
Klinický obraz spinální muskulární atrofie u dospělých pacientů
Clinical manifestations of spinal muscular atrophy in adult patients
Spinal muscular atrophy (SMA) is a phenotypically heterogeneous disease. Although the life expectancy and motor capability is significantly reduced in most patients, SMA types III and IV patients live to adulthood. In some cases, the association with a higher number of copies of the SMN2 gene and other protective factors are responsible for minimal motor impairment only. However, precise data on the prevalence of this disease in adult patients remain unknown. In the adult age, in addition to the classical 5q13 form of SMA, there are other rare genetic forms of late onset SMA with dominant disability in the lower limbs, which must be distinguished because of different clinical and therapeutic approaches. Since the development of SMA therapy, there has been a need for a clinical evaluation of therapeutic response and a definition of multidisciplinary care standards that have some specifics in adult SMA patients.
Keywords:
spinal muscular atrophy – Adults – motor neuron – Kennedy‘s disease – differential diagnostics
Authors:
T. Horák 1; J. Bednařík 1; M. Horáková 1; D. Botiková 2; S. Voháňka 1
Authors‘ workplace:
Neurologická klinika, ERN-EURO NMD Centrum, LF MU a FN Brno
1; Rehabilitační oddělení, LF MU a FN Brno
2
Published in:
Cesk Slov Neurol N 2020; 83/116(Supplementum 2): 13-16
doi:
https://doi.org/10.48095/cccsnn20202S13
Overview
Spinální muskulární atrofie (SMA) je fenotypicky heterogenní onemocnění. Přestože u většiny pacientů významně zkracuje délku dožití a omezuje jejich motorické schopnosti, pacienti se SMA typu III a IV se dožívají dospělého věku. Díky asociaci s vyšším počtem kopií SMN2 genu a jinými protektivními faktory mají v některých případech minimální motorické postižení. Přesné údaje o prevalenci tohoto onemocnění u dospělých pacientů však dosud nejsou známé. V dospělém věku existují kromě klasické 5q13 formy SMA i další vzácné genetické formy SMA s pozdním začátkem a dominantním postižením na dolních končetinách, které je z klinického a terapeutického hlediska nutné odlišit. S rozvojem terapie SMA vznikla potřeba klinického hodnocení úspěšnosti léčby a definice standardů multidisiciplinární péče, které mají u dospělých pacientů se SMA svá specifika.
Klíčová slova:
spinální svalová atrofie – dospělí – motorický neuron – Kennedyho choroba – diferenciální diagnostika
Úvod
Spinální muskulární atrofie (SMA) se řadí mezi tzv. vzácná onemocnění. Epidemiologické údaje jsou napříč literaturou velmi variabilní s rozmezím udávané prevalence 1,4–13,0 na 100 000 obyvatel, nicméně nejčastěji je prevalence je udávána průměrně 2 na 100 000 [1]. V ČR tak zřejmě žije okolo 200 pacientů s tímto onemocněním. Přesné údaje o prevalenci SMA u dospělých však neexistují. Z incidence tohoto onemocnění 10 (5–18) na 100 000 živě narozených dětí – SMA typ I (58 %), typ II (29 %), typ III (13 %), typ IV (1 %) – a průměrné délky dožití do dospělosti u jednotlivých typů SMA – typ I (1 %), typ II (75–93 %), typ III (99,9 %), typ IV (100 %) – můžeme odvodit, že dospělého věku se každoročně dožijí 3–4 pacienti s touto chorobou. Můžeme tak tedy odhadovat, že v ČR žije okolo 80 dospělých pacientů se SMA (typu II 44 %, typu III 49 % a typu IV 7 %) (obr. 1). Přesné údaje o prevalenci SMA u dospělých v ČR v budoucnu poskytne projekt REaDY (REgistry of muscular DYstrophy) zahrnující také registr pacientů se SMA [2].
Fig. 1. Distribution of SMA types in the adult population estimated from the incidence,
prevalence and life expectancy data.

Vzhledem k absenci kauzální terapie do roku 2016 (FDA [Food and Drug Administration] schválila nusinersen pro léčbu všech typů 5q SMA v prosinci 2016 a EMA [European Medicines Agency] v červnu 2017) byli zejména dospělí pacienti dispenzarizováni mimo nervosvalová centra a jednotliví lékaři, obvykle ambulantní neurologové a praktiční lékaři, měli ve své péči nanejvýš jednotky pacientů. SMA je však komplexní onemocnění, které postihuje pacienta multiorgánově a již dlouhou dobu je známá skutečnost, že komplexní multidisciplinární péče a dodržování standardů péče (standards of care; SoC) zlepšuje kvalitu života a prodlužuje dožití u všech typů SMA [3,4]. S rozvojem léčby se situace změnila a vznikla potřeba důslednější organizace a hierarchizace péče vč. hodnocení efektivity finančně nákladné terapie s nutností použití hodnotících škál v rámci centrové péče.
Spinální muskulární atrofie a její specifika u dospělých
Jedná se o dědičné autozomálně recesivní, progresivní neurodegenerativní onemocnění způsobené nejčastěji mutací genu SMN1 (survival motor neuron 1) s odlišnými klinickými podtypy [5]. Je způsobeno degenerací alfa motoneuronů míchy, což má za následek progredující atrofii svalů, svalovou slabost predominantně na dolních končetinách a nakonec paralýzu [6]. Široká škála klinické manifestace u dospělých pacientů je dána různými faktory, z nichž dominantní roli zřejmě hraje počet kopií genu SMN2 [7], který je běžně odpovědný za 10 % produkce proteinu SMN. U nejběžnějšího genetického typu SMA podmíněného mutací genu SMN1 rozlišujeme 4, resp. 5 klinických forem (0–IV) na podkladě maximální dosažené motorické funkce a věku rozvoje prvních klinických příznaků [8]. Typy II–IV se běžně dožívají dospělého věku.
Z klinického hlediska je výhodné dělení na SMA s časným a pozdním začátkem. Zatímco přirozený průběh choroby je u pacientů se SMA s časným začátkem (early onset SMA; typ I–IIIa) velmi dobře popsán, u pozdních forem onemocnění (late onset SMA; typy IIIb a IV) je informací o přirozeném průběhu podstatně méně (tab. 1) [9].
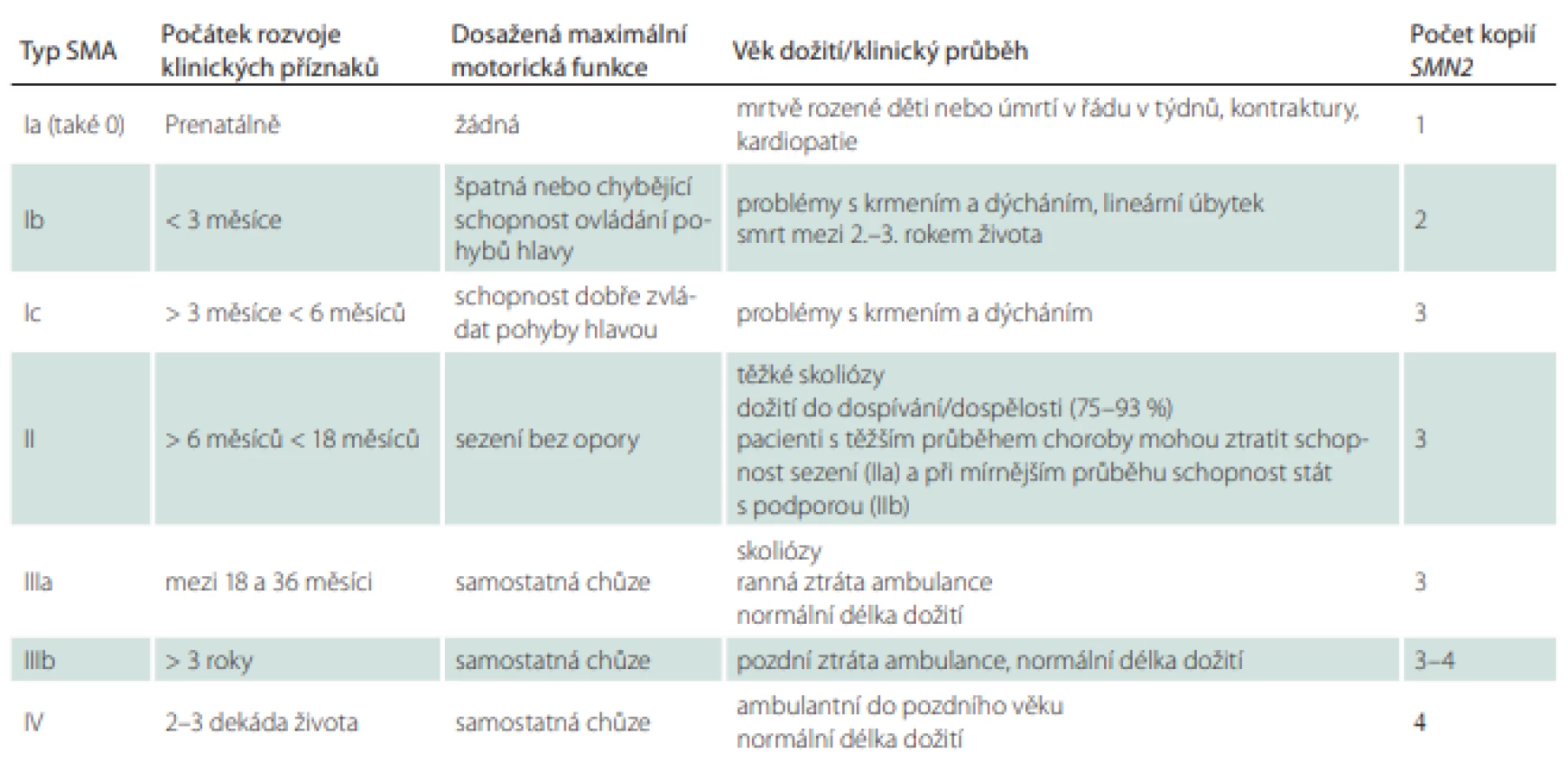
Spinální muskulární atrofie s časným začátkem
Pacienti se SMA typu I se dospělého věku dožívají jen vzácně (1 % případů, typ Ic) [10,11]. Předpokladem je intenzivní pulmonologická péče s využitím moderních respiračních pomůcek, umělé plicní ventilace nebo také kvalitní péče o výživu pomocí gastrostomie.
Pacienti klinické formy typu II (Dubowitz) se dožívají dospělého věku v 75–93 % [10]. Klinický průběh se vyznačuje obdobím zpomalení progrese. Progredující skolióza a kontraktury jsou však u této skupiny pacientů velmi časté, vyvíjejí se již od počátku a téměř vždy v dospělosti vyžadují zvláštní ortopedickou péči. Často se vyskytuje slabost žvýkacích svalů, potíže s polykáním a časné respirační onemocnění se širokým spektrem závažnosti.
SMA typu III (choroba Kugelberg-Welander) je mírnější formou SMA charakteristickou pomalou progresí. Pacienti s tímto typem SMA si často zachovávají schopnost stát a chodit až do dospělosti [12], přestože někteří mohou tuto schopnost ztratit, a někdy tak využívají mechanický invalidní vozík. Tuto skupinu dělíme na typy IIIa a IIIb. Typ IIIa s rozvojem klinických příznaků nemoci do 3 let věku má mírně sníženou délku života se zpožděným motorickým vývojem. Pacienti se SMA typu IIIa mají ortopedické problémy, které jsou srovnatelné s těmi, které se vyskytují u dospělých pacientů se SMA typu II, i když obecně s pozdějším nástupem a sníženou závažností nemoci. Typ IIIb již řadíme k tzv. SMA s pozdním začátkem.
Spinální muskulární atrofie s pozdním začátkem
Typ IIIb jsou spolu se SMA typu IV asociovány s vyšším počtem kopií SMN2, obvykle v počtu 4 kopií [9]. SMA typu IIIb se vyznačuje nástupem po 3. roce života, normální délkou života a normálním vývojem motoriky. Pacienti se SMA typu IIIb mají ortopedické potíže pouze mírné. Vyskytuje se obvykle mírná slabost dolních končetin projevující se obtížemi při stoupání do schodů a nejistou „chvějící se“ chůzí.
Dospělý typ SMA IV je charakterizován pozdním nástupem nemoci během druhé nebo spíše třetí dekády a normální délkou života. Mírný průběh nemoci se projevuje plně zachovanou schopností chůze. Respirační a nutriční problémy jsou velmi vzácné a tito pacienti obvykle nemají potřebu respirační asistence [13].
Existují i další vzácné genetické formy SMA s pozdním začátkem a dominantním postižením na dolních končetinách, které je z klinického a terapeutického hlediska nutné odlišit od klasické 5q13 formy SMA. Ty byly v posledních letech popsány zejména díky pokroku v lékařské genetice (sekvenace nové generace), který umožnil zkoumání molekulárních mechanizmů, jež způsobují degeneraci motorických neuronů.
Diferenciální diagnostika SMA s pozdním začátkem
Druhým nejčastějším genetickým onemocněním, jež způsobuje degeneraci motorických neuronů a je nutné jej diferenciálně diagnosticky odlišit od jiných SMA typů, které se mohou objevit během dospívání či v dospělosti, je bulbospinální muskulární atrofie (SBMA) známá také jako Kennedyho choroba. Genetickým podkladem je expanze CAG tripletů androgenového receptorového genu chromozomu X, jejíž velikost koreluje se závažností a časností rozvoje klinických příznaků. Dědičnost je gonozomálně recesivní a na rozdíl od SMA se projevuje pouze u mužů. Klinický obraz se rozvíjí mezi 20. a 70. rokem života a zahrnuje svalové atrofie, slabost a fascikulace, zvláště v bulbární oblasti (jazyk, mimické svaly) a dále u proximálních svalů na končetinách. Dalšími příznaky jsou gynekomastie, tremor rukou, svalové bolesti a krampi. Jeho prevalence se odhaduje na 1–2/ 100 000 obyvatel [14].
Ostatní geneticky podmíněné typy SMA s pozdním začátkem:
Spinální svalová atrofie typ Jokela (SMAJ) – spinální motorická neuronopatie s pozdním rozvojem (LOSMoN). Onemocnění je způsobené mutací v genu CHCHD10 (kódující protein působící v mitochondriální membráně), při jehož dysfunkci dochází k narušení procesu oxidativní fosforylace. Onemocnění je charakteristické pozdním rozvojem klinických potíží okolo 40. roku života (14–70 let). V klinickém obraze dominují křeče, fascikulace a areflexie. Vzácně se mohou vyskytovat bulbární nebo respirační příznaky. Mutace v genu CHCHD10 byla zatím ve spojení se SMA popsána pouze u finské populace [15].
SMA typ Finkel (SMA-FK) byla popsána u populace v Brazílii. Je způsobená mutací v genu VAPB, který za normálních okolností kóduje protein působící v endoplazmatickém retikulu motoneuronů. Při disfunkci endoplazmatického retikula tak dochází k nahromadění proteinů uvnitř nervových buněk a následně k jejich poškození a smrti. Onemocnění se rozvíjí typicky okolo 45. roku života [16,17].
SMA s predominantní postižením dolních končetin (SMA-LED) je způsobené mutací genu DYNC1H1. Je velmi variabilní co se týká věku rozvoje klinických příznaků, ale v dospělosti se rozvíjí pouze minoritně. Jak odkazuje název, postihuje převážně dolní končetiny, a způsobuje tak charakteristickou postavu tvarem připomínající postavu kulturisty [18,19].
SMA s postižením srdce s mutací v genu LMNA s proximální svalovou slabostí s atrofií bylo popsáno u dvou rodin [20,21] a způsobuje srdeční arytmii.
V neposlední řadě některé klasifikace řadí pod SMA určité typy hereditárních motorických neuropatií (HMN) někdy též nazývané distální SMA (dSMA) [22,23]. Patří sem např. kongenitální distální SMA (scapuloperoneální SMA [SPSMA]), která je způsobená mutací v genu TRPV4. Jedná se o vzácné autozomálně dominantní onemocnění, pro které jsou charakteristické pes cavus, distální oslabení končetin, oslabení hlasivkových svalů (dysfonie) a skolióza. Mutace se vyskytují převážně v doméně proteinu, která je významnou součástí iontového kanálu pro Ca2+ a další kationty.
Mimo onemocnění s postižením periferního motorického neuronu se známou genetickou podstatou je potřeba diferenciálně diagnosticky odlišit progresivní svalovou atrofii (PMA). Ta je často nomenklaturně řazená jako varianta amyotrofické laterální sklerózy (ALS). Od ALS se liší absencí postižení centrálního motorického neuronu v počátku onemocnění, které ale většina pacientů s postupující progresí onemocnění rozvine [24,25]. I z tohoto důvodu je stále předmětem diskuzí a kontroverze, zda se jedná o formu ALS či samostatnou nozologickou jednotku.
Postižení motorických a dechových funkcí a jejich hodnocení
Nedílnou součástí klinického obrazu SMA u dospělých pacientů jsou onemocnění pohybového aparátu, kdy pozorujeme zejména omezený rozsah pohybu v kloubech [26]. Zatímco SMA typu I má velmi závažný průběh, který je ortopedickou léčbou jen velmi obtížně ovlivnitelný, pacienti se SMA dospělého typu IV bývají mnohem méně postiženi a konzervativní terapie často postačuje ke zlepšení kvality života [27]. Největší pozornost pohybovému aparátu je tudíž nutné věnovat podskupinám SMA typu II a IIIa, protože adekvátní a včasná léčba vč. provedení preventivních chirurgických zákroků může významně zpomalit progresi onemocnění a zlepšit kvalitu života. Charakteristické příznaky SMA (zejména typu II a IIIa) zahrnují kontraktury především na dolních končetinách (s kyčelními subluxacemi a dislokacemi). Loketní klouby jsou převážně postiženy ohybovými kontrakturami a omezenou supinací následovanou kontrakturami ramen a zápěstí. Na dolních končetinách jsou časté výrazné flekční kontraktury kyčelního a kolenního kloubu a silně omezená pohyblivost kotníku. Indikace pro chirurgickou léčbu jsou na horních končetinách vzácné, oproti tomu chirurgická léčba kontraktur dolních končetin mívá velmi dobrý efekt. Kontinuální fyzioterapie a pracovní terapie jsou nezbytné pro zachování zbývajících schopností v každodenním životě a mohou pomoci snížit progresi kontraktur a optimalizovat zbytkový rozsah pohybu [26,28]. U SMA pacientů jsou rovněž pozorovány hypermobilní klouby postihující nejčastěji zápěstí [29]. Skolióza se objevuje u téměř 100 % pacientů se SMA v pozdějším stadiu onemocnění a zůstává jedním z hlavních problémů ortopedické terapie. Mezi následky skoliózy patří deformity hrudního koše, vzájemný útlak žeber či ovlivnění sklonu pánve. Důsledkem respiračních problémů je masivní snížení vitální kapacity. Zejména v rámci nemocniční péče je nutné mít na paměti omezenou dechovou kapacitu, problémy s anestézií během perioperační péče, problémy s výživou a růstem, ale také potřebu zvláštních rehabilitačních a integračních postupů [27]. Kromě toho existuje vysoké riziko spontánních zlomenin v důsledku osteopenie.
Na rozdíl od dětských pacientů nejsou u dospělých používány některé škály a kritéria specifické pro novorozence. Vzhledem k rozmanitosti závažnosti fenotypického projevu u dospělých pacientů se SMA není možné uplatnit jednu škálu u všech pacientů, ale je vždy nutné specifikovat jejich užití individuálně dle typu SMA a věku pacienta. Vzhledem k predominantnímu postižení na dolních končetinách zůstává zachována motorická funkce primárně na horních končetinách. U dospělých pacientů se SMA všech typů proto provádíme klinimetrické vyšetření motorické funkce ramene, lokte, zápěstí i ruky pomocí škál RULM (Revised Upper Limb Module) [30] a dále vyšetření respiračních funkcí. U pacientů se SMA typu III a IV se dále provádí škála HFMSE (Hammersmith Functional Motor Scale Expanded) nebo RHS (Revised Hammersmith Scale for SMA) [31] hodnotící motorické tělesné funkce a 6MWT (test 6minutové chůze) u pacientů se zachovanou schopností chůze. Klinimetrické hodnocení je tedy důležité pro hodnocení efektu léčby a také k posouzení kvality multidisciplinární péče a mělo by být u dospělých pacientů prováděno v rámci péče v neuromuskulárních centrech spolu s organizací multidisciplinární péče podle SoC.
Grantová podpora
Ministerstvo školství, mládeže a tělovýchovy České republiky (SVMUNI/A/1325/2019). Ministerstvo zdravotnictví České republiky (MH CZ – DRO, FNBr, 65269705).
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
MUDr. Magda Horáková
Neurologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: magda.horakova@mail.muni.cz
Sources
1. Verhaart IE, Robertson A, Wilson IJ et al. Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis 2017; 12(1): 124. doi: 10.1186/ s13023-017-0671-8.
2. Strenkova J, Vohanka S, Haberlova J et al. REaDY – Czech Registry of Muscular Dystrophies. Cesk Slov Neurol N 2014; 77/ 110(2): 230–234.
3. Mercuri E, Mazzone E, Finkel R et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018; 28(2): 103–115. doi: 10.1016/ j.nmd.2017.11.005.
4. Finkel RS, Mercuri E, Meyer OH et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord 2018; 28(3): 197–207. doi: 10.1016/ j.nmd.2017.11.004.
5. Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther 2017; 24(9): 529–533. doi: 10.1038/ gt.2017.52.
6. Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol 2012; 11(5): 443–452. doi: 10.1016/ S1474-4422(12)70061-3.
7. Wirth B, Brichta L, Schrank B et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet 2006; 119(4): 422–428. doi: 10.1007/ s00439-006-0156-7.
8. Munsat TL, Davies KE. International SMA consortium meeting. (26-28 June 1992, Bonn, Germany). Neuromuscul Disord 1992; 2(5–6): 423–428. doi: 10.1016/ s0960-8966(06)80015-5.
9. Piepers S, van den Berg LH, Brugman F et al. A natural history study of late onset spinal muscular atrophy types 3b and 4. J Neurol 2008; 255(9): 1400–1404. doi: 10.1007/ s00415-008-0929-0.
10. Farrar MA, Vucic S, Johnston HM et al. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr 2013; 162(1): 155–159. doi: 10.1016/ j.jpeds.2012.05.067.
11. Park HB, Lee SM, Lee JS et al. Survival analysis of spinal muscular atrophy type I. Korean J Pediatr 2010; 53(11): 965–970. doi: 10.3345/ kjp.2010.53.11.965.
12. Zerres K, Rudnik-Schöneborn S, Forrest E et al. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci 1997; 146(1): 67–72. doi: 10.1016/ s0022-510x(96)00284-5.
13. Juntas Morales R, Pageot N, Taieb G et al. Adult-onset spinal muscular atrophy: an update. Rev Neurol (Paris) 2017; 173(5): 308–319. doi: 10.1016/ j.neurol.2017.03.015.
14. Finsterer J. Bulbar and spinal muscular atrophy (Kennedy’s disease): a review. Eur J Neurol 2009; 16(5): 556–561. doi: 10.1111/ j.1468-1331.2009.02591.x.
15. Penttilä S, Jokela M, Bouquin H et al. Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann Neurol 2015; 77(1): 163–172. doi: 10.1002/ ana.24319.
16. Nishimura AL, Mitne-Neto M, Silva HC et al. A mutation in the vesicle-trafficking protein VAPB causes late--onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 2004; 75(5): 822–831. doi: 10.1086/ 425287.
17. Kosac V, de Freitas MR, Prado FM et al. Familial adult spinal muscular atrophy associated with the VAPB gene: report of 42 cases in Brazil. Arq Neuropsiquiatr 2013; 71(10): 788–790. doi: 10.1590/ 0004-282X20130123.
18. Rossor AM, Oates EC, Salter HK et al. Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2. Brain J Neurol 2015; 138(Pt 2): 293–310. doi: 10.1093/ brain/ awu356.
19. Neveling K, Martinez-Carrera LA, Hölker I et al. Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am J Hum Genet 2013; 92(6): 946–954. doi: 10.1016/ j.ajhg.2013.04.011.
20. Iwahara N, Hisahara S, Hayashi T et al. A novel lamin A/ C gene mutation causing spinal muscular atrophy phenotype with cardiac involvement: report of one case. BMC Neurol 2015; 15: 13. doi: 10.1186/ s12883-015-0269-5.
21. Rudnik-Schöneborn S, Botzenhart E, Eggermann T et al. Mutations of the LMNA gene can mimic autosomal dominant proximal spinal muscular atrophy. Neurogenetics 2007; 8(2): 137–142. doi: 10.1007/ s10048-006-0070-0.
22. Devic P, Petiot P. [Distal hereditary motor neuropathy]. Rev Neurol (Paris) 2011; 167(11): 781–790. doi: 10.1016/ j.neurol.2011.03.003.
23. Mazanec R, Potočková V, Laššuthová P et al. Hereditární motorické neuropatie. Neurol praxi 2016; 17(6): 354–358. doi: 10.36290/ neu.2016.074.
24. Ikeda K, Iwasaki Y. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology 2010; 74(23): 1926. doi: 10.1212/ WNL.0b013e3181e03ac0.
25. Liewluck T, Saperstein DS. Progressive Muscular Atrophy. Neurol Clin 2015; 33(4): 761–773. doi: 10.1016/ j.ncl.2015.07.005.
26. Fujak A, Kopschina C, Gras F et al. Contractures of the upper extremities in spinal muscular atrophy type II. Descriptive clinical study with retrospective data collection. Ortop Traumatol Rehabil 2010; 12(5): 410–419.
27. Haaker G, Fujak A. Proximal spinal muscular atrophy: current orthopedic perspective. Appl Clin Genet 2013; 6(11): 113–120. doi: 10.2147/ TACG.S53615.
28. Willig TN, Bach JR, Rouffet MJ et al. Correlation of flexion contractures with upper extremity function and pain for spinal muscular atrophy and congenital myopathy patients. Am J Phys Med Rehabil 1995; 74(1): 33–38. doi: 10.1097/ 00002060-199501000-00006.
29. de Groot IJM, de Witte LP. Physical complaints in ageing persons with spinal muscular atrophy. J Rehabil Med 2005; 37(4): 258–262. doi: 10.1080/ 16501970510030156.
30. Mazzone E, Mayhew A, Montes J et al. Revised upper limb module for spinal muscular atrophy: Development of a new module: RULM in SMA. Muscle Nerve 2016; 55(6). doi: 10.1002/ mus.25430.
31. Ramsey D, Scoto M, Mayhew A et al. Revised Hammersmith Scale for spinal muscular atrophy: A SMA specific clinical outcome assessment tool. PLoS ONE 2017; 12(2): e0172346. doi: 10.1371/ journal.pone.0172346.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2020 Issue Supplementum 2
Most read in this issue
- Klinický obraz spinální muskulární atrofie u dospělých pacientů
- Genetika spinální muskulární atrofie
- Léčba spinální svalové atrofie
- Rehabilitace u spinální muskulární atrofie