
Přehled difuzních gliomů dle klasifikace WHO 2021, 2. část – difuzní gliomy dětského typu
Authors:
M. Hendrych 1; M. Barák 2; H. Valeková 2; T. Kazda 3; P. Pospíšil 3
![]() ; R. Lakomý 4; J. Šána 4,5; R. Jančálek 2; M. Hermanová 1
; R. Lakomý 4; J. Šána 4,5; R. Jančálek 2; M. Hermanová 1
Authors‘ workplace:
I. ústav patologie, LF MU a FN u sv. Anny v Brně
1; Neurochirurgická klinika LF MU a FN U sv. Anny v Brně
2; Klinika radiační onkologie LF MU a MOÚ, Brno
3; Klinika komplexní onkologické péče LF MU a MOÚ, Brno
4; CEITECH – Středoevropský technologický institut, MU, Brno
5
Published in:
Cesk Slov Neurol N 2024; 87(1): 9-17
Category:
Review Article
doi:
https://doi.org/10.48095/cccsnn20249
Overview
Pátá edice klasifikace WHO nádorů centrálního nervového systému rozděluje difuzní gliomy do dvou hlavních skupin dle typického věku výskytu: dětský typ, který postihuje především dětské pacienty, a dospělý typ, který se dominantně vyskytuje u pacientů v dospělém věku. Do skupiny difuzních gliomů dětského typu byly recentně zařazeny nové jednotky poprvé definované dle klasifikace WHO 2021. Současně i etablované gliomy prodělaly změny v diagnostických kritériích na podkladě recentních poznatků molekulárně-genetického podkladu. Tato druhá část přehledové práce předkládá souhrn jednotek zařazených do skupiny difuzních gliomů dětského typu dle 5. edice klasifikace WHO nádorů centrálního nervového systému z roku 2021.
Klíčová slova:
difuzní gliom – WHO CNS 2021 – integrovaná diagnostika – PLNTY – angiocentrický gliom – difuzní astrocytom – difuzní středočarový gliom – difuzní hemisférický gliom – low-grade gliomy – high-grade gliomy
Úvod
Aktuální klasifikace dle 5. edice klasifikace WHO nádorů centrálního nervového systému z roku 2021 [1,2] reflektuje odlišnou biologickou povahu difuzních gliomů primárně se vyskytujících u dospělých a pediatrických pacientů a zavádí jejich dělení do dvou věkových skupin – difuzní gliomy dětského typu a difuzní gliomy dospělého typu. Odlišení těchto dvou skupin je založeno na podkladě molekulárně-genetických alterací rekurentně se vyskytujících v daných jednotkách, přičemž gliomy dětského typu se mohou vyskytovat i u dospělých pacientů a obráceně. Difuzní gliomy dětského typu jsou dále rozděleny na nádory nízkého stupně (low-grade) a vysokého stupně malignity (high-grade) [1] a obě tyto skupiny jsou shrnuty v této druhé části přehledové práce (tab. 1).
Obdobně jako diagnostika se významně rozvíjí i terapeutické možnosti. V současnosti se cílená terapie a imunoterapie doporučují jako preferované postupy v případech recidivujícího nebo progredujícího onemocnění. Přibližně u 10–15 % dětských gliomů je nalézána bodová mutace BRAF V600E, která vede ke konstitutivní aktivaci dráhy MEK/ERK. Kombinovaná léčba zaměřená na BRAF a následnou dráhu MEK (dabrafenib/trametinib) se ukázala jako úspěšná v několika klinických studiích u high-grade gliomů dospělých. U pediatrické populace jsou zatím zkušenosti omezené, i když v malých případových studiích byly zaznamenány slibné výsledky [3]. Aktuálně probíhá u pediatrických rekurentních nebo progredujících high-grade gliomů studie fáze II testující blokátor BRAF vemurafenib [4] a studie fáze III testující dordaviprone u pacientů s difuzním středočarovým gliomem, H3 K27-alterovaným [5].
Genové fúze zahrnující NTRK1, NTRK2 a NTRK3 kódující fúzní proteiny TRK (TRKA, TRKB, TRKC) lze ovlivnit inhibitory TRK (larotrectinib a entrectinib). Sledovaná míra objektivní odpovědi u nádorů pozitivních na fúzi TRK je poměrně vysoká (objektivní odpověď na léčbu [overall response rate; ORR] až 93 %) a zaznamenaná toxicita je přijatelná. V současné době testování u obou preparátů postoupilo již do fáze II klinických hodnocení [6].
Gliomy, u kterých byla prokázána vysoká mutační nálož (hypermutantní nádory), mohou reagovat na imunoterapii pomocí checkpoint inhibitorů (nivolumab, pembrolizumab). Nicméně důkazy o jejich účinnosti jsou v současné době omezeny pouze na malé retrospektivní soubory a kazuistiky.
Hlubší molekulární porozumění pediatrickým gliomům vedlo k testování mnoha dalších preparátů zaměřených na dráhy MAPK/ERK či mTOR. Jde např. o inhibitory MEK (selumetinib, binimetinib) nebo o inhibitory RAF druhé generace (tovorafenib). Zkoumány jsou látky, které se zaměřují na dráhu mTOR (everolimus). Probíhající studie pediatrického neuro-onkologického konzorcia (PNOC) hodnotí efekt kombinace trametinibu a everolimu u recidivujících dětských gliomů [7]. U nádorů obsahujících aktivační změny FGFR byl zkoumán inhibitor FGFR (erdafitinib). U nízce stupňových gliomů se jeví jako slibná terapie duálním inhibitorem isocitrátdehydrogenáza (IDH) (vorasidenib), přestože důkazy o jeho efektivitě u pediatrické populace zatím nebyly předloženy.
Ačkoli cílená léčba již významně ovlivňuje léčebná paradigmata dětských gliomů, je třeba konstatovat, že pokud pomineme subependymální obrovskobuněčný astrocytom asociovaný s tuberózní sklerózou (everolimus) a gliom s mutací BRAF V600E (dabrafenib + trametinib), tak cílená terapie zatím nemá své místo v primární léčbě. V současné době je však předmětem hodnocení v řadě probíhajících prospektivních klinických studií [8,9].

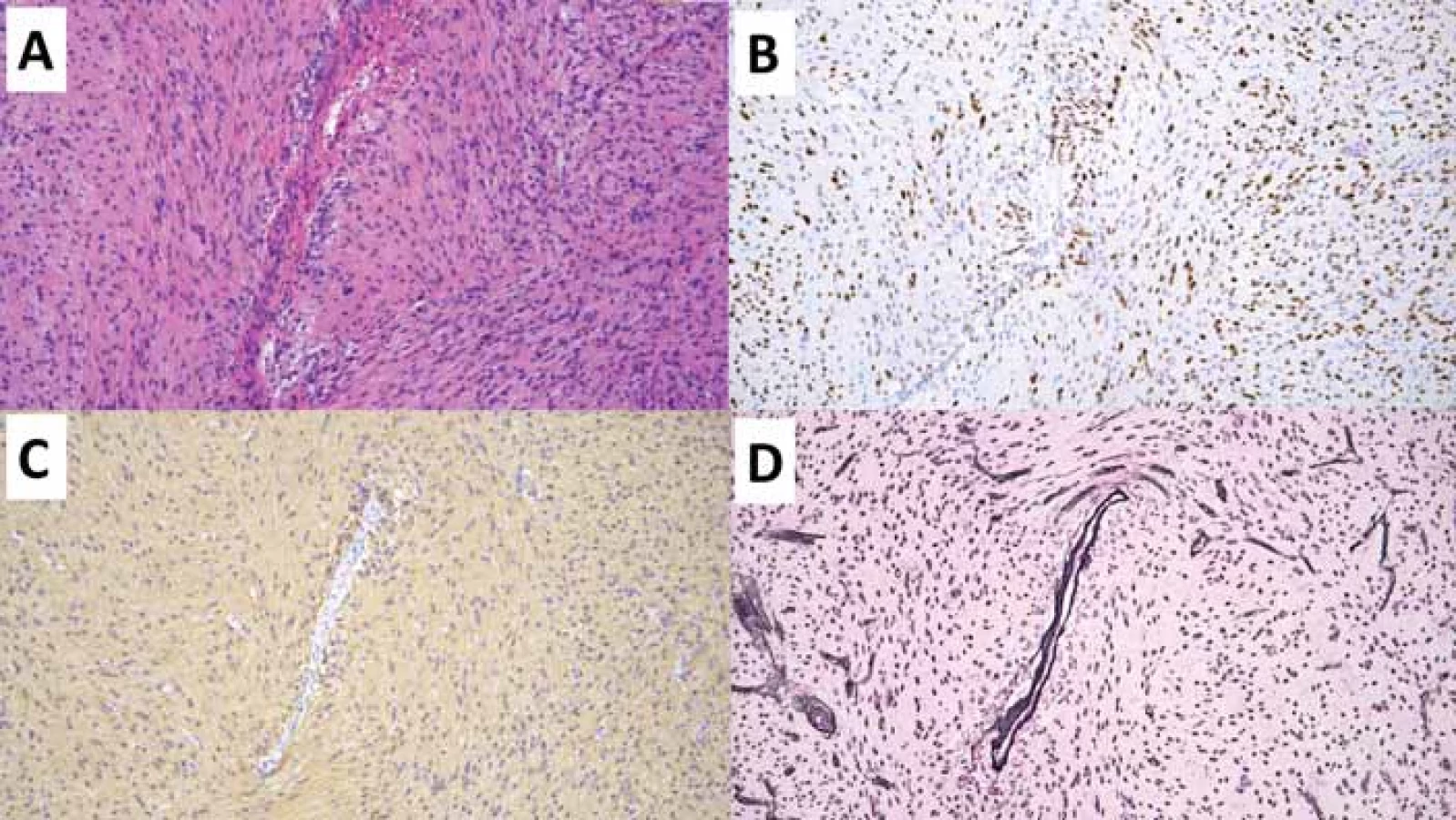
(B) Imunohistochemické vyšetření OLIG2 hnědě zvýrazňující jádra nádorových gliálních buněk centrálně se agregujících okolo cévy.
(C) Imunohistochemické vyšetření GFAP zobrazující gliální elementy agregující se centrálně okolo cévy.
(D) Speciální barvení Gömöri černě zvýrazňující stěnu cév a jádra buněk.
GFAP – glial fi brillary acidic protein; OLIG2 – oligodendrocyte transcription factor 2
Fig. 1. Angiocentric glioma. Original magnifi cation 100×.
(A) Hematoxylin-eosin staining displaying a larger blood vessel in the center, where tumor cells of the angiocentric glioma aggregate around it.
(B) Immunohistochemical examination of OLIG2 highlighting the nuclei of tumor glial cells (brown) centrally aggregating around the vessel.
(C) Immunohistochemical examination of GFAP showing glial elements aggregating centrally around the vessel.
(D) Special Gömöri stain highlighting the vessel wall and cell nuclei (black).
GFAP – glial fi brillary acidic protein; OLIG2 – oligodendrocyte transcription factor 2
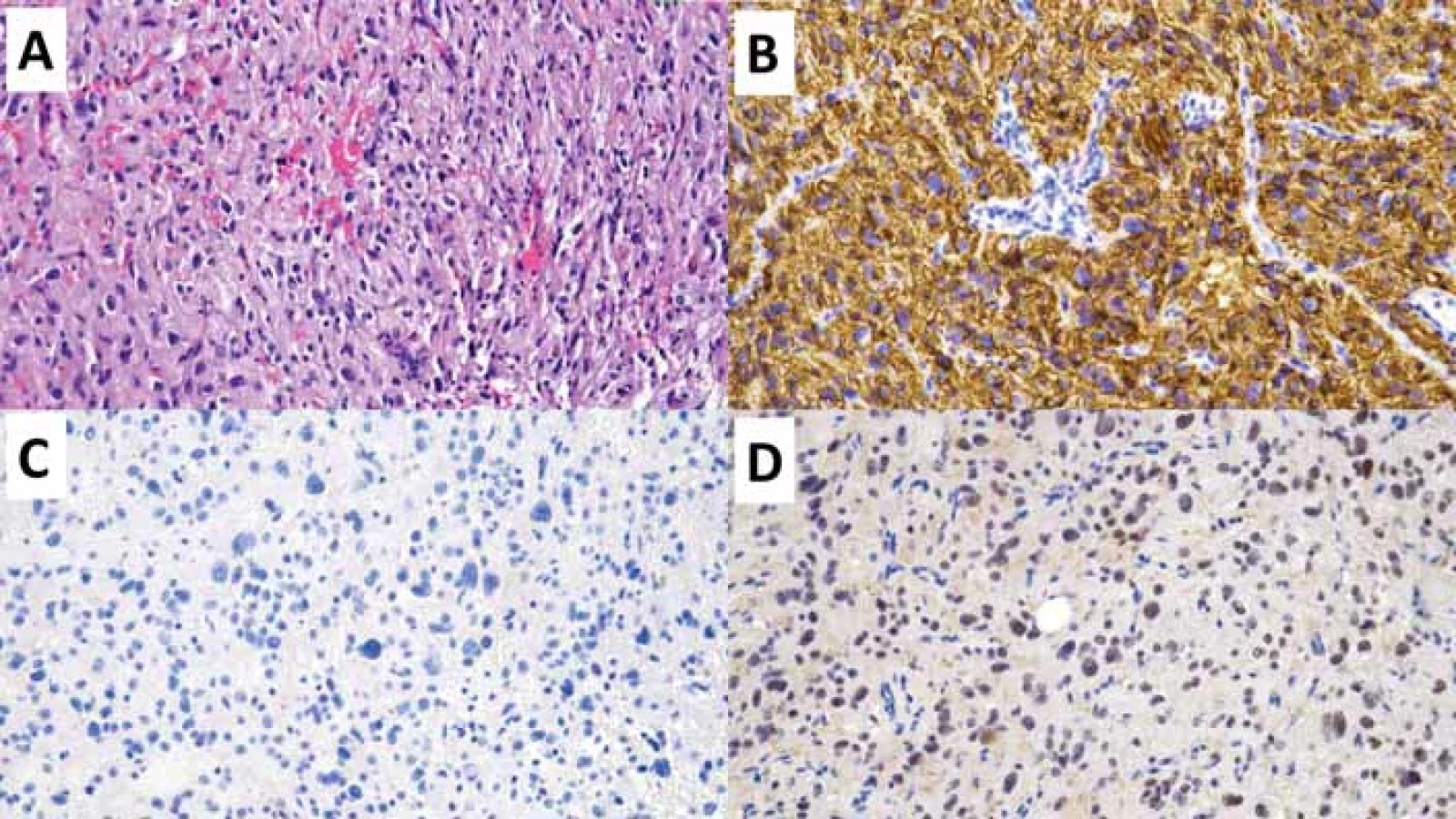
(B) Imunohistochemivyšetření CD34 s difuzní silnou expresí.
(C) V imunohistochemickém vyšetření mutačně specifi ckou protilátkou IDH1 R132H nebyla prokázána mutace R132H genu IDH1.
(D) Imunohistochemické vyšetření ARTX se zachovanou wildtype expresí ARTX.
ARTX – alpha thalassemia/mental retardation syndrome X-linked; IDH – isocitrátdehydrogenáza
Fig. 2. Polymorphic low-grade neuroepithelial tumor of the young (PLNTY). Original magnifi cation 200×.
(A) Diff use glial proliferation of oligodendroglial diff erentiation; hematoxylin-eosin staining.
(B) Immunohistochemical examination of CD34 with diff use strong expression.
(C) In the immunohistochemical examination with the mutation specifi c IDH1 R132H antibody, the R132H mutation of the IDH1 gene
was not demonstrated.
(D) Immunohistochemical examination of ARTX with preserved wildtype expression of ARTX.
ARTX – alpha thalassemia/mental retardation syndrome X-linked; IDH – isocitrate dehydrogenase
Dětský typ difuzních low-grade gliomů
Difuzní astrocytom, MYB - nebo MYBL1-alterovaný, WHO G1
Difuzní astrocytom, MYB - nebo MYBL1-alterovaný je vzácný difuzně infiltrující astrocytárně diferencovaný gliom s alterací v genech MYB nebo MYBL1 a současně bez průkazu IDH mutace či alterace histonu H3 (tab. 2). Typicky se vyskytuje u dětí a mladých dospělých s farmakorezistentní epilepsií [2,10]. Charakteristická je supratentoriální lokalizace, predominantně v temporálním laloku, a to s postižením kortexu či subkortikální oblasti. Ojediněle byly publikovány případy i s lokalizací v mozkovém kmeni [11]. Jedná se o indolentní tumor s dobrou prognózou [12,13] a pouze ojedinělou anaplastickou transformací [14].
Charakteristický mikroskopický obraz zahrnuje unimorfní proliferaci cytologicky blandních gliálních buněk astrocytární diferenciace, pro který byl iniciálně nazýván jako isomorfní astrocytom [13]. Vlastní tumor má jen mírně zvýšenou buněčnou denzitu v porovnání s mozkovou tkání, kterou infiltruje. Charakteristický je imunofenotyp s absencí exprese OLIG2, CD34 a MAP2 [12].
Angiocentrický gliom, WHO G1
Angiocentrický gliom je vzácný difuzně rostoucí tumor predominantně lokalizovaný v oblasti kortexu temporálního či frontálního laloku a bývá typicky asociovaný s farmakorezistentní epilepsií [15]. Pro angiocentrický gliom je charakteristická alterace genu MYB, nejčastěji fúze MYB:: QKI (tab. 3) [16]. Prognóza pacientů s angiocentrickým gliomem je příznivá, naprostá většina pacientů je vyléčena kompletní resekcí, přičemž rekurence byla zastižena pouze u minima případů [15].
Vlastní neoplazie je tvořena variabilně uspořádanou, především vřetenobuněčnou proliferací s minimálně úsekovitým uspořádáním do charakteristických angiocentrických formací s radiálním řazením buněk okolo cév (obr. 1). Vřetenité nádorové buňky obsahují pravidelná útlá jádra bez signifikantních cytonukleárních atypií či signifikantní mitotické aktivity. Specifický je i imunofenotyp neoplazie s expresí astrocytárních markerů, tak i s luminální nebo tečkovitou expresí epiteliálního membránového antigenu (EMA), charakteristickou pro neoplazie ependymální diferenciace [17].
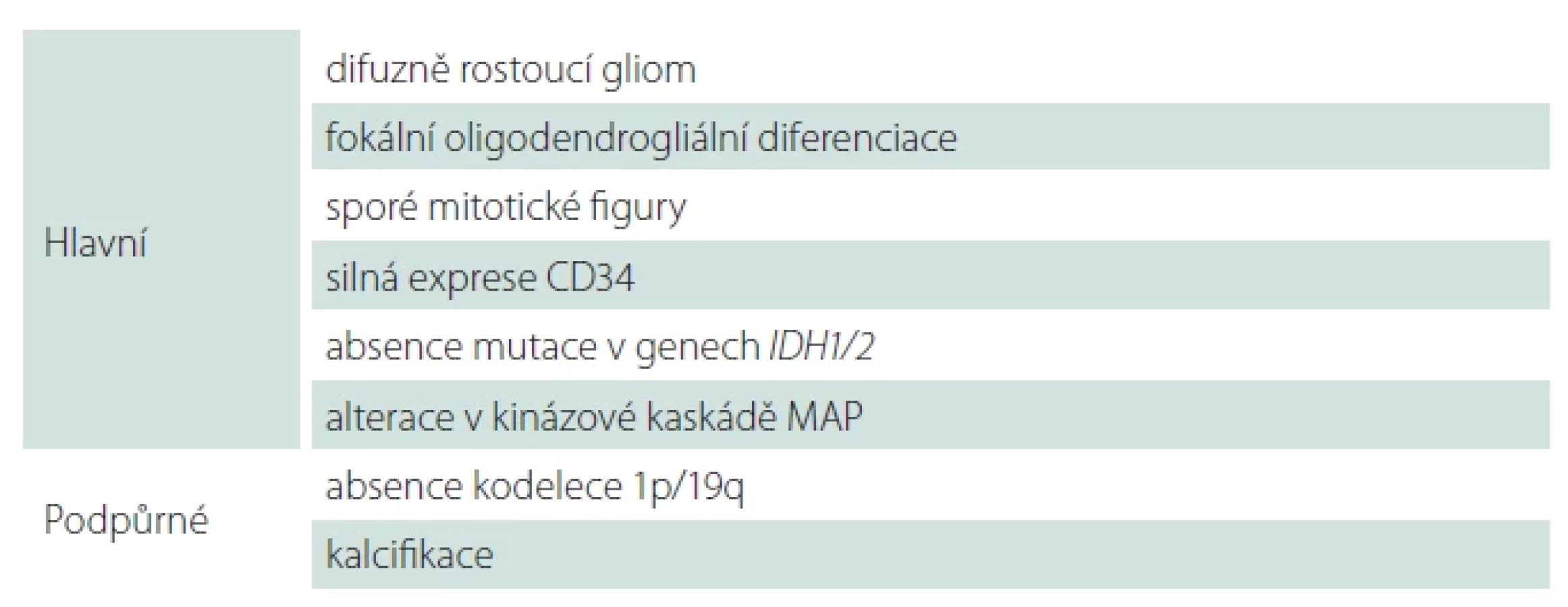
Polymorfní low-grade neuroepiteliální tumor mladistvých (PLNTY), WHO G1
Polymorfní low-grade neuroepiteliální tumor mladistvých (PLNTY) je recentně popsaná neoplazie asociovaná s farmakorezistentní epilepsií typicky dětských pacientů [18]. PLNTY se obvykle vyskytuje v oblasti temporálního laloku, a to kortikálně či subkortikálně. Charakteristicky je PLNTY tvořený solidní a cystickou komponentou s četnými kalcifikacemi [19]. Obdobně jako další tumory asociované s farmakorezistentní epilepsií se i PLNTY může vyskytovat asociovaný s fokální kortikální dysplazií (FCD IIIB dle ILAE) [20,21].
Morfologicky je pro PLNTY typická variabilně heterogenní diferenciace s konstantní, alespoň fokální přítomností oligodendrogliální diferenciace (obr. 2) a přítomné kalcifikace. Molekulárně-geneticky je PLNTY charakterizován jako IDH-wildtype gliom se silnou difuzní expresí CD34 a alteracemi v MAP kinázové kaskádě (tab. 4) [18,22]. Jedná se o potenciálně vyléčitelné onemocnění s nízkou tendencí k rekurenci po kompletní resekci, přičemž v současnosti je popsán pouze jeden případ maligního zvratu PLNTY v glioblastoma multiformu (GBM) [23].
Difuzní low-grade gliom s alterací MAPK dráhy
Skupina difuzního low-grade gliomu s alterací MAPK dráhy zahrnuje difuzně rostoucí gliální neoplazie geneticky definované alterací v genech MAPK cesty při současné absenci mutace v genech IDH1/2, genech pro histon H3 a absencí homozygotní delece genu CDKN2A [1,2,10]. Predominantně se vyskytují v supratentoriální oblasti, nicméně jsou popsány případy v rozsahu celé kraniospinální osy [11,24]. Nejčastějšími detekovanými alteracemi jsou alterace genu FGFR1 a mutace BRAF V600E (tab. 5) [25]. Difuzní low-grade gliom s alterací MAPK dráhy je skupina low-grade gliomů s predikovanou dobrou prognózou odvíjející se především od lokalizace, morfologie a molekulárních alterací. Grade jednotce v současné době nebyl přidělen [2].
Morfologicky se jedná o nízce celulární gliální proliferaci astrocytární či oligodendrogliální diferenciace s mírnými cytonukleárními atypiemi a absencí jiných morfologických známek anaplazie, přičemž tumory s mutací BRAF V600E jsou morfologicky spíše astrocytárně diferencované, zatímco tumory s FGFR1 alterací se diferencují oligodendrogliálně [24,25]. Imunohistochemicky jsou nádorové elementy OLIG2 pozitivní, s variabilní expresí GFAP a ojedinělou reakcí s CD34.


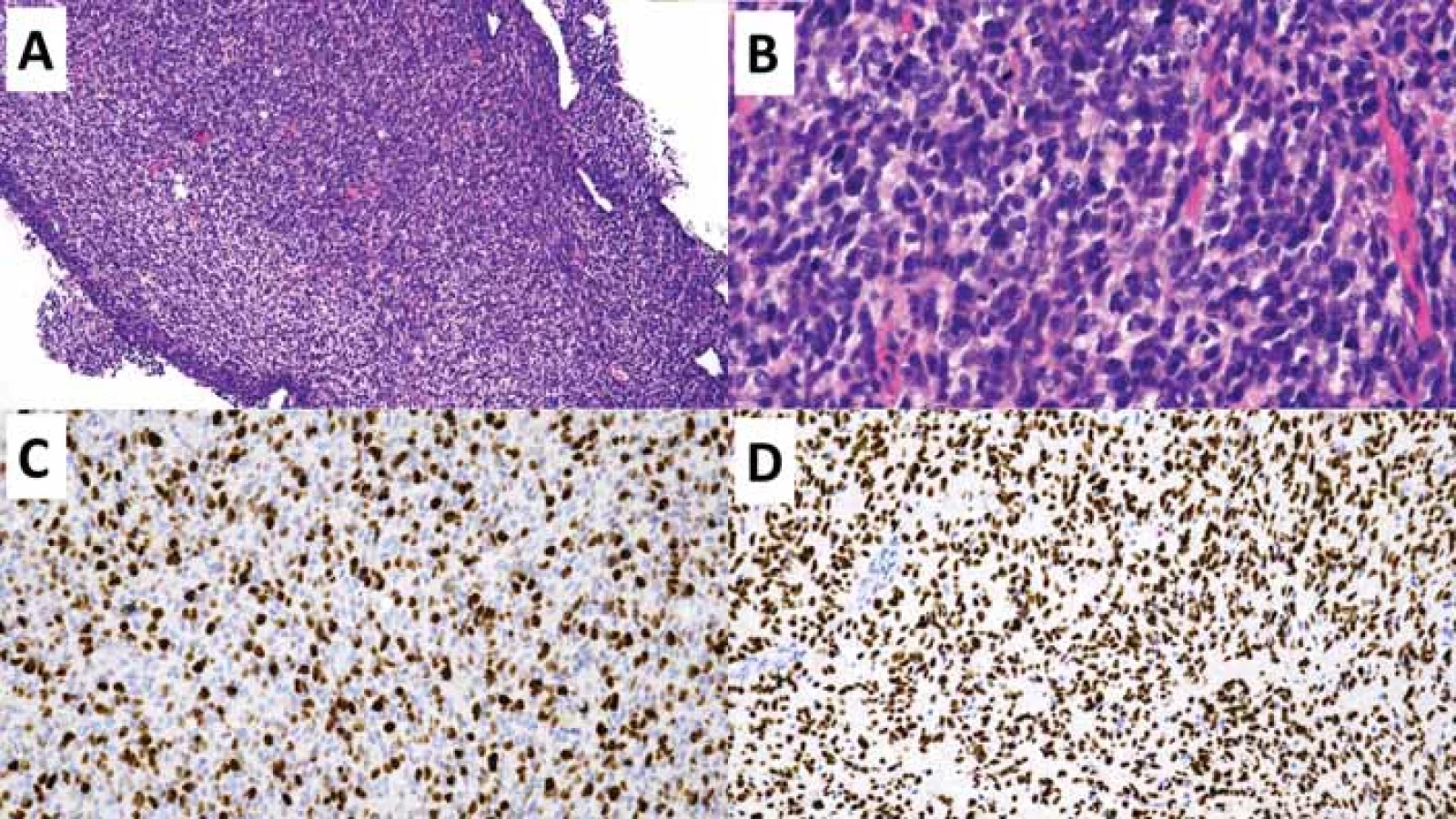
(B) Detailní snímek zobrazující atypické gliální buňky s briskní mitotickou aktivitou. Původní zvětšení 400×.
(C) Imunohistochemické vyšetření proliferační aktivity Ki-67 s pozitivní hnědou reakcí v proliferujících buňkách - buňkách mimo G0
fázi buněčného cyklu. Původní zvětšení 200×.
(D) Imunohistochemické vyšetření s mutačně specifi ckou protilátkou H3 K27M s pozitivní reakcí v nádorových buňkách (hnědé), negativní
jsou buňky cév (modré). Původní zvětšení 200×.
Fig. 3. Diff use midline altered glioma H3-K27.
(A) Overview of a stereotactic biopsy specimen with infi ltrating hypercellular glial proliferation. Original magnifi cation 100×.
(B) Close-up image showing atypical glial cells with brisk mitotic activity. Original magnifi cation 400×.
(C) Immunohistochemical examination of the proliferation activity of Ki-67 with a positive brown reaction in proliferating cells - cells beyond the G0 phase of the cell cycle. Original magnifi cation 200×.
(D) Immunohistochemical examination with the mutation-specifi c antibody H3 K27M with a positive reaction in tumor cells (brown), negative in vascular cells (blue). Original magnifi cation 200×.


Dětský typ difuzních high-grade gliomů
Difuzní středočarový gliom, H3 K27-alterovaný, WHO G4
Difuzní středočarový gliom, H3 K27-alterovaný (diffuse midline glioma; DMG) je geneticky heterogenní skupina grade 4 IDH-wildtype gliomů s průkaznou ztrátou exprese H3 K27me3 na základě mutace H3 K27M [26], alterací EGFR [27] nebo overexpresí EZHIP (tab. 6) [28]. DMG z definice postihuje primárně středočarové struktury – mozkový kmen, talamus, míchu, mozeček, hypotalamus a glandula pinealis, nicméně postmortem studie prokázala u významné části pacientů s DMG i infiltraci tumoru do mozkových hemisfér a leptomeningeální diseminaci [29]. Přestože se jedná o neoplazii vyskytující se především u pediatrických pacientů, jsou popsány i nečetné případy DMG v dospělé populaci [30]. DMG mohou mít low-grade i high-grade morfologické rysy, nicméně z definice se jedná o grade 4 neoplazii s velmi špatnou prognózou. Dvouletého přežití dosahuje méně než 10 % pacientů [26]. Prognosticky příznivější jsou varianty DMG s geneticky charakteristickými subtypy H3.1 a H3.2 K27M či overexpresí EZHIP [28,31].
Histologický obraz zahrnuje široké spektrum diferenciací s minimálně fokální astrocytární diferenciací (obr. 3). Mezi dalšími možnými diferenciačními komponentami se lze setkat s obrovskobuněčnými elementy, pilomyxoidní komponentou, oligodendrogliální, sarkomatoidní, ependymální, epiteloidní, rabdoidní, embryonální/primitivní neuroektodermální komponentou, tak i komponentou atypických gangliových buněk [30]. Právě výrazná morfologická heterogenita podtrhuje nutnost testování alterací definujících DMG u všech IDH-wildtype difuzních gliomů postihujících středočarové struktury [32]. Samotná ztráta exprese H3 K27me3 však není pro DMG specifická, jelikož byla popsána také u IDH-mutovaných gliomů, především oligodendrogliomu s kanonickou mutací IDH1 R132H a nesčetně i u IDH-wildtype gliomů [33,34], což podmiňuje další specifikaci genetických alterací (tab. 6) pro diskriminaci DMG a GBM.
Difuzní hemisferický gliom, H3 G34-mutovaný, WHO G4
Difuzní hemisferický gliom, H3 G34-mutovaný (G34-DHG), je vzácná high-grade difuzní gliální neoplazie vznikající v mozkových hemisférách. Jedná se o neoplazii definovanou na podkladě specifické genetické mutace G34R/V v genu H3F3A, která ji vyčleňuje ze skupiny morfologicky neodlišitelných IDH-wildtype difuzních gliomů (tab. 7) [35]. Tato specifická alterace predikuje agresivní biologické chování bez ohledu na histologickou morfologii [32]. Přestože se jedná o neoplazii vyskytující se u pediatrických pacientů či mladých dospělých s mediánem věku pacientů 15,8 roku [36], její výskyt u dospělých pacientů není vyloučený [37]. Prognóza pacientů diagnostikovaných s G34-DHG je v porovnání s GBM i DMG mírně lepší, přičemž medián přežití v recentní systematické přehledové práci dosahuje 17,3 měsíce. Zároveň prognóza dospělých pacientů s G34-DHG je obdobně jako u pacientů s DMG lepší v porovnání s pediatrickými pacienty [36]. Obdobně jako u pacientů s GBM a DMG i u části pacientů s G34-DHG dochází v průběhu progrese onemocnění k leptomeningeální diseminaci [36,38,39].
Morfologicky je G34-DHG velmi heterogenní. Na jedné straně spektra je morfologicky neodlišitelný od GBM, na druhé může imitovat i embryonální tumory/primitivní neuroektodermální tumory, přičemž morfologický vzhled nemá vliv na prognózu. Naopak charakteristický je imunofenotyp neoplazie s průkaznou ztrátou exprese ATRX asociovanou s mutací genu ATRX, nukleární hyperakumulací p53 asociovanou s mutací genu TP53 a absencí nukleární exprese OLIG2. V současné době nejsou jednoznačně ustanovena indikační kritéria pro racionální vyšetření mutace G34R/V genu H3F3A [32], nicméně první práce navrhuje vyšetření provádět u pacientů mladších 50 let, především u tumorů s PNET-like komponentou a abnormální expresí ATRX, p53 a OLIG2 [40].
Difuzní high-grade gliom dětského typu, H3-wildtype a IDH-wildtype
Difuzní high-grade gliom dětského typu, H3-wildtype a IDH-wildtype (pHGG) je difuzní gliální neoplazie vyskytující se typicky u děti i adolescentů a je geneticky definována absencí mutace v genech IDH1/2 a současně i v genech pro histon H3 (tab. 8). pHGG byl dříve nazývaný pediatrický glioblastom, nově však je od používání termínu glioblastom v pediatrické populaci opuštěno pro odlišení rozdílné biologické povahy pediatrických a adultních gliomů [1,2]. pHGG se vyskytují nejčastěji supratentoriálně, méně četně i v oblasti mozkového kmene či mozečku. Histologický obraz osciluje od morfologie charakteristické pro GBM po primitivní neuroektodermální diferenciaci, přičemž oba morfologické extrémy se mohou vzájemně prolínat [41,42]. pHGG jsou agresivní neoplazie se špatnou prognózou lišící se v rámci molekulárních podskupin. Nejhorší prognóza je u pHGG MYCN s mediánem přežití 14 měsíců, mírně příznivější u RTK1 s mediánem přežití 21 měsíců a nejpříznivější u RTK2 s mediánem přežití 44 měsíců [41].
pHGG zahrnuje tři distinktní molekulární podskupiny lišící se molekulárními alteracemi, rozdílným metylačním profilem i prognózou – pHGG RTK1, RTK2 a MYCN [41]. Pro pHGG RTK1 je charakteristická amplifikace PDGFRA (průkazná u cca 33 % případů), pro pHGG RTK 2 amplifikace EGFR (cca 50 % případů) a mutace promotoru TERT (cca 64 % případů) a u pHGG MYCN je častá amplifikace MYCN (cca 50 % případů), přičemž tyto nejsou podskupinově specifické a v nízkém procentu se mohou vyskytovat i v jiné podskupině, než pro kterou jsou charakteristické [32,42].
Hemisférický gliom infantilního typu
Hemisferický gliom infantilního typu (iHG) je nová jednotka supratentoriálních difuzních gliomů vyskytujících se v časném dětském věku, především do 1 roku, a obsahující charakteristické fúze v genech pro receptor tyrosinkináz – NTRK, ROS1, ALK a MET1 vedoucí k aberantní expresi kinázové domény řídící tumorogenezi (tab. 9) [43,44]. Pro senzitivitu k cílené terapii je doporučeno testování těchto charakteristických alterací diagnosticky provádět [32]. Vlastní neoplazie může infiltrovat přiléhající leptomeningy i leptomeningeálně diseminovat [43,44]. Grading není u tohoto typu gliomu v současnosti aplikován (WHO).
Histologický obraz iHG je značně heterogenní, zahrnující morfologicky low-grade i high-grade neoplazie, především s astrocytární vřetenobuněčnou, méně často gemistocytární i ependymální diferenciací. Ojediněle je popisována gangliocytární komponenta či primitivní neuroektodermální diferenciace [2].

Závěr
Difuzní gliomy pediatrického typu jsou skupina nádorů, jejichž klasifikace zaznamenala v poslední době revoluční progres. Jednak byla jednoznačně vyčleněna v podobě samostatné skupiny tumorů od morfologicky příbuzných tumorů dospělého věku. Zároveň byly definovány nové diagnostické jednotky na podkladě pochopení jejich gliomageneze a průkazu charakteristických genetických alterací, které přinášejí terapeutický potenciál pro použití cílené terapie s možným pozitivním dopadem na prognózu.
Grantová podpora
Tato práce byla podpořena Grantovou agenturou MU (MUNI/A/1379/2022) a Agenturou pro zdravotnický výzkum ministerstva zdravotnictví (NU23-03-00100).
Prohlášení o konfliktu zájmů
Autoři deklarují, že v souvislosti s předmětem studie nemají žádný konflikt zájmů.
Sources
1. Louis DN, Perry A, Wesseling P et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 2021; 23 (8): 1231–1251. doi: 10.1093/neuonc/noab106.
2. WHO Classification of Tumours Editorial Board. World Health Organization Classification of Tumours of the Central Nervous System. Lyon: International Agency for Research on Cancer 2021.
3. Toll SA, Tran HN, Cotter J et al. Sustained response of three pediatric BRAFV600E mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget 2019; 10 (4): 551–557. doi: 10.18632/oncotarget.26560.
4. Nicolaides T, Nazemi KJ, Crawford J et al. Phase I study of vemurafenib in children with recurrent or progressive BRAFV600E mutant brain tumors: Pacific Pediatric Neuro-Oncology Consortium study (PNOC-002). Oncotarget 2020; 11 (21): 1942–1952. doi: 10.18632/oncotarget.27600.
5. Jackson ER, Persson ML, Fish CJ et al. A review of the anti-tumor potential of current therapeutics targeting the mitochondrial protease ClpP in H3K27-altered, diffuse midline glioma. Neuro Oncol 2023; noad144. doi: 10.1093/neuonc/noad144.
6. Laetsch TW, DuBois SG, Mascarenhas L et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 2018; 19 (5): 705–714. doi: 10.1016/S1470-2045 (18) 30119-0.
7. Manoharan N, Liu KX, Mueller S et al. Pediatric low-grade glioma: targeted therapeutics and clinical trials in the molecular era. Neoplasia 2023; 36 : 100857. doi: 10.1016/j.neo.2022.100857.
8. Krueger DA, Care MM, Holland K et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 2010; 363 (19): 1801–1811. doi: 10.1056/NEJMoa1001671.
9. Bouffet E, Hansford JR, Garrè ML et al. Dabrafenib plus trametinib in pediatric glioma with BRAF V600 mutations. N Engl J Med 2023; 389 (12): 1108–1120. doi: 10.1056/NEJMoa2303815.
10. Ellison DW, Hawkins C, Jones DTW et al. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF V600E mutation. Acta Neuropathol 2019; 137 (4): 683–687. doi: 10.1007/s00401-019-01987-0.
11. Ryall S, Zapotocky M, Fukuoka K et al. Integrated molecular and clinical analysis of 1,000 pediatric low - -grade gliomas. Cancer Cell 2020; 37 (4): 569–583.e5. doi: 10.1016/j.ccell.2020.03.011.
12. Wefers AK, Stichel D, Schrimpf D et al. Isomorphic diffuse glioma is a morphologically and molecularly distinct tumour entity with recurrent gene fusions of MYBL1 or MYB and a benign disease course. Acta Neuropathol 2020; 139 (1): 193–209. doi: 10.1007/s00401-019-02078-w.
13. Blümcke I, Luyken C, Urbach H et al. An isomorphic subtype of long-term epilepsy-associated astrocytomas associated with benign prognosis. Acta Neuropathol 2004; 107 (5): 381–388. doi: 10.1007/s00401-004-0833-3.
14. Chiang J, Harreld JH, Tinkle CL et al. A single-center study of the clinicopathologic correlates of gliomas with a MYB or MYBL1 alteration. Acta Neuropathol 2019; 138 (6): 1091–1092. doi: 10.1007/s00401-019-02081-1.
15. Ampie L, Choy W, Didomenico JD et al. Clinical attributes and surgical outcomes of angiocentric gliomas. J Clin Neurosci 2016; 28 : 117–122. doi: 10.1016/ j.jocn.2015.11.015.
16. Bandopadhayay P, Ramkissoon LA, Jain P et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 2016; 48 (3): 273–282. doi: 10.1038/ng.3500.
17. Lellouch-Tubiana A, Boddaert N, Bourgeois M et al. Angiocentric neuroepithelial tumor (ANET): a new epilepsy-related clinicopathological entity with distinctive MRI. Brain Pathology 2005; 15 (4): 281–286. doi: 10.1111/j.1750-3639.2005.tb00112.x.
18. Huse JT, Snuderl M, Jones DTW et al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): an epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol 2017; 133 (3): 417–429. doi: 10.1007/s00401-016-1639-9.
19. Johnson DR, Giannini C, Jenkins RB et al. Plenty of calcification: imaging characterization of polymorphous low-grade neuroepithelial tumor of the young. Neuroradiology 2019; 61 (11): 1327–1332. doi: 10.1007/s00234-019-02269-y.
20. Gupta VR, Giller C, Kolhe R et al. Polymorphous low-grade neuroepithelial tumor of the young: a case report with genomic findings. World Neurosurg 2019; 132 : 347–355. doi: 10.1016/j.wneu.2019.08.221.
21. Blümcke I, Coras R, Busch RM et al. Toward a better definition of focal cortical dysplasia: an iterative histopathological and genetic agreement trial. Epilepsia 2021; 62 (6): 1416–1428. doi: 10.1111/epi.16899.
22. Hendrych M, Hemza J, Kočvarová J et al. Polymorphous low-grade neuroepithelial tumor of the young. Cesk Slov Neurol N 2021; 84/117 (3): 282–285. doi: 10.48095/cccsnn2021282.
23. Bale TA, Sait SF, Benhamida J et al. Malignant transformation of a polymorphous low grade neuroepithelial tumor of the young (PLNTY). Acta Neuropathol 2021; 141 (1): 123–125. doi: 10.1007/s00401-020-02245-4.
24. Zhang J, Wu G, Miller CP et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013; 45 (6): 602–612. doi: 10.1038/ng.2611.
25. Qaddoumi I, Orisme W, Wen J et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 2016; 131 (6): 833–845. doi: 10.1007/s00401-016-1539-z.
26. Louis DN, Giannini C, Capper D et al. cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol 2018; 135 (4): 639–642. doi: 10.1007/s00401-018 - 1826-y.
27. Sievers P, Sill M, Schrimpf D et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro Oncol 2021; 23 (1): 34–43. doi: 10.1093/neuonc/noaa251.
28. Castel D, Kergrohen T, Tauziède-Espariat A et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol 2020; 139 (6): 1109–1113. doi: 10.1007/s00401-020-02142-w.
29. Buczkowicz P, Bartels U, Bouffet E et al. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol 2014; 128 (4): 573–581. doi: 10.1007/s00401-014-1319-6.
30. Solomon DA, Wood MD, Tihan T et al. Diffuse midline gliomas with histone H3-K27M mutation: a series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathology 2016; 26 (5): 569–580. doi: 10.1111/bpa. 12336.
31. Hoffman LM, Van Zanten SEMV, Colditz N et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of Diffuse Intrinsic Pontine Glioma (DIPG): a collaborative report from the International and European Society for Pediatric Oncology DIPG registries. J Clin Oncol 2018; 36 (19): 1963–1972. doi: 10.1200/JCO.2017.75.9308.
32. Brat DJ, Aldape K, Bridge JA et al. Molecular biomarker testing for the diagnosis of diffuse gliomas. Arch Pathol Lab Med 2022; 146 (5): 547–574. doi: 10.5858/arpa.2021-0295-CP.
33. Habiba U, Sugino H, Yordanova R et al. Loss of H3K27 trimethylation is frequent in IDH1-R132H but not in non-canonical IDH1/2 mutated and 1p/19q codeleted oligodendroglioma: a Japanese cohort study. Acta Neuropathol Commun 2021; 9 (1): 95. doi: 10.1186/s40478-021-01194-7.
34. Filipski K, Braun Y, Zinke J et al. Lack of H3K27 trimethylation is associated with 1p/19q codeletion in diffuse gliomas. Acta Neuropathol 2019; 138 (2): 331–334. doi: 10.1007/s00401-019-02025-9.
35. Louis DN, Wesseling P, Aldape K et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol 2020; 30 (4): 844–856. doi: 10.1111/bpa.12 832.
36. Crowell C, Mata-Mbemba D, Bennett J et al. Systematic review of diffuse hemispheric glioma, H3 G34-mutant: outcomes and associated clinical factors. Neurooncol Adv 2022; 4 (1): vdac133. doi: 10.1093/noajnl/vdac 133.
37. Lim KY, Won JK, Park CK et al. H3 G34-mutant high-grade glioma. Brain Tumor Pathol 2021; 38 (1): 4–13. doi: 10.1007/s10014-020-00378-8.
38. Korshunov A, Capper D, Reuss D et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol 2016; 131 (1): 137–146. doi: 10.1007/s00401-015-1493-1.
39. Gessi M, Gielen GH, Hammes J et al. H3.3 G34R mutations in pediatric primitive neuroectodermal tumors of central nervous system (CNS-PNET) and pediatric glioblastomas: possible diagnostic and therapeutic implications? J Neurooncol 2013; 112 (1): 67–72. doi: 10.1007/s11060-012-1040-z.
40. Trejo-Lopez JA, Praska CE, Zepeda Mendoza C et al. H3 G34 mutation assessment for diffuse gliomas in adults: when would testing be most diagnostically useful? J Neuropathol Exp Neurol 2022; 82 (1): 93–95. doi: 10.1093/jnen/nlac102.
41. Korshunov A, Schrimpf D, Ryzhova M et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol 2017; 134 (3): 507–516. doi: 10.1007/s00401-017-1710-1.
42. Tauziède-Espariat A, Debily MA, Castel D et al. An integrative radiological, histopathological and molecular analysis of pediatric pontine histone-wildtype glioma with MYCN amplification (HGG-MYCN). Acta Neuropathol Commun 2019; 7 (1): 87. doi: 10.1186/s40478-019-0738-y.
43. Clarke M, Mackay A, Ismer B et al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov 2020; 10 (7): 942–963. doi: 10.1158/2159-8290.CD-19-1030.
44. Guerreiro Stucklin AS, Ryall S, Fukuoka K et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 2019; 10 (1): 4343. doi: 10.1038/s41467-019-12187-5.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2024 Issue 1
Most read in this issue
- Diffuse glioma overview based on the 2021 WHO classifi cation part 2 – pediatric type
- Tonsilla cerebelli – anatomy, function and its significance for neurosurgery
- Standardization of MRI in Multiple Sclerosis Management Consensus by the Czech Expert Radiology-Neurology Panel
- Adaptation and validation of the Czech version of the Addenbrooke‘s Cognitive Examination (ACE-III-CZ) – pilot study