systém |
Poděkování: Autoři děkují prof. MUDr. Pavlu Martáskovi, DrSc., za cenné rady a kritické připomínky.Publikace byla podpořena výzkumným záměrem MŠMT VZ 0021620849 a grantem IGA MZ NR9215-3.
prof. MUDr. Evžen Růžička, DrSc.
Neurologická klinika
1. LF UK a VFN
Kateřinská 30
120 00 Praha 2
e-mail: eruzi@lf1.cuni.cz
Zdroje
1. de La u LM, Breteler MM. Epidemi ology of Parkinson‘s dise ase. Lancet Ne urol 2006; 5(6): 525– 535.
2. Jankovic J. Parkinson‘s dise ase: clinical fe atures and di agnosis. J Ne urol Ne urosurg Psychi atry 2008; 79(4): 368– 376.
3. Schulz JB, Falkenburger BH. Ne uronal pathology in Parkinson‘s dise ase. Cell Tissue Res 2004; 318(1): 135– 147.
4. Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson‘s dise ase: molecules implicated in the formati on and degradati on of alpha‑ synuclein aggregates. Ne uropathology 2007; 27(5): 494– 506.
5. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Ste ur EN, Braak E. Staging of brain pathology related to sporadic Parkinson‘s dise ase. Ne urobi ol Aging 2003; 24(2): 197– 211.
6. McNa ught KS, Olanow CW. Protein aggregati on in the pathogenesis of famili al and sporadic Parkinson‘s dise ase. Ne urobi ol Aging 2006; 27(4): 530– 545.
7. McNa ught KS, Jackson T, JnoBaptiste R, Kapustin A,Olanow CW. Prote asomal dysfuncti on in sporadic Parkinson‘s dise ase. Ne urology 2006; 66 (10 Suppl 4): S37– S49.
8. Onyango IG. Mitochondri al dysfuncti on and oxidative stress in Parkinson‘s dise ase. Ne urochem Res 2008; 33(3): 589– 597.
9. Tansey MG, McCoy MK, Frank‑ Cannon TC. Ne uro inflammatory mechanisms in Parkinson‘s dise ase: potenti al environmental triggers, pathways, and targets for e arly therape utic interventi on. Exp Ne urol 2007; 208(1): 1– 25.
10. Kim YS, Joh TH. Microgli a, major player in the brain inflammati on: their roles in the pathogenesis of Parkinson‘s dise ase. Exp Mol Med 2006; 38(4): 333– 347.
11. Herrup K, Yang Y. Cell cycle regulati on in the postmitotic ne uron: oxymoron or new bi ology? Nat Rev Ne urosci 2007; 8(5): 368– 378.
12. Burke RE. Programmed cell de ath and new discoveri es in the genetics of parkinsonism. J Ne urochem 2008; 104(4): 875– 890.
13. Dick FD, De Palma G, Ahmadi A, Scott NW, Prescott GJ, Bennett J et al. Environmental risk factors for Parkinson‘s dise ase and parkinsonism: the Ge oparkinson study. Occup Environ Med 2007; 64(10): 666– 672.
14. Tro ulinaki K, Tavernarakis N. Ne urodegenerative conditi ons associ ated with ageing: a molecular interplay? Mech Ageing Dev 2005; 126(1): 23– 33.
15. Tan EK, Skipper LM. Pathogenic mutati ons in Parkinson dise ase. Hum Mutat 2007; 28(7): 641– 653.
16. Schrag A, Schott JM. Epidemi ological, clinical, and genetic characteristics of e arly‑onset parkinsonism. Lancet Ne urol 2006; 5(4): 355– 363.
17. Lero ux P. Contributi on à l‘étude des ca uses de la paralysi e agitante. Paris: in Thesis 1890.
18. Gowers WR. A Manu al of Dise ases of the Nervo us System. Philadelphi a: Blakiston‘s Son 1900.
19. Polymeropo ulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iori o G et al. Mapping of a Gene for Parkinson‘s Dise ase to Chromosome 4q21– q23. Sci ence 1996; 274(5290): 1197– 1199.
20. Polymeropo ulos MH, Lavedan C, Leroy E, Ide SE, Deheji a A, Dutra A et al. Mutati on in the alpha‑ Synuclein Gene Identifi ed in Famili es with Parkinson‘s Dise ase. Sci ence 1997; 276(5321): 2045– 2047.
21. Krüger R, Kuhn W, Müller T, Wo italla D, Graeber M,Kösel S et al. Ala30Pro mutati on in the gene encoding alpha‑ synuclein in Parkinson‘s dise ase. Nat Genet 1998; 18(2): 106– 108.
22. Zarranz JJ, Alegre J, Gómez‑ Esteban JC, Lezcano E,Ros R, Ampuero I et al. The new mutati on, E46K, of alpha‑ synuclein ca uses Parkinson and Lewy body dementi a. Ann Ne urol 2004; 55(2): 164– 173.
23. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J et al. Alpha‑ Synuclein Locus Triplicati on Ca uses Parkinson‘s Dise ase. Sci ence 2003; 302(5646): 841.
24. Ibáñez P, Bonnet AM, Débarges B, Lohmann E,Tison F, Pollak P et al. Ca usal relati on between alpha‑ synuclein gene duplicati on and famili al Parkinson‘s dise ase. Lancet 2004; 364(9440): 1169– 1171.
25. Pals P, Lincoln S, Manning J, Heckman M, Skipper L,Hulihan M et al. Alpha‑ Synuclein promoter confers susceptibility to Parkinson‘s dise ase. Ann Ne urol 2004; 56(4): 591– 595.
26. Uversky VN, Li J, Fink AL. Evidence for a parti ally folded intermedi ate in alpha‑ synuclein fibril formati on. J Bi ol Chem 2001; 276(14): 10737– 10744.
27. Yu S, Uéda K, Chan P. Alpha‑ synuclein and dopamine metabolism. Mol Ne urobi ol 2005; 31(1– 3): 243– 254.
28. Clayton DF, Ge orge JM. Synucleins in synaptic plasticity and ne urodegenerative disorders. J Ne urosci Res 1999; 58(1): 120– 129.
29. Uversky VN. A protein‑chamele on: conformati onal plasticity of alpha‑ synuclein, a disordered protein involved in ne urodegenerative disorders. J Bi omol Struct Dyn 2003; 21(2): 211– 234.
30. Uversky VN, Li J, Fink AL. Metal‑ triggered structural transformati ons, aggregati on, and fibrillati on of human alpha‑ synuclein. A possible molecular NK between Parkinson‘s dise ase and he avy metal exposure. J Bi ol Chem 2001; 276(47): 44284– 44296.
31. Munishkina LA, Phelan C, Uversky VN, Fink AL. Conformati onal behavi or and aggregati on of alpha‑ synuclein in organic solvents: modeling the effects of membranes. Bi ochemistry 2003; 42(9): 2720– 2730.
32. Manning‑ Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraqu at ca uses up‑ regulati on and aggregati on of alpha‑ synuclein in mice: paraqu at and alpha‑ synuclein. J Bi ol Chem 2002; 277(3): 1641– 1644.
33. Junn E, Ronchetti RD, Quezado MM, Kim SY, Mo uradi an MM. Tissue transglutaminase‑induced aggregati on of alpha‑ synuclein: Implicati ons for Lewy body formati on in Parkinson‘s dise ase and dementi a with Lewy bodi es. Proc Natl Acad Sci USA 2003; 100(4): 2047– 2052.
34. Smith WW, Margolis RL, Li X, Troncoso JC, Lee MK, Dawson VL et al. Alpha‑ Synuclein Phosphorylati on Enhances Eosinophilic Cytoplasmic Inclusi on Formati on in SH‑ SY5Y Cells. J Ne urosci 2005; 25(23): 5544– 5552.
35. Greenba um EA, Graves CL, Mishizen‑ Eberz AJ, Lupoli MA, Lynch DR, Englander SW et al. The E46K mutati on in alpha‑ synuclein incre ases amylo id fibril formati on. J Bi ol Chem 2005; 280(9): 7800– 7807.
36. Vogi atzi T, Xilo uri M, Vekrellis K, Stefanis L. Wild type alpha‑ synuclein is degraded by chaperone‑ medi ated a utophagy and macro a utophagy in ne uronal cells. J Bi ol Chem 2008; 283(35): 23542– 23556.
37. Bennett MC. The role of alpha‑ synuclein in ne urodegenerative dise ases. Pharmacol Ther 2005; 105(3): 311– 331.
38. Goldberg MS, Lansbury PT jr. Is there a ca use--and‑ effect relati onship between alpha‑ synuclein fibrillizati on and Parkinson‘s dise ase? Nat Cell Bi ol 2000; 2(7): E115– E119.
39. Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT et al. Pharmacological promoti on of inclusi on formati on: a therape utic appro ach for Huntington‘s and Parkinson‘s dise ases. Proc Natl Acad Sci USA 2006; 103(11): 4246– 4251.
40. Lee SJ. Origins and effects of extracellular alpha‑ synuclein: implicati ons in Parkinson‘s dise ase. J Mol Ne urosci 2008; 34(1): 17– 22.
41. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S et al. Mutati ons in the parkin gene ca use a utosomal recessive juvenile parkinsonism. Nature 1998; 392(6676): 605– 608.
42. Hedrich K, Kann M, Lanthaler AJ, Dalski A, Eskelson C, Landt O et al. The importance of gene dosage studi es: mutati onal analysis of the parkin gene in e arly‑onset parkinsonism. Hum Mol Genet 2001; 10(16): 1649– 1656.
43. Tan EK, Puong KY, Chan DK, Yew K, Fo ok‑ Chong S,Shen H et al. Impaired transcripti onal upregulati on of Parkin promoter vari ant under oxidative stress and prote asomal inhibiti on: clinical associ ati on. Hum Genet 2005; 118(3– 4): 484– 488.
44. Klein C, Lohmann‑Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygo us mutati ons in genes associ ated with parkinsonism. Lancet Ne urol 2007; 6(7): 652– 662.
45. Sun M, Lato urelle JC, Wo oten GF, Lew MF, Klein C,Shill HA et al. Influence of Heterozygosity for Parkin Mutati on on Onset Age in Famili al Parkinson Dise ase: The GenePD Study. Arch Ne urol 2006; 63(6): 826– 832.
46. Khan NL, Bro oks DJ, Pavese N, Sweeney MG, Wo od NW, Lees AJ et al. Progressi on of nigrostri atal dysfuncti on in a parkin kindred: an [18F]dopa PET and clinical study. Brain 2002; 125(10): 2248– 2256.
47. Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S et al. Famili al Parkinson dise ase gene product, parkin, is a ubiquitin‑protein ligase. Nat Genet 2000; 25(3): 302– 305.
48. Imai Y, Soda M, Ino ue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can le ad to endoplasmic reticulum stress, is a substrate of Parkin. Cell 2001; 105(7): 891– 902.
49. Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R et al. Ubiquitinati on of a New Form of alpha-Synuclein by Parkin from Human Brain: Implicati ons for Parkinson‘s Dise ase. Sci ence 2001; 293(5528): 263– 269.
50. Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB et al. Parkin negatively regulates JNK pathway in the dopaminergic ne urons of Drosophila. Proc Natl Acad Sci USA 2005; 102(29): 10345– 10350.
51. Machida Y, Chiba T, Takayanagi A, Tanaka Y, Asanuma M, Ogawa N et al. Common anti‑apoptotic roles of parkin and alpha‑ synuclein in human dopaminergic cells. Bi ochem Bi ophys Res Commun 2005; 332(1): 233– 240.
52. Kitao Y, Imai Y, Ozawa K, Kataoka A, Ikeda T, Soda Met al. Pael receptor induces de ath of dopaminergic ne urons in the substanti a nigra vi a endoplasmic reticulum stress and dopamine toxicity, which is enhanced under conditi on of parkin inactivati on. Hum Mol Genet 2007; 16(1): 50– 60.
53. Henn IH, Bo uman L, Schlehe JS, Schli erf A, Schramm JE, Wegener E et al. Parkin Medi ates Ne uroprotecti on thro ugh Activati on of IkappaB Kinase/ Nucle ar Factor‑ kappaB Signaling. J Ne urosci 2007; 27(8): 1868– 1878.
54. Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M,Azuma H et al. Parkin enhances mitochondri al bi ogenesis in proliferating cells. Hum Mol Genet 2006; 15(6): 883– 895.
55. Hampe C, Ardila‑ Osori o H, Fo urni er M, Brice A,Corti O. Bi ochemical analysis of Parkinson‘s disease‑ ca using vari ants of Parkin, an E3 ubiquitin‑protein ligase with mono ubiquitylati on capacity. Hum Mol Genet 2006; 15(13): 2059– 2075.
56. Wang C, Tan JM, Ho MW, Zaiden N, Wong SH, Chew CL et al. Alterati ons in the solubility and intracellular localizati on of parkin by several famili al Parkinson‘s dise ase‑linked po int mutati ons. J Ne urochem 2005; 93(2): 422– 431.
57. Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP et al. Stress‑induced alterati on in parkin solubility promotes parkin aggregati on and compromise parkin‘s protective functi on. Hum Mol Genet 2005; 14: 3885– 3897.
58. Gasser T, Müller‑ Myhsok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V et al. A susceptibility locus for Parkinson‘s dise ase maps to chromosome 2p13. Nat Genet 1998; 18(3): 262– 265.
59. Sharma M, Mueller JC, Zimprich A, Lichtner P, Hofer A, Leitner P et al. The sepi apterin reductase gene regi on reve als associ ati on in the PARK3 locus: analysis of famili al and sporadic Parkinson‘s dise ase in Europe an populati ons. J Med Genet 2006; 43(7): 557– 562.
60. Tobin JE, Cui J, Wilk JB, Lato urelle JC, Larami e JM, McKee AC et al. Sepi apterin reductase expressi on is incre ased in Parkinson‘s dise ase brain tissue. Brain Res 2007; 1139: 42– 47.
61. Leroy E, Boyer R, Auburger G, Le ube B, Ulm G, Mezey E et al. The ubiquitin pathway in Parkinson‘s dise ase. Nature 1998; 395(6701): 451– 452.
62. Carmine Belin A, Westerlund M, Bergman O, Nissbrandt H, Lind C, Sydow O et al. S18Y in ubiquitin carboxy‑terminal hydrolase L1 (UCH‑ L1) associ ated with decre ased risk of Parkinson‘s dise ase in Sweden. Parkinsonism Relat Disord 2007; 13(5): 295– 298.
63. Lowe J, McDermott H, Landon M, Mayer RJ, Wilkinson KD. Ubiquitin carboxyl‑terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusi on bodi es characteristic of human ne urodegenerative dise ases. J Pathol 1990; 161(2): 153– 160.
64. Li u Y, Fallon L, Lashuel HA, Li u Z, Lansbury PT Jr. The UCH‑ L1 gene encodes two opposing enzymatic activiti es that affect alpha‑ synuclein degradati on and Parkinson‘s dise ase susceptibility. Cell 2002; 111(2): 209– 218.
65. Kyratzi E, Pavlaki M, Stefanis L. The S18Y polymorphic vari ant of UCH‑ L1 confers an anti oxidant functi on to ne uronal cells. Hum Mol Genet 2008; 17(14): 2160– 2171.
66. Valente EM, Bentivogli o AR, Dixon PH, Ferraris A,Ialongo T, Frontali M et al. Localizati on of a novel locus for a utosomal recessive e arly‑onset parkinsonism, PARK6, on human chromosome 1p35– p36. Am J Hum Genet 2001; 68(4): 895– 900.
67. Valente EM, Abo u‑ Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S et al. Hereditary Early‑onset Parkinson‘s Dise ase Ca used by Mutati ons in PINK1. Sci ence 2004; 304(5674): 1158– 1160.
68. Ishihara‑Pa ul L, Hulihan MM, Kachergus J, Upmanyu R, Warren L, Amo uri R et al. PINK1 mutati ons and parkinsonism. Ne urology 2008; 71(12): 896– 902.
69. Khan NL, Valente EM, Bentivogli o AR, Wo od NW, Albanese A, Bro oks DJ et al. Clinical and subclinical dopaminergic dysfuncti on in PARK6‑linked parkinsonism: an 18F‑ dopa PET study. Ann Ne urol 2002; 52(6): 849– 853.
70. Murakami T, Moriwaki Y, Kawarabayashi T, Nagai M,Ohta Y, Deguchi K et al. PINK1, a gene product of PARK6, accumulates in alpha‑ synucleinopathy brains. J Ne urol Ne urosurg Psychi atry 2007; 78(6): 653– 654.
71. Petit A, Kawarai T, Paitel E, Sanjo N, Maj M, Scheid Met al. Wild‑type PINK1 Prevents Basal and Induced Ne uronal Apoptosis, a Protective Effect Abrogated by Parkinson Dise ase‑related Mutati ons. J Bi ol Chem 2005; 280(40): 34025– 34032.
72. Yang Y, Gehrke S, Imai Y, Hu ang Z, Ouyang Y, Wang JW et al. Mitochondri al pathology and muscle and dopaminergic ne uron degenerati on ca used by inactivati on of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA 2006; 103(28): 10793– 10798.
73. Po ole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/ Parkin pathway regulates mitochondri al morphology. Proc Natl Acad Sci USA 2008; 105(5): 1638– 1643.
74. Mills RD, Sim CH, Mok SS, Mulhern TD, Culvenor JG, Cheng HC. Bi ochemical aspects of the ne uroprotective mechanism of PTEN‑induced kinase‑ 1 (PINK1). J Ne urochem 2008; 105(1): 18– 33.
75. van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Ho uwing‑ Duistermaat JJ, Snijders PJ et al. Park7, a novel locus for a utosomal recessive e arly‑onset parkinsonism, on chromosome 1p36. Am J Hum Genet 2001; 69(3): 629– 634.
76. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Kri eger E et al. Mutati ons in the DJ‑ 1 Gene Associ ated with Autosomal Recessive Early‑onset Parkinsonism. Sci ence 2003; 299(5604): 256– 259.
77. Dekker MC, Eshuis SA, Maguire RP, Veenma‑ van der Duijn L, Pruim J, Snijders PJ et al. PET ne uro imaging and mutati ons in the DJ‑ 1 gene. J Ne ural Transm 2004; 111(12): 1575– 1581.
78. Nagakubo D, Taira T, Kita ura H, Ikeda M, Tamai K, Iguchi‑ Ariga SM et al. DJ‑ 1, a novel oncogene which transforms mo use NIH3T3 cells in co operati on with ras. Bi ochem Bi ophys Res Commun 1997; 231(2): 509– 513.
79. Lev N, Ickowicz D, Melamed E, Offen D. Oxidative insults induce DJ‑ 1 upregulati on and redistributi on: Implicati ons for ne uroprotecti on. Ne urotoxicology 2008; 29(3): 397– 405.
80. Kinumi T, Kimata J, Taira T, Ariga H, Niki E. Cysteine‑ 106 of DJ‑ 1 is the most sensitive cysteine residue to hydrogen peroxide‑ medi ated oxidati on in vivo in human umbilical vein endotheli al cells. Bi ochem Bi ophys Res Commun 2004; 317(3): 722– 728.
81. Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ‑ 1, a cancer‑ and Parkinson‘s dise ase‑associ ated protein, stabilizes the anti oxidant transcripti onal master regulator Nrf2. Proc Natl Asad Sci USA 2006; 103(41): 15091– 15096.
82. Zho u W, Freed CR. DJ‑ 1 Up‑ regulates Glutathi one Synthesis during Oxidative Stress and Inhibits A53T alpha‑ Synuclein Toxicity. J Bi ol Chem 2005; 280(52): 43150– 43158.
83. Junn E, Taniguchi H, Je ong BS, Zhao X, Ichijo H, Mo uradi an MM. Interacti on of DJ‑ 1 with Daxx inhibits apoptosis signal‑ regulating kinase 1 activity and cell de ath. Proc Natl Asad Sci USA 2005; 102(27): 9691– 9696.
84. Anderson PC, Daggett V. Molecular basis for the structural instability of human DJ‑ 1 induced by the L166P mutati on associ ated with Parkinson‘s dise ase. Bi ochemistry 2008; 47(36): 9380– 9393.
85. Görner K, Holtorf E, Waak J, Pham T‑ T, Vogt‑ Weisenhorn DM, Wurst W et al. Structural Determinants of the C‑terminal Helix‑ Kink‑ Helix Motif Essenti al for Protein Stability and Survival Promoting Activity of DJ‑ 1. J Bi ol Chem 2007; 282(18): 13680– 13691.
86. Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson‘s dise ase (PARK8) maps to chromosome 12p11.2– q13.1. Ann Ne urol 2002; 51(3): 296– 301.
87. Paisán‑ Ruíz C, Jain S, Evans EW, Gilks WP, Simón J,van der Brug M et al. Cloning of the gene containing mutati ons that ca use PARK8‑linked Parkinson‘s dise ase. Ne uron 2004; 44(4): 595– 600.
88. Clark LN, Wang Y, Karlins E, Saito L, Meji a‑ Santana H, Harris J et al. Frequency of LRRK2 mutati ons in e arly‑ and late‑onset Parkinson dise ase. Ne urology 2006; 67(10): 1786– 1791.
89. Lesage S, Dürr A, Tazir M, Lohmann E, Le utenegger AL, Janin S et al. LRRK2 G2019S as a Ca use of Parkinson‘s Dise ase in North African Arabs. N Engl J Med 2006; 354(4): 422– 423.
90. Ozeli us LJ, Senthil G, Sa unders‑ Pullman R, Ohmann E, Deligtisch A, Tagli ati M et al. LRRK2 G2019S as a Ca use of Parkinson‘s Dise ase in Ashkenazi Jews. N Engl J Med 2006; 354(4): 424– 425.
91. Tan EK, Shen H, Tan LC, Farrer M, Yew K, Chu a E et al. The G2019S LRRK2 mutati on is uncommon in an Asi an cohort of Parkinson‘s dise ase pati ents. Ne urosci Lett 2005; 384(3): 327– 329.
92. Farrer MJ, Stone JT, Lin CH, Dächsel JC, Hulihan MM, Ha ugarvoll K et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson‘s dise ase in Asi a. Parkinsonism Relat Disord 2007; 13(2): 89– 92.
93. Ross OA, Wu YR, Lee MC, Funayama M, Chen ML, Soto AI et al. Analysis of Lrrk2 R1628P as a risk factor for Parkinson‘s dise ase. Ann Ne urol 2008; 64(1): 88– 92.
94. Gi asson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ et al. Bi ochemical and pathological characterizati on of Lrrk2. Ann Ne urol 2006; 59(2): 315– 322.
95. Häbig K, Walter M, Poths S, Ri ess O, Bonin M. RNA interference of LRRK2- micro array expressi on analysis of a Parkinson‘s dise ase key player. Ne urogenetics 2008; 9(2): 83– 94.
96. Gandhi PN, Wang X, Zhu X, Chen SG, Wilson‑ Delfosse AL. The Roc domain of le ucine‑ rich repe at kinase 2 is suffici ent for interacti on with microtubules. J Ne urosci Res 2008; 86(8): 1711– 1720.
97. Iaccarino C, Crosi o C, Vitale C, Sanna G, Carri MT, Barone P. Apoptotic mechanisms in mutant LRRK2– medi ated cell de ath. Hum Mol Genet 2007; 16(11): 1319– 1326.
98. Wang L, Xi e C, Greggi o E, Parisi ado u L, Shim H,Sun L et al. The chaperone activity of he at shock protein 90 is critical for maintaining the stability of le ucine‑ rich repe at kinase 2. J Ne urosci 2008; 28(13): 3384– 3391.
99. Smith WW, Pei Z, Ji ang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 medi ates ne uronal toxicity. Nat Ne urosci 2006; 9(10): 1231– 1233.
100. Deng J, Lewis PA, Greggi o E, Sluch E, Beilina A, Co okson MR. Structure of the ROC domain from the Parkinson‘s dise ase‑associ ated le ucine‑ rich repe at kinase 2 reve als a dimeric GTPase. Proc Natl Acad Sci USA 2008; 105(5): 1499– 1504.
101. Hampshire DJ, Roberts E, Crow Y, Bond J, Mubaidin A, Wri ekat AL et al. Kufor‑ Rakeb syndrome, pallido‑ pyramidal degenerati on with supranucle ar upgaze paresis and dementi a, maps to 1p36. J Med Genet 2001; 38(10): 680– 682.
102. Willi ams DR, Hadeed A, al‑ Din AS, Wreikat AL, Lees AJ. Kufor Rakeb dise ase: a utosomal recessive, levodopa‑ responsive parkinsonism with pyramidal degenerati on, supranucle ar gaze palsy, and dementi a. Mov Disord 2005; 20(10): 1264– 1271.
103. Ramirez A, Heimbach A, Gründemann J, Stiller B,Hampshire D, Cid LP et al. Hereditary parkinsonism with dementi a is ca used by mutati ons in ATP13A2, encoding a lysosomal type 5 P‑type ATPase. Nat Genet 2006; 38(10): 1184– 1191.
104. Di Fonzo A, Chi en HF, Socal M, Gira udo S, Tassorelli C, Iliceto G et al. ATP13A2 missense mutati ons in juvenile parkinsonism and yo ung onset Parkinson dise ase. Ne urology 2007; 68(19): 1557– 1562.
105. Hicks AA, Pétursson H, Jónsson T, Stefánsson H, Jóhannsdóttir HS, Sainz J et al. A susceptibility gene for late‑onset idi opathic Parkinson‘s dise ase. Ann Ne urol 2002; 52(5): 549– 555.
106. No ureddine MA, Li YJ, van der Walt JM, Walters R,Jewett RM, Xu H et al. Genomic convergence to identify candidate genes for Parkinson dise ase: SAGE analysis of the substanti a nigra. Mov Disord 2005; 20(10): 1299– 1309.
107. Anderson LR, Betarbet R, Ge aring M, Gulcher J,Hicks AA, Stefánsson K et al. PARK10 candidate RNF11 is expressed by vulnerable ne urons and localizes to Lewy bodi es in Parkinson dise ase brain. J Ne uropathol Exp Ne urol 2007; 66(10): 955– 964.
108. Connor MK, Seth A. A central role for the ring finger protein RNF11 in ubiquitin‑medi ated prote olysis vi a interacti ons with E2s and E3s. Oncogene 2004; 23(11): 2089– 2095.
109. Azmi P, Seth A. RNF11 is a multifuncti onal modulator of growth factor receptor signalling and transcripti onal regulati on. Eur J Cancer 2005; 41(16): 2549– 2560.
110. La uti er C, Goldwurm S, Dürr A, Gi ovannone B,Tsi aras WG, Pezzoli G et al. Mutati ons in the GIGYF2 (TNRC15) gene at the PARK11 locus in famili al Parkinson dise ase. Am J Hum Genet 2008; 82(4): 822– 833.
111. Gi ovannone B, Lee E, Lavi ola L, Gi orgino F, Cleveland KA, Smith RJ. Two novel proteins that are linked to insulin‑like growth factor (IGF‑I) receptors by the Grb10 adapter and modulate IGF‑I signaling. J Bi ol Chem 2003; 278(34): 31564– 31573.
112. Schulingkamp RJ, Pagano TC, Hung D, Raffa RB. Insulin receptors and insulin acti on in the brain: revi ew and clinical implicati ons. Ne urosci Bi obehav Rev 2000; 24(8): 855– 872.
113. Offen D, Shtaif B, Hadad D, Weizman A, Melamed E,Gil‑ Ad I. Protective effect of insulin‑like‑ growth‑ factor‑ 1 against dopamine‑induced ne urotoxicity in human and rodent ne uronal cultures: possible implicati ons for Parkinson‘s dise ase. Ne urosci Lett 2001; 316(3): 129– 132.
114. Pankratz N, Nichols WC, Uni acke SK, Halter C, Murrell J, Rudolph A et al. Genome‑ wide linkage analysis and evidence of gene‑ by‑ gene interacti ons in a sample of 362 multiplex Parkinson dise ase famili es. Hum Mol Genet 2003; 12(20): 2599– 2608.
115. Stra uss KM, Martins LM, Plun‑ Favre a u H, Marx FP, Ka utzmann S, Berg D et al. Loss of functi on mutati ons in the gene encoding Omi/ HtrA2 in Parkinson‘s dise ase. Hum Mol Genet 2005; 14(15): 2099– 2111.
116. Simón‑ Sánchez J, Singleton AB. Sequencing analysis of OMI/ HTRA2 shows previ o usly reported pathogenic mutati ons in ne urologically normal controls. Hum Mol Genet 2008; 17(13): 1988– 1993.
117. Martins LM, Iaccarino I, Tenev T, Gschmeissner S,Totty NF, Lemo ine NR et al. The serine prote ase Omi/ HtrA2 regulates apoptosis by binding XIAP thro ugh a re aper‑like motif. J Bi ol Chem 2002; 277(1): 439– 444.
118. Spi ess C, Beil A, Ehrmann M. A temperature‑dependent switch from chaperone to prote ase in a widely conserved he at shock protein. Cell 1999; 97(3): 339– 347.
119. Horvath R, Kley RA, Lochmüller H, Vorgerd M. Parkinson syndrome, ne uropathy, and myopathy ca u-sed by the mutati on A8344G (MERRF) in tRNALys. Ne u-rology 2007; 68(1): 56– 58.
120. van der Walt JM, Nicodemus KK, Martin ER, Scott WK, Nance MA, Watts RL et al. Mitochondri al polymorphisms significantly reduce the risk of Parkinson dise ase. Am J Hum Genet 2003; 72(4): 804– 811.
121. Autere J, Mo ilanen JS, Finnilä S, So ininen H, Mannermaa A, Hartikainen P et al. Mitochondri al DNA polymorphisms as risk factors for Parkinson‘s dise ase and Parkinson‘s dise ase dementi a. Hum Genet 2004; 115(1): 29– 35.
122. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH et al. High levels of mitochondri al DNA deleti ons in substanti a nigra ne urons in aging and Parkinson dise ase. Nat Genet 2006; 38(5): 515– 517.
123. Kraytsberg Y, Kudryavtseva E, McKee AC, Ge ula C,Kowall NW, Khrapko K. Mitochondri al DNA deleti ons are abundant and ca use functi onal impairment in aged human substanti a nigra ne urons. Nat Genet 2006; 38(5): 518– 520.
124. Luoma PT, Eerola J, Ahola S, Hakonen AH, Hellström O, Kivistö KT et al. Mitochondri al DNA polymerase gamma vari ants in idi opathic sporadic Parkinson dise ase. Ne urology 2007; 69(11): 1152– 1159.
125. Benmoyal‑ Segal L, Soreq H. Gene‑ environment interacti ons in sporadic Parkinson‘s dise ase. J Ne urochem 2006; 97(6): 1740– 1755.
126. Yang YX, Latchman DS. Nurr1 transcripti onally regulates the expressi on of alpha‑ synuclein. Ne uroreport 2008; 19(8): 867– 871.
127. Zheng K, Heydari B, Simon DK. A common NURR1 polymorphism associ ated with Parkinson dise ase and diffuse Lewy body dise ase. Arch Ne urol 2003; 60(5): 722– 725.
128. Bembi B, Zambito Marsala S, Sidransky E, Ci ana G, Carrozzi M, Zorzon M et al. Ga ucher‘s dise ase with Parkinson‘s dise ase: clinical and pathological aspects. Ne urology 2003; 61(1): 99– 101.
129. Sato C, Morgan A, Lang AE, Salehi‑ Rad S, Kawarai T, Meng Y et al. Analysis of the glucocerebrosidase gene in Parkinson‘s dise ase. Mov Disord 2005; 20(3): 367– 370.
130. Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Cle arance of alpha‑ synuclein oligomeric intermedi ates vi a the lysosomal degradati on pathway. J Ne urosci 2004; 24(8): 1888– 1896.
131. Braak H, Rüb U, Jansen Ste ur EN, Del Tredici K, de Vos RA. Cognitive status correlates with ne uropathologic stage in Parkinson dise ase. Ne urology 2005; 64(8): 1404– 1410.
132. van de Warrenburg BP, Lammens M, Lücking CB, Denèfle P, Wesseling P, Bo o ij J et al. Clinical and pathologic abnormaliti es in a family with parkinsonism and parkin gene mutati ons. Ne urology 2001; 56(4): 555– 557.
133. Zabeti an CP, Hutter CM, Factor SA, Nutt JG, Higgins DS, Griffith A et al. Associ ati on analysis of MAPT H1 haplotype and subhaplotypes in Parkinson‘s dise ase. Ann Ne urol 2007; 62(2): 137– 144.
134. Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ et al. Synphilin‑1 associ ates with alpha‑ synuclein and promotes the formati on of cytosolic inclusi ons. Nat Genet 1999; 22(1): 110– 114.
135. Marx FP, Holzmann C, Stra uss KM, Li L, Eberhardt O,Gerhardt E et al. Identificati on and functi onal characterizati on of a novel R621C mutati on in the synphilin‑1 gene in Parkinson‘s dise ase. Hum Mol Genet 2003; 12(11): 1223– 1231.
136. Myhre R, Klungland H, Farrer MJ, Aasly JO. Genetic associ ati on study of synphilin‑1 in idi opathic Parkinson‘s dise ase. BMC Med Genet 2008; 9: 19.
137. Gilgun‑ Sherki Y, Djaldetti R, Melamed E, Offen D. Polymorphism in candidate genes: implicati ons for the risk and tre atment of idi opathic Parkinson‘s dise ase. Pharmacogenomics J 2004; 4(5): 291– 306.
138. Tan EK, Jankovic J. Genetic testing in Parkinson dise ase: promises and pitfalls. Arch Ne urol 2006; 63(9): 1232– 1237.
139. Ishihara L, Warren L, Gibson R, Amo uri R, Lesage S,Dürr A et al. Clinical Fe atures of Parkinson Dise ase Pati ents With Homozygo us Le ucine‑ Rich Repe at Kinase 2 G2019S Mutati ons. Arch Ne urol 2006; 63(9): 1250– 1254.
140. Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I et al. Lewy body Parkinson‘s dise ase in a large pedigree with 77 Parkin mutati on carri ers. Ann Ne urol 2005; 58(3): 411– 422.
141. Weiner WJ. There is no Parkinson dise ase. Arch Ne urol 2008; 65(6): 705– 708.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2009 Číslo 5
Nejčtenější v tomto čísle
- Lumbální spinální stenóza a neurogenní klaudikace
- Doporučený postup při zahájení léčby Parkinsonovy nemoci
- Disoci ativní křeče
- Indikace dekompresivní kraniektomie u traumat mozku