
Multisystémová atrofie
Multiple system atrophy
Multiple system atrophy (MSA) is relatively rare neurodegenerative disease with fatal prognosis affecting middle-aged and elderly individuals. There is combination of several motor and non-motor symptoms in MSA. Causal treatment does not exist and symptomatic treatment effect is unsatisfactory. The article briefly discusses genetics and pathophysiology of MSA with special focus on the clinical picture, practical aspects of diagnostics and current therapeutic options of MSA in the Czech Republic.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Keywords:
multiple system atrophy – atypical parkinsonian syndrome – alpha synuclein – REM sleep behavioral disorders – autonomic dysfunction – orthostatic hypotension – inspiratory stridor
:
J. Klempíř 1,2,3; T. Bartošová 1,2
:
Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN v Praze
1; Anatomický ústav, 1. LF UK v Praze
2; Evropská referenční síť pro vzácná neurologická onemocnění
3
:
Cesk Slov Neurol N 2019; 82(4): 370-380
:
Review Article
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amcsnn2019370
Multisystémová atrofie (MSA) je relativně vzácné neurodegenerativní onemocnění středního a vyššího věku s fatální prognózou. U MSA se kombinují různé motorické i nonmotorické příznaky. Kauzální léčba neexistuje a účinky symptomatické léčby jsou neuspokojivé. Článek stručně pojednává o patofyziologii a genetice MSA. Detailně se zaměřuje na klinický obraz, praktické aspekty diagnostiky a aktuální terapeutické možnosti MSA v ČR
Klíčová slova:
multisystémová atrofie – atypický parkinsonský syndrom – alfa synuklein – abnormní chování v REM spánku – autonomní dysfunkce – ortostatická hypotenze – inspirační stridor
Úvod
Multisystémová atrofie (MSA) byla poprvé popsána Dejerinem a Thomasem v roce 1900 jako olivopontocerebelární atrofie. U MSA se objevují příznaky autonomní dysfunkce, parkinsonského, mozečkového a pyramidového syndromu. K nonmotorickým projevům MSA patří zejména bolest, ortostatická hypotenze, poruchy urogenitální, gastrointestinální, respirační, poruchy spánku, chování a kognitivní deficit.
Epidemiologie
Četnost výskytu nemoci v ČR není známa. V Evropě je odhadována incidence 0,6/
100 000 a prevalence 1,9– 4,9/ 100 000 [1– 3]. MSA se v Evropě iniciálně manifestuje 2– 4× častěji parkinsonským než mozečkovým syndromem [4]. Statistické údaje jsou pravděpodobně zkresleny přehlížením manifestace nemoci ve stáří, nedostatečnou diagnostickou zkušeností s parkinsonskými a mozečkovými syndromy, netypickým průběhem nemoci, úmrtím pacienta ještě před rozvojem typického klinického obrazu a nedůsledným používáním klasifikačních kódů pro pojišťovny [5– 7]. Rozdíly ve výskytu v závislosti na rase nebyly doloženy. Podle některých pozorování jsou častěji postiženi muži [3]. Průměrný věk počátku nemoci se pohybuje mezi 55.– 60. rokem (31– 78 let) a medián přežití je 7– 10 let. Přesvědčivé důkazy o vlivu prostředí na vznik nemoci neexistují a velmi vzácně je zdokumentován familiární výskyt [8].
Patofyziologie
U neurodegenerativních onemocnění obecně dochází k hromadění patologických proteinů v nervovém systému a zániku specifických populací nervových buněk. MSA je řazena mezi tzv. synukleinopatie, podobně jako Parkinsonova nemoc (PN) a nemoc s Lewyho tělísky (diffuse Lewy body disease; DLBD). Mutace alfa synukleinu (αS) v oblasti genu (4q22.1) je považována za příčinu vzniku některých autozomálně dominantních variant PN (mutace PARK 1, PARK 4)
a DLBD. Alfa synuklein je protein, který se v nerozpustné formě ukládá do inkluzí u MSA přednostně se akumulujících v cytoplazmě oligodendrocytů. Inkluze obsahují hyperfosforylovaný αS, tau protein, ubikvitin a další proteiny. Existuje předpoklad, že zvýšená přítomnost (koncentrace) těchto inkluzí narušuje funkci glie a myelinu a vede také k zánětlivým změnám, které sekundárně podporují neurodegeneraci [9,10]. Někteří vědci v etiopatogenezi MSA spatřují analogické procesy, jež jsou popisovány u prionových onemocnění [10].
Inkluze se u MSA mohou vyskytovat v neuronech i axonech, ale průkaz gliálních cytoplazmatických inkluzí (Pappova-Lantosova tělíska) je pro MSA patognomický [11]. U MSA s parkinsonským fenotypem (MSA-P)
je popisována atrofie kaudata, putamen, substantia nigra, locus coeruelus a nucleus dorsalis nervi vagi. Ve striatu dochází k selektivnímu úbytku postsynaptických D1 receptorů. Postiženy jsou však i ostatní neurotransmiterové systémy. U MSA s cerebelárním fenotypem (MSA-C) dochází k významnému úbytku Purkyňových buněk mozečku, atrofii pedunculi cerebellares medii, baze pontu a nucleus olivares inferiores. Rozvoj autonomní dysfunkce souvisí s postižením nucleus intermediolateralis, Onufova jádra a katecholaminergních neuronů ve ventrolaterální části oblongaty [12].
Genetika
Wenning et al publikovali vyšší výskyt parkinsonizmu v první a druhé generaci u 38 pacientů s histopatologicky verifikovanou MSA, ale jasný důkaz o dědičnosti podán nebyl [13]. V Japonsku byl u dvou rodin popsán výskyt MSA s mutací genu COQ2 (chromozom 4q21) pro koenzym Q [14]. Pokles hladiny koenzymu Q10 a snížení aktivity mitochondriálního komplexu II byly zjištěny v různých tkáních i u nemocných s MSA bez mutace pro COQ2 [15,16]. Při studiu monozygotních dvojčat byla u pacientů s MSA popsána deleční mutace genu SHC2 (Src homology 2 domain containing-transforming protein 2) [17,18]. Polymorfizmy v lokusu pro synuklein (4q22.1) (abnormally phosphorylated alpha-synuclein; SNCA) pravděpodobně zvyšují riziko rozvoje MSA [19,20]. Vztah mezi výskytem mutace COQ2, SNCA a rozvojem onemocnění nebyl nalezen [21].
V evropské populaci byly identifikovány nejméně čtyři polymorfizmy (EDN1 rs16872704, MAPT rs9303521, FBXO47 rs78523330, a ELOVL7
rs7715147) spojené s rizikem rozvoje MSA, ale v čínské populaci (906 nemocných a 941 nekonsagvinních kontrol) se zvýšené riziko pro tyto polymorfizmy nepotvrdilo [22]. Naopak jiná studie provedená v Číně ukazuje na možné vyšší riziko vzniku MSA u osob s polymorfizmem rs1799964 pro TNF-α (tumor necrosis factor-α) a až o 5 let časnějším počátkem nemoci s polymorfizmem pro IL-1β rs16944 [23]. Polymorfizmy pro gen LRRK2 (enzym „Leucine-rich repeat kinase 2“), který může způsobit PN, nebyly v čínské populaci zjištěny jako rizikové pro vznik MSA [24]. Další pozorování naznačují asociaci mutací genu pro glukocerebrosidázu, která je patognomická pro Gaucherovu nemoc, se vznikem synukleopatií vč. MSA [25]. Uvažuje se o tom, že variabilita počtu kopií segmentů DNA (copy number variants) v nekódujících oblastech může prostřednictvím modulace transkripční aktivity zvyšovat riziko vzniku MSA [26].
Z výše několika málo uvedených pozorování se lze oprávněně domnívat, že genetické faktory mohou přispívat k rozvoji MSA, ale současná úroveň poznání neumožňuje využití těchto poznatků v klinické
praxi.
Klinický obraz
Pro MSA je typická kombinace autonomní dysfunkce, parkinsonského anebo mozečkového syndromu s příznaky postižení kortikospinální dráhy. Variabilita klinického obrazu způsobila, že v minulosti byla u nemocných s dominantními mozečkovými příznaky obvykle stanovena diagnóza olivopontocerebelární degenerace, s převahou parkinsonského syndromu (PS) striatonigrální degenerace a u pacientů s výraznou autonomní dysfunkcí Shyův-Dragerův syndrom. Z hlediska histopatologické klasifikace se však jedná o stejné onemocnění. Podle iniciální manifestace rozlišujeme dva základní typy: MSA-P a MSA-C. S progresí se obvykle oba fenotypy kombinují.
Poruchy hybnosti
Parkinsonský syndrom je obecně definován poruchou iniciace pohybu (akineze), zpomalením pohybu (bradykineze) a snížením rozsahu pohybu (hypokineze), zvýšeným svalovým napětím (rigiditou), třesem a posturální instabilitou. Pro MSA jsou příznačné hypomimie, monotonní hypofonie, hypokinetická dysatrie a dysfagie. Na končetinách dominují poruchy volní motoriky, rigidita nebývá výrazná a asi třetinu pacientů postihuje klidový třes [27]. Na rozdíl od PN lateralizace postižení končetin nemusí být nápadná. Poruchy volní motoriky a rigidita jsou typicky výraznější na krku a trupu. Objevují se problémy s iniciací chůze, krok je zkrácený, u freezingu nepomáhají senzorické triky, váznou synkineze, otočky a v pokročilých stadiích se přidává i apraxie chůze. Proaktivní i reaktivní posturální instabilita je častou příčinou pádů bez varovných příznaků a bez poruchy vědomí. Pull test a tandemová chůze jsou mnohdy pozitivní do 3 let od začátku nemoci.
Dystonie různou měrou postihuje mimické, žvýkací, laryngeální, trupové i končetinové svaly vč. haluxů. Typické jsou cervikální dystonie (obvykle antecollis) a inklinace s lateralizací axiálního svalstva (syndrom šikmé věže v Pise). Rovněž výrazný předklon hlavy a trupu (kamptokormie) přispívá k instabilitě a pádům. Kamptokormie se chůzí zhoršuje, zatímco v sedu a lehu se mírní. Postura axiálního svalstva je alespoň v některých případech ovlivněna myopatickými změnami paravertebrálního svalstva [28]. Abnormní postavení a deformity rukou a nohou (striatální ruka, noha, palec) jsou vysvětlovány poškozením nucleus caudatus a putamen [29]. Na rukou vzniká mírná flexe v metakarpofalangeálních kloubech, s extenzí v interfalangeálních kloubech a někdy i s ulnární deviací a dorzální flexí prstů. Dystonie a chorea se někdy objevují po nasazení dopaminergní medikace, a to především v kraniocervikální oblasti, ale vzácně mohou postihnout i končetiny. Vzácně se u MSA objevují stimulus-senzitivní kortikální myoklonus, hemibalizmus a chorea bez souvislosti s dopaminergní léčbou [30– 32].
Mozečkový syndrom se projevuje ataktickou dysartrií, nepravidelným posturálním nebo akčním třesem, ataxií trupu a končetin. Mozečkové příznaky postihnou dvě třetiny pacientů bez ohledu na iniciální symptomy [4].
Z pyramidových příznaků se objevují hlavně hyperreflexie a pozitivní pyramidové jevy iritační na dolních končetinách. Spasticita postihuje kolem 10 % pacientů, ale jen vzácně je chůze spastická nebo spasticko-ataktická. Pyramidové příznaky se vyskytují až v 50 % případů a častěji u MSA-C.
Poměrně časté jsou různé poruchy sledovacích a sakadických pohybů, vestibulookulárního reflexu a nystagmus [33].
Sexuální dysfunkce
Časný a závažný výskyt autonomní dysfunkce patří k hlavním projevům MSA. Erektilní dysfunkce u většiny mužů předchází ostatní projevy až o několik let [34]. U žen se vykytuje snížená genitální citlivost před dobou nástupu neurologických symptomů nebo v ní [35].
Poruchy mikce
Poruchy dolních močových cest se často pojí se sexuální dysfunkcí a rovněž mohou být iniciálním projevem MSA až v 18 % případů [36]. Jedná se však o nespecifický projev, protože různé poruchy mikce mohou být přítomny i u jiných atypických PS a v pokročilých stadiích PN. V počátečních stadiích MSA se podobně jako u PN objevuje urgentní inkontinence pro hyperaktivitu detruzoru. V pokročilých fázích nemoci se přidružují i další mikční poruchy (vezikouretrální dysfunkce, denervace zevního svěrače, hypoaktivita detruzoru, dyssynergie detruzoru a svěračů). S délkou trvání nemoci narůstá postmikční reziduum a je žádoucí provést urodynamické vyšetření [37]. Nykturie narušuje kvalitu nočního spánku. Dyssynergie detruzoru a svěračů může způsobit i močovou retenci. Mezi další urologická vyšetření patří cystouretroskopie, videourodynamika, elektromyografie sfinkterů a ultrasonografie. Recidivující uroinfekty, zejména v pokročilých stadiích, mohou být příčinou urosepse s fatálními následky.
Poruchy regulace krevního tlaku a srdečního rytmu
Ortostatická hypotenze obvykle následuje rozvoj urogenitálních příznaků. V různé formě postihuje většinu pacientů [3,38,39]. Neurodegenerace a depozita αS postihují pregangliové autonomní neurony, proto při vertikalizaci nedochází k adekvátní aktivaci postgangliových sympatických vláken, uvolnění noradrenalinu a vazokonstrikci při vertikalizaci [40].
Krevní tlak měříme nejprve po 15 min klidu v horizontální poloze těsně před vertikalizací. Pro ortostatickou hypotenzi svědčí pokles systolického tlaku nejméně o 30 mm Hg nebo 15 mm Hg během 3 min po postavení. U pacientů s MSA rovněž často dochází i k poklesu srdeční frekvence a variabilitě srdečního rytmu. Pokles krevního tlaku může být způsoben nebo zhoršen hypovolemií, anemií a nežádoucími účinky farmakoterapie. K závažnému poklesu krevního tlaku může docházet i u chronicky ležících pacientů. V diagnostice ortostatické hypotenze může pomoci měření tlaku při Valsalvově manévru. Fyziologickou reakcí je vzestup krevního tlaku i tepové frekvence. Při ortostatické hypotenzi pro postižení sympatiku krevní tlak klesá [41]. Někteří pacienti nejsou schopni pro dechovou insuficienci vyvinout dostatečné zvýšení nitrohrudního tlaku.
U pacientů s MSA se častěji objevuje i postprandiální hypotenze. Ataky hypotenze mohou způsobit synkopu. Poruchy regulace krevního tlaku podmiňují i rozvoj otoků dolních končetin. U MSA se však také běžně vyskytují asymptomatické hypertenzní špičky, typicky v horizontální poloze a ve spánku. Změny srdečního rytmu mohou mít fatální následky. Ke komplexní diagnostice a před nasazením léčby je vhodné holterovské monitorování.
Gastrointestinální poruchy
Dysfagie patří u MSA k běžným projevům progrese nemoci. Podobně jako u PN je postižena iniciace polykacího aktu – formování sousta a jeho posun v dutině ústní. Zbytky stravy se mohou hromadit v epiglotických valekulách, a proto pacienti pokašlávají a mají pocit cizího tělesa v krku. Později nemohou nemocní odkašlat a mají vymizelý dávivý reflex. Na dysfagii se také podílí obleněná peristaltika trávicí trubice. Dysfagie je nezřídka diagnostikována až při manifestaci tichých aspirací bronchopneumonií, protože pacienti sami často o prodlouženém příjmu stravy aktivně nereferují. Velmi častým příznakem je obstipace, méně častá je fekální inkontinence.
Poruchy termoregulace
U většiny pacientů je sníženo pocení a v polovině případů dochází až k anhidróze [42]. Poruchy termoregulace rovněž souvisí s pregangliovým postižením autonomního systému.
Poruchy spánku a dýchání
K prvním projevům MSA již v premotorické fázi patří abnormní chování v rapid eye movement (REM) spánku podobně jako i u jiných synukleinopatií a některých dalších neurodegenerací vč. spinocerebelárních ataxií. Živé a nepříjemné sny postihují přibližně polovinu pacientů. Častá je nadměrná denní spavost způsobená špatnou kvalitou nočního spánku nejen pro poruchu chování v REM spánku, ale i pro ronchopatii (chrápání), apnoické pauzy obstrukčního i centrálního typu, noční inspirační stridor, syndrom neklidných nohou a periodické pohyby dolních končetin [43,44]. Inspirační laryngeální stridor vzniká při oslabení m. cricoarytenoideus posterior a je považován za prognosticky negativní příznak pro zvýšené riziko náhlého úmrtí [45,46]. Paréza hlasivek může vzniknout spontánně a náhle, ale i v souvislosti s instrumentálním vyšetřením nebo intubací. V průběhu dne se mohou objevit mimovolní povzdechnutí a lapavé dýchání.
Kognitivní deficit
Kognitivní deficit postihuje v různé míře až 75 % nemocných s MSA. Nejprve se projevuje ve formě izolovaných deficitů, které se mohou s progresí měnit na multidoménové a vzácně dosahovat různého stupně demence (12– 18 %). Poruchy frontálních funkcí a poruchy pozornosti postihují třetinu pacientů [47]. Různou měrou mohou být postiženy exekutivní funkce (zejména iniciace, perseverace), vizuospaciální a vizuokonstrukční funkce [48]. Kognitivní profil u MSA vykazuje podobnosti s deficity u PN a progresivní supranukleární obrny (PSP) [49]. U PSP se kognitivní postižení objevuje časněji a rychleji progreduje, u PN nastává až v pokročilých stadiích. Úzkostné poruchy a deprese mohou negativně ovlivňovat kognitivní výkon.
Poruchy chování
K nejčastějším poruchám chování u MSA patří různě závažné stupně deprese, úzkostné stavy a apatie. Méně časté příznaky jsou iritabilita, poruchy vnímání a myšlení, agitace a desinhibice [50]. Téměř vždy se časem projevuje emoční inkontinence [51].
Bolest
Bolest postihuje 47– 72 % pacientů s MSA, z toho více nemocné s MSA-P a zhruba ve 30 % je přítomna již v iniciálních stadiích [52,53]. Bolesti jsou nejčastěji referovány na končetinách a pak v cervikální a lumbosakrální oblasti. Podle původu se jedná o bolesti nociceptivní (myoskeletální, artrotické, v souvislosti s dystonií, rigiditou a dyskinezemi indukovanými dopaminergní léčbou) a centrální neuropatické (pocity pálivé, mrazivé, elektrického výboje, brnění, mravenčení, píchání, svědění, znecitlivění). Na vzniku bolesti, jakož i na jejím abnormním vnímání a prožívání se pravděpodobně podílí jak nigrostriatální degenerace, tak atrofie noradrenergního locus coeruleus, které moduluje zpracování bolestivých signálů prostřednictvím descendentního inhibičního systému. Práh pro percepci a toleranci bolesti může být negativně ovlivněn současnými depresivními a úzkostnými příznaky [53].
Ostatní poruchy
Poruchy čichu se u MSA rovněž vyskytují. Jsou však méně závažné v porovnání s PN a nemají v klinické praxi valnou diagnostickou hodnotu [54]. Popisovány jsou i recidivující záchvaty zblednutí a bolestí periferních částí těla, zejména pak prstů na rukou (sekundární Raynaudův fenomén).
Stanovení klinické diagnózy
Klinická diagnóza MSA se stanovuje pomocí revidovaných kritérií publikovaných v roce 2008 (tab. 1) [55]. I přes jejich relativně vysokou senzitivitu a specificitu potvrzují postmortální studie klinickou diagnózu přibližně jen ve dvou třetinách případů [56]. MSA-P je nejčastěji zaměňována s DLBD, PSP a PN a v případě MSA-C s hereditární ataxií (viz diferenciální diagnostiku). Také iniciální syndrom čistého autonomního selhání může konvertovat do obrazu MSA, PN a DLBD [57,58]. Diagnóza MSA je u většiny pacientů stanovena do 5 let od počátku příznaků ortostatické hypotenze [58].
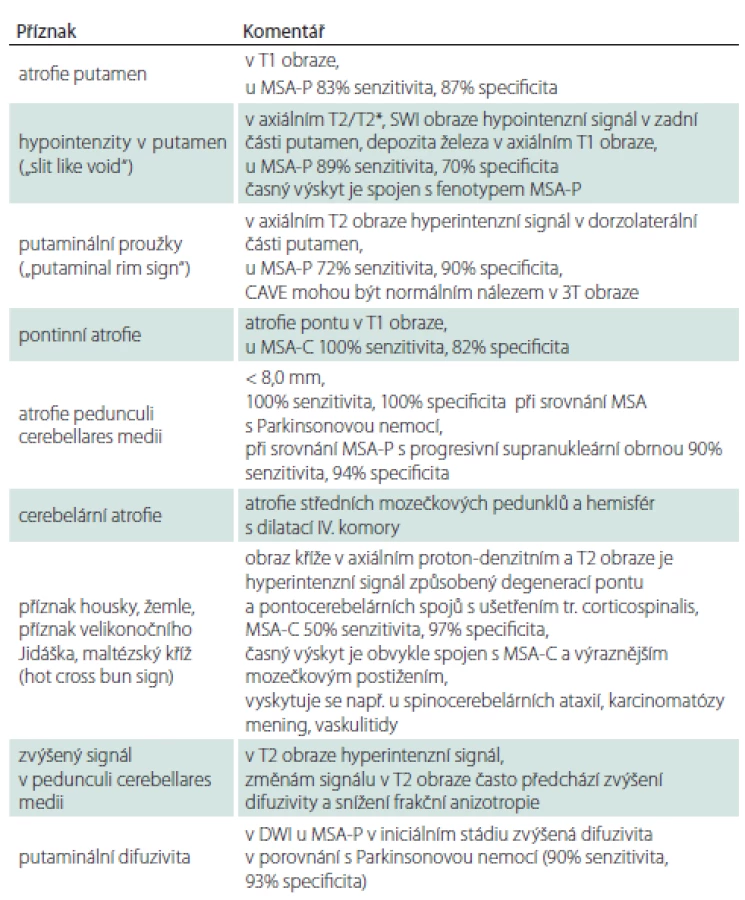
Průběh nemoci
K prvním příznakům nemoci patří různé formy autonomní dysfunkce a abnormní chování v REM spánku (obr. 1). Poruchy chůze a stability rychle progredují a většina pacientů do 3 let od začátku motorických příznaků potřebuje oporu, do 5 let jsou upoutáni na vozík a kolem 6.– 8. roku bývají imobilní. Mezi časté příčiny úmrtí patří bronchopneumonie, urosepse, sepse z dekubitů a náhlá smrt. K úmrtí často dochází v noci pro akutní parézu hlasivek nebo akutní dysfunkci kardiorespiračních center mozkového kmene. Mezi další příčiny patří sufokace sputem nebo stravou, obstrukce horních dýchacích cest při neinvazivní ventilaci s přetlakem při uzávěru dýchacích cest příklopkou hrtanovou (syndrom vlající epiglottis), kardiální autonomní dysfunkce nebo jejich kombinace [59]. U agresivních forem může dojít k úmrtí již po 3 letech. PS, inspirační stridor a časný rozvoj těžké autonomní dysfunkce jsou prognosticky nepříznivé. Mozečkové formy a pozdní počátek jsou spojovány s pomalejší progresí. U MSA s časným počátkem (před 40. rokem) byly v porovnání s pozdním nástupem pozorovány častější výskyt dystonie, výraznější zlepšení po levodopě, vyšší výskyt levodopou indukovaných dyskinéz, pyramidové příznaky a delší doba přežití [60]. Pozdní forma MSA se manifestuje po 75. roce života. Pozdní nástup autonomní dysfunkce je považován za prognosticky příznivý faktor [61]. U některých pacientů s velmi pozdním nástupem autonomní dysfunkce (po 11 letech) bylo popsáno přežití delší 15 let, neodpovídavost na levodopu a častý výskyt levodopou indukovaných dyskinéz [62,63].
Diferenciální diagnostika
V rámci diferenciální diagnostiky je MSA-P zaměňována s PN, která se však mnohdy projevuje asymetrickým PS a instabilita i autonomní dysfunkce nepatří k jejím iniciálním projevům. Klidový třes typu „počítání peněz“ diagnózu MSA spíše zpochybňuje. Akutní test s levodopou je neprůkazný. Přínos z podávání levodopy ověřujeme nejdříve po 4 týdnech léčby na vysoké stabilní dávce jejím náhlým vysazením. Pokud se hybný stav do týdne nezhorší, je diagnóza PN spolehlivě vyloučena.
V případě časné kombinace instability, pádů, autonomní dysfunkce, kognitivního deficitu a psychotické produkce je nutno myslet i na DLBD.
U většiny případů PSP je podobně jako u MSA časná instabilita, ale chybí projevy výraznější autonomní dysfunkce, dále se rozvíjí paréza vertikálního pohledu, frontální poruchy chování a rychle progredující subkortikální deficit. Relativně vzácně se PSP manifestuje po několik let jen akineticko-rigidním syndromem.
Brzký rozvoj afázie, apraxie končetin, myoklonu a asymetrického klinického nálezu korespondujícího s hemisferální atrofií svědčí pro syndrom kortikobazální ganglionické degenerace.
Anamnéza a zobrazení mozku obvykle postačí k vyloučení normotenzního hydrocefalu (instabilita, apraxie chůze, kognitivní deficit, inkontinence moči), leukodystrofie, vaskulárního a polékového PS.
Multisystémová atrofie s cerebelárním fenotypem se řadí mezi sporadické ataxie s pozdním začátkem. Podobný klinický obraz mohou mít různé autozomálně dominantní dědičné spinocerebelární ataxie (zejména SCA 2, 3, 6, 8, 17), autozomálně recesivní Friedreichova ataxie s pozdním nástupem a X-vázaný syndrom premutace fragilního chromozomu X. Hereditární spastické paraparézy s různým typem dědičnosti mohou mít i mozečkové
postižení.
Syndrom čistého autonomního selhání (pure autonomic failure; PAF), který se vyznačuje pozvolnější progresí, patří mezi relativně vzácné primární neurogenní ortostatické hypotenze. Vyznačuje se ztrátou periferních noradrenergních vláken s poruchou funkce baroreceptorů a intaktními centrálními autonomními dráhami. Klinicky se PAF manifestuje těžkou arteriální hypotenzí ve vzpřímené poloze a obvykle současnou arteriální hypertenzí vleže. Laboratorně jsou typické velmi nízká plazmatická hladina noradrenalinu a relativně pomalá a neměnná srdeční frekvence, zatímco v případě MSA mohou být postiženy pouze centrální části autonomního systému a integrita periferního sympatiku, tedy i hladiny noradrenalinu, zůstávají často neporušeny. Pro PAF jsou typické snížené pocení, erektilní dysfunkce, mikční obtíže a obstipace bez příznaků degenerace centrálního nervového systému.
Laboratorní vyšetření
Magnetická rezonance
Přestože na počátku klinické manifestace MSA může být morfologické vyšetření mozku normální, zobrazení MR 1,5T u MSA vykazuje 50% senzitivitu a 90% specificitu [64]. Změny signálu a trofické změny v putamen a v zadní jámě lební podporují diagnózu MSA (tab. 2, obr. 2), ale nevyskytují se současně u všech pacientů a mohou být přítomny i u PN a jiných PS [65,66]. Nalezené změny nemusí korelovat s iniciálním fenotypem MSA. Ve volumometrických studiích byly popsány lokální atrofické změny v oblasti ncl. caudatus, substantia nigra, oliva inferior, bulbus et tractus olfactorius, v prefrontální, motorické, suplementární motorické a inzulární kůře [64].

možná MSA – klinická diagnóza možné multisystémové atrofie podle kritérií v tab. 1; pravděpodobná MSA – klinická diagnóza pravděpodobné
multisystémové atrofie podle kritérií v tab. 1
Fig. 1. Development of symptoms in multiple system atrophy. The graph shows the period of the usual onset of selected symptoms in
multiple system atrophy.
possible MSA – clinical diagnosis of possible multiple system atrophy according to the criteria in tab. 1; probable MSA – clinical diagnosis of
probable multiple system atrophy according to the criteria in tab. 1
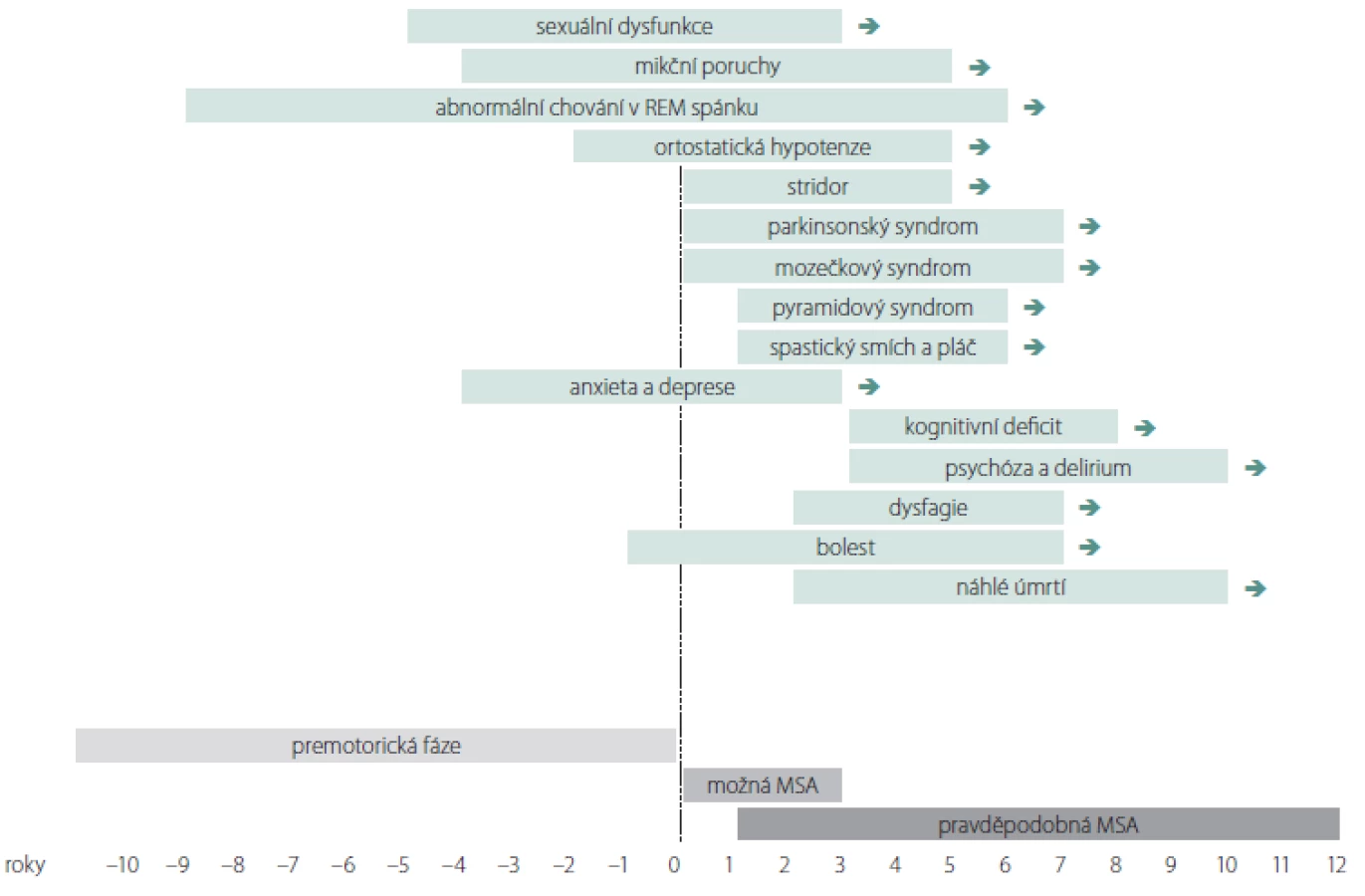
a) V T2 obraze atrofie pedunculi cerebellares medii; b) snížený signál v T2 obraze svědčící pro akumulaci železa v dorzální a laterální části
put a men bilaterálně, atrofie putamen; c) v T1 obraze atrofie pontu a cerebela, * rozšíření IV. mozkové komory; d) zvýšení signálu v T2 obraze
v pedunculi cerebellares medii bilaterálně, extrémní atrofi e pontu; e) v T2 obraze hot cross bun sign (příznak žemle).
Fig. 2. Changes in MRI supporting the diagnosis of multiple system atrophy.
a) In T2 image atrophy of pedunculi cerebellares medii; b) reduced signal in T2 image was indicative of accumulation of iron in the dorsal
and lateral parts of the putamen bilaterally and atrophy of the putamen; c) pontine and cerebellar atrophy in a T1 image, * enlargement of
the IVth ventricle; d) increase of T2 image signal in pedunculi cerebellares medii bilaterally and extreme pontine atrophy; and e) hot cross bun
sign in a T2 image.

Na MR difuzí vážených obrazech (diffusion weighted imaging; DWI) se u MSA ukazuje zvýšená difuzivita v putamen, a to již v časných stadiích na rozdíl od PN [67]. U MSA má lokálně zvýšený aparentní difuzní koeficient (apparent diffusion coefficient; ADC) v pedunculli cerebellares medii a pontu při srovnání s PSP senzitivitu 91 % a specificitu 84 % [68].
Jednofotonová emisní výpočetní tomografie
Vyšetření jednofotonovou emisní výpočetní tomografií (single photon emission computed tomography; SPECT) s ligandy vážícími se na dopaminový transportér jako [123I]beta-CIT umožňují v časném stadiu PS diagnostikovat presynaptickou poruchu ve striatu. Výrazná asymetrie signálu korespondující s druhostranným výraznějším postižením končetin podporuje diagnózu PN. SPECT vyšetření postsynaptických D2 receptorů s [123I]IBMZ může být přínosné v diferenciální diagnostice mezi PN a atypickým PS. Obě vyšetření jsou v porovnání s MR nákladná a neposkytují diagnostickou jistotu. Výsledky mohou být zkresleny lokálními atrofickými a vaskulárními změnami a rovněž medikací ovlivňující dopaminový systém.
Pozitronová emisní tomografie
Funkční vyšetření PET při použití ligandu 18-fluorodeoxyglukóza ukazuje u MSA zvýšenou aktivitu v pallidotalamických a pontocerebelárních spojeních a sníženou aktivitu v premotorické kůře, suplementární motorické a parietální asociační oblasti (senzitivita 85 %, specificita 96 %) [69]. Vyšetření PET se dosud v běžné klinické praxi neprovádí.
Transkraniální sonografie
Podle některých studií může UZ vyšetření mozku napomoci v diferenciální diagnostice PS. Zvýšená echogenicita substantia nigra je častým nálezem u nemocných s PN, zejména před 60. rokem života. Při klinicky manifestním PS a současně normoechogenní substantia nigra a nedilatované III. komoře je PN méně pravděpodobná. Rozšíření III. komory je častější u PSP. Nález normoechogenní substantia nigra a hyperechogenního ncl. lenticularis se vyskytuje u MSA-P i PSP [70– 72]. Toto levné vyšetření i přes svoji neinvazivní povahu rovněž nepatří k diagnostickým standardům.
Terapie
Léčba MSA je pouze symptomatická a v nejbližších letech nelze očekávat zavedení nové nebo dokonce kauzální léčby do rutinní praxe (tab. 3). Přibližně 40 % pacientů je responzivních na levodopu, ale efekt je většinou krátkodobý, nedostatečný a ke zlepšení stavu je zapotřebí vyšších dávek než u PN (1 000– 2 500 mg/ den ve 4– 6 dávkách). Agonisty dopaminu pro svoji malou a jen občasnou účinnost u MSA obvykle nenasazujeme a ponecháváme je pouze v případě, že u pacienta byla původně diagnostikována PN a po vysazení agonisty došlo ke zhoršení stavu. V některých případech se mohou chůze a stabilita zlepšit po amantadinu. Vzhledem k rychlosti progrese nemoci a riziku brzké ztráty dopaminergní odpovídavosti je žádoucí co nejdříve titrovat levodopu a amantadin podle efektu a tolerance. V pokročilých stadiích je vhodné ověřovat přetrvávající přínos této léčby. PS a dystonie se mohou zlepšit po anticholinergní léčbě (biperiden), která je relativně riziková pro mikční obtíže a psychické komplikace. Při aplikaci botulotoxinu pro cervikální dystonii je poměrně vysoké riziko zhoršení dysfagie. Pro oddálení ztráty soběstačnosti a prevenci komplikací (kontraktury, dekubity, sarkopenie) má dosud nedoceněný význam časně zahájená fyzioterapie a ergoterapie.


Inkontinenční pomůcky a farmakologickou léčbu mikčních obtíží zajištuje urolog. Na erektilní dysfunkci bez výskytu ortostatické hypotenze lze opatrně vyzkoušet inhibitory fosfodiesterázy. Pro léčbu ortostatické hypotenze se osvědčil pravidelný ranní příjem 4– 5 dl studené vody [73,74]. Kromě vydatné hydratace zkoušíme také zvýšený přísun soli, elastické kompresní punčochy až do třísel, jídlo v malých porcích, odpočinek a spánek se zvýšenou polohou hlavy a trupu, a pak teprve podávání sympatomimetika anebo mineralokortikoidu. Droxidopa (prekurzor noradrenalinu) ani oktreotid (analog somatostatinu, 25– 50 ug s.c. 30 min před jídlem) užívaný při postprandiální hypotenzi nejsou v ČR dostupné. Blokátor noradrenergního transportéru atomoxetin nemá v ČR pro léčbu ortostatické hypotenze schválenu úhradu [75]. Pro riziko závažné arteriální hypertenze ve spánku tyto léky podáváme nejpozději 4 h před usnutím.
Na abnormní chování v REM spánku je lékem první volby klonazepam a v zahraničí jsou doporučovány i melatonin (2 mg), gabapentin (300– 800 mg), pregabalin (75– 150 mg) a oxybát sodný (4,5– 9 g). Noční inspirační stridor a apnoické pauzy verifikované polysomnografickým vyšetřením je možno kompenzovat trvalým přetlakem v dýchacích cestách (continuous positive airway pressure; CPAP). Při těžké spánkové apnoi lze vyzkoušet dvouúrovňový přetlak v dýchacích cestách (bi-level positive airways pressure; BPAP). Parézu hlasivek a závažný stridor je možné řešit paliativní tracheostomií. Pro léčbu syndromu neklidných nohou je lékem první volby agonista dopaminu v malé dávce a v případě nedostatečného účinku oxykodon v kombinaci s naloxonem. Použít lze i jiné opioidy (např. tramadol).
Poruchy artikulace, fonace a dysfagie se mohou zlepšit při respirační terapii a cílené logopedické péči. V pokročilých stadiích je však často možná jen nonverbální komunikace. Pro únik slin (drooling) jsou používány anticholinergní preparáty a v zahraniční botulotoxin injektovaný do slinných žláz. V ČR pro tuto indikaci není schválena úhrada. Pro dysfagii jsou nutná zahušťovadla tekutin na bázi přírodní gumy, modifikovaná strava, popíjení (sipping) a později perkutánní endoskopická gastrostomie. Denní příjem energie, který je individuální (2 400– 3 000 kcal), stanovuje nutriční terapeut nebo nutricionista. Peristaltiku jícnu a žaludku je možné zlepšit prokinetiky a obstipaci laxativy nebo aplikací botulotoxinu do análního
sfinkteru.
Bolest může souviset s hypodopaminergním stavem. Analgetickou léčbu volíme podle typu bolesti, ale často je zapotřebí vyzkoušet paracetamol, metamizol, nesteroidní analgetika, tramadol nebo kodein v kombinaci s paracetamolem, koanalgetika i botulotoxin.
Anxiózní a depresivní symptomy reagují dobře na antidepresiva. Obvykle začínáme poloviční dávkou nejnižší terapeutické dávky, kterou po týdnu zvyšujeme na dvojnásobek, a pokud se nedostaví dostatečné zlepšení do 3 týdnů, dávku ještě zdvojnásobíme. V případě psychotických příznaků a deliriózních stavů jsou podobně jako u PN preferována antipsychotika s nižší afinitou k postsynaptickým D2 receptorům ve striatu. Kognitiva (inhibitory acetylcholinestrázy) jsou u MSA neúčinná.
Na zlepšení diagnostických i terapeutických standardů u MSA se zaměřují Evropská referenční síť pro vzácná neurologická onemocnění [76] a Evropská skupina pro studium multisystémové atrofie [77]. V roce 2018 byla v ČR založena pacientská organizace – Spolek pro atypické PS.
Podpořeno Progres Q27.
Autoři děkují za cenné připomínky prof. MU Dr. Janu Rothovi, CSc., MU Dr. Petru Duškovi, Ph.D. za snímky z MR, MU Dr. Pavlu Duškovi Jr. a MU Dr. Pavlu Duškovi, Ph.D. za konzultaci urologické problematiky.
Přijato k recenzi: 9. 4. 2019
Přijato do tisku: 4. 7. 2019
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Jiří Klempíř, Ph.D.
Centrum extrapyramidových onemocnění
Neurologická klinika a Centrum klinických neurověd 1. LF UK a VFN v Praze
Kateřinská 30 120 00 Praha
e-mail jiri.klempir@vfn.cz
Sources
1. Vanacore N, Bonifati V, Fabbrini G et al. Epidemiology of multiple system atrophy. ESGAP Consortium. European Study Group on Atypical Parkinsonisms. Neurol Sci 2001; 22(1): 97– 99.
2. Vanacore N. Epidemiological evidence on multiple system atrophy. J Neural Transm (Vienna) 2005; 112(12): 1605– 1612. doi: 10.1007/ s00702-005-0380-7.
3. Wenning GK, Geser F, Krismer F et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013; 12(3): 264– 274. doi: 10.1016/ S1474-4422(12)70327-7.
4. Kollensperger M, Geser F, Ndayisaba JP et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord 2010; 25(15): 2604– 2612. doi: 10.1002/ mds.23192.
5. Mensikova K, Tuckova L, Ehrmann J et al. Unusual phenotype of pathologically confirmed progressive supranuclear palsy with autonomic dysfunction and cerebellar ataxia: case report. Medicine (Baltimore) 2016; 95(46): e5237. doi: 10.1097/ MD.0000000000005
237.
6. Batla A, Stamelou M, Mensikova K et al. Markedly asymmetric presentation in multiple system atrophy. Parkinsonism Relat Disord 2013; 19(10): 901– 905. doi: 10.1016/ j.parkreldis.2013.05.004.
7. Mensikova K, Matej R, Tuckova L et al. Progressive supranuclear palsy phenotype mimicking synucleinopathies. J Neurol Sci 2013; 329(1– 2): 34– 37. doi: 10.1016/ j.jns.2013.03.008.
8. Nussbaum RL. Genetics of synucleinopathies. Cold Spring Harb Perspect Med 2018; 8(6): pii: a024109. doi: 10.1101/ cshperspect.a024109.
9. Jellinger KA. Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Mov Disord 2014; 29(14): 1720– 1741. doi: 10.1002/ mds.26052.
10. Wong YC, Krainc D. Alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 2017; 23(2): 1– 13. doi: 10.1038/ nm.4269.
11. Valera E, Masliah E. The neuropathology of multiple system atrophy and its therapeutic implications. Auton Neurosci 2018; 211: 1– 6. doi: 10.1016/ j.autneu.2017.11.002.
12. Benarroch EE. Brainstem in multiple system atrophy: clinicopathological correlations. Cell Mol Neurobiol 2003; 23(4– 5): 519– 526.
13. Wenning GK, Wagner S, Daniel S et al. Multiple system atrophy: sporadic or familial? Lancet 1993; 342(8872): 681. doi: 10.1016/ 0140-6736(93)91789-o.
14. Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med 2013; 369(3): 233– 244. doi: 10.1056/ NEJMoa1212115.
15. Monzio Compagnoni G, Kleiner G, Bordoni A et al. Mitochondrial dysfunction in fibroblasts of multiple system atrophy. Biochim Biophys Acta Mol Basis Dis 2018; 1864(12): 3588– 3597. doi: 10.1016/ j.bbadis.2018.09.018.
16. Du J, Wang T, Huang P et al. Clinical correlates of decreased plasma coenzyme Q10 levels in patients with multiple system atrophy. Parkinsonism Relat Disord 2018; 57: 58– 62. doi: 10.1016/ j.parkreldis.2018.07.017.
17. Sasaki H, Emi M, Iijima H et al. Copy number loss of (src homology 2 domain containing) – transforming protein 2 (SHC2) gene: discordant loss in monozygotic twins and frequent loss in patients with multiple system atrophy. Mol Brain 2011; 4: 24. doi: 10.1186/ 1756-6606-
4-24.
18. Ferguson MC, Garland EM, Hedges L et al. SHC2 gene copy number in multiple system atrophy (MSA). Clin Auton Res 2014; 24(1): 25– 30. doi: 10.1007/ s10286-013-0216-8.
19. Scholz SW, Houlden H, Schulte C et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 2009; 65(5): 610– 614. doi: 10.1002/ ana.21685.
20. Federoff M, Schottlaender LV, Houlden H et al. Multiple system atrophy: the application of genetics in understanding etiology. Clin Auton Res 2015; 25(1): 19– 36. doi: 10.1007/ s10286-014-0267-5.
21. Sailer A, Scholz SW, Nalls MA et al. A genome-wide association study in multiple system atrophy. Neurology 2016; 87(15): 1591– 1598. doi: 10.1212/ WNL.0000000000003221.
22. Gu X, Chen Y, Zhou Q et al. Analysis of GWAS-linked variants in multiple system atrophy. Neurobiol Aging 2018; 67: 201 e1– e4. doi: 10.1016/ j.neurobiolaging.2018.03.018.
23. Zhou X, Wang C, Chen Z et al. Association of TNF-alpha rs1799964 and IL-1beta rs16944 polymorphisms with multiple system atrophy in Chinese Han population. Int J Neurosci 2018; 128(8): 761– 764. doi: 10.1080/ 00207454.2017.1418346.
24. Yuan X, Chen Y, Cao B et al. An association analysis of the R1628P and G2385R polymorphisms of the LRRK2 gene in multiple system atrophy in a Chinese population. Parkinsonism Relat Disord 2015; 21(2): 147– 149. doi: 10.1016/ j.parkreldis.2014.11.022.
25. Sklerov M, Kang UJ, Liong C et al. Frequency of GBA variants in autopsy-proven multiple system atrophy. Mov Disord Clin Pract 2017; 4(4): 574– 581. doi: 10.1002/ mdc3.12481.
26. Hama Y, Katsu M, Takigawa I et al. Genomic copy number variation analysis in multiple system atrophy. Mol Brain 2017; 10(1): 54. doi: 10.1186/ s13041-017-0335-6.
27. Geser F, Seppi K, Stampfer-Kountchev M et al. The European Multiple System Atrophy-Study Group (EMSA-SG). J Neural Transm (Vienna) 2005; 112(12): 1677– 1686. doi: 10.1007/ s00702-005-0328-y.
28. Margraf NG, Wrede A, Deuschl G et al. Pathophysiological concepts and treatment of camptocormia. J Parkinsons Dis 2016; 6(3): 485– 501. doi: 10.3233/ JPD-160836.
29. Ashour R, Tintner R, Jankovic J. Striatal deformities of the hand and foot in Parkinson‘s disease. Lancet Neurol 2005; 4(7): 423– 431. doi: 10.1016/ S1474-4422(05)70
119-8.
30. Steiger MJ, Pires M, Scaravilli F et al. Hemiballism and chorea in a patient with parkinsonism due to a multisystem degeneration. Mov Disord 1992; 7(1): 71– 77. doi: 10.1002/ mds.870070115.
31. Salazar G, Valls-Sole J, Marti MJ et al. Postural and action myoclonus in patients with parkinsonian type multiple system atrophy. Mov Disord 2000; 15(1): 77– 83.
32. Chen R, Ashby P, Lang AE. Stimulus-sensitive myoclonus in akinetic-rigid syndromes. Brain 1992; 115 (Pt 6): 1875– 1888. doi: 10.1093/ brain/ 115.6.1875.
33. Anderson T, Luxon L, Quinn N et al. Oculomotor function in multiple system atrophy: clinical and laboratory features in 30 patients. Mov Disord 2008; 23(7): 977– 984. doi: 10.1002/ mds.21999.
34. Wenning GK, Colosimo C, Geser F et al. Multiple system atrophy. Lancet Neurol 2004; 3(2): 93– 103.
35. Oertel WH, Wachter T, Quinn NP et al. Reduced genital sensitivity in female patients with multiple system atrophy of parkinsonian type. Mov Disord 2003; 18(4): 430– 432. doi: 10.1002/ mds.10384.
36. Sakakibara R, Panicker J, Simeoni S et al. Bladder dysfunction as the initial presentation of multiple system atrophy: a prospective cohort study. Clin Auton Res 2018. [in press]. doi: 10.1007/ s10286-018-0550-y.
37. Ito T, Sakakibara R, Yasuda K et al. Incomplete emptying and urinary retention in multiple-system atrophy: when does it occur and how do we manage it? Mov Disord 2006; 21(6): 816– 823. doi: 10.1002/ mds.20815.
38. Roncevic D, Palma JA, Martinez J et al. Cerebellar and parkinsonian phenotypes in multiple system atrophy: similarities, differences and survival. J Neural Transm (Vienna) 2014; 121(5): 507– 512. doi: 10.1007/ s00702-013-1133-7.
39. Low PA, Reich SG, Jankovic J et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 2015; 14(7): 710– 719. doi: 10.1016/ S1474-4422(15)00058-7.
40. Orimo S, Suzuki M, Inaba A et al. 123I-MIBG myocardial scintigraphy for differentiating Parkinson‘s disease from other neurodegenerative parkinsonism: a systematic review and meta-analysis. Parkinsonism Relat Disord 2012; 18(5): 494– 500. doi: 10.1016/ j.parkreldis.2012.01.009.
41. Goldstein DS. Dysautonomia in Parkinson disease. Compr Physiol 2014; 4(2): 805– 826. doi: 10.1002/ cphy.c130026.
42. Iodice V, Lipp A, Ahlskog JE et al. Autopsy confirmed multiple system atrophy cases: Mayo experience and role of autonomic function tests. J Neurol Neurosurg Psychiatry 2012; 83(4): 453– 459. doi: 10.1136/ jnnp-2011-301068.
43. Moreno-Lopez C, Santamaria J, Salamero M et al. Excessive daytime sleepiness in multiple system atrophy (SLEEMSA study). Arch Neurol 2011; 68(2): 223– 230. doi: 10.1001/ archneurol.2010.359.
44. Cochen De Cock V. Sleep Abnormalities in MultipleSystem Atrophy. Curr Treat Options Neurol 2018; 20(6): 16. doi: 10.1007/ s11940-018-0503-8.
45. Ozawa T, Sekiya K, Aizawa N et al. Laryngeal stridor in multiple system atrophy: clinicopathological features and causal hypotheses. J Neurol Sci 2016; 361: 243– 249. doi: 10.1016/ j.jns.2016.01.007.
46. Ohshima Y, Nakayama H, Matsuyama N et al. Natural course and potential prognostic factors for sleep-disordered breathing in multiple system atrophy. Sleep Med 2017; 34: 13– 17. doi: 10.1016/ j.sleep.2017.01.020.
47. Stankovic I, Krismer F, Jesic A et al. Cognitive impairment in multiple system atrophy: a position statement by the Neuropsychology Task Force of the MDS Multiple System Atrophy (MODIMSA) study group. Mov Disord 2014; 29(7): 857– 867. doi: 10.1002/ mds.25
880.
48. Gerstenecker A. The neuropsychology (broadly conceived) of multiple system atrophy, progressive supranuclear palsy, and corticobasal degeneration. Arch Clin Neuropsychol 2017; 32(7): 861– 875. doi: 10.1093/ arclin/ acx093.
49. Brown RG, Lacomblez L, Landwehrmeyer BG et al. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain 2010; 133 (Pt 8): 2382– 2393. doi: 10.1093/ brain/ awq158.
50. Kao AW, Racine CA, Quitania LC et al. Cognitive and neuropsychiatric profile of the synucleinopathies: Parkinson disease, dementia with Lewy bodies, and multiple system atrophy. Alzheimer Dis Assoc Disord 2009; 23(4): 365– 370. doi: 10.1097/ WAD.0b013e3181b5065d.
51. Kollensperger M, Geser F, Seppi K et al. Red flags for multiple system atrophy. Mov Disord 2008; 23(8): 1093– 1099. doi: 10.1002/ mds.21992.
52. Tison F, Wenning GK, Volonte MA et al. Pain in multiple system atrophy. J Neurol 1996; 243(2): 153– 156.
53. Schrag A, Sheikh S, Quinn NP et al. A comparison of depression, anxiety, and health status in patients with progressive supranuclear palsy and multiple system atrophy. Mov Disord 2010; 25(8): 1077– 1081. doi: 10.1002/ mds.22794.
54. Katzenschlager R, Lees AJ. Olfaction and Parkinson‘s syndromes: its role in differential diagnosis. Curr Opin Neurol 2004; 17(4): 417– 423.
55. Gilman S, Wenning GK, Low PA et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71(9): 670– 676. doi: 10.1212/ 01.wnl.0000324625.00404.15.
56. Koga S, Aoki N, Uitti RJ et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology 2015; 85(5): 404– 412. doi: 10.1212/ WNL. 0000000000001807.
57. Singer W, Berini SE, Sandroni P et al. Pure autonomic failure: predictors of conversion to clinical CNS involvement. Neurology 2017; 88(12): 1129– 1136. doi: 10.1212/ WNL.0000000000003737.
58. Kaufmann H, Norcliffe-Kaufmann L, Palma JA et al. Natural history of pure autonomic failure: a United States prospective cohort. Ann Neurol 2017; 81(2): 287– 297. doi: 10.1002/ ana.24877.
59. Shimohata T, Aizawa N, Nakayama H et al. Mechanisms and prevention of sudden death in multiple system atrophy. Parkinsonism Relat Disord 2016; 30: 1– 6. doi: 10.1016/ j.parkreldis.2016.04.011.
60. Batla A, De Pablo-Fernandez E, Erro R et al. Young--onset multiple system atrophy: Clinical and pathological features. Mov Disord 2018; 33(7): 1099– 1107. doi: 10.1002/ mds.27450.
61. Calandra-Buonaura G, Guaraldi P, Sambati L et al. Multiple system atrophy with prolonged survival: is late onset of dysautonomia the clue? Neurol Sci 2013; 34(10): 1875– 1878. doi: 10.1007/ s10072-013-1470-1.
62. Petrovic IN, Ling H, Asi Y et al. Multiple system atrophy-parkinsonism with slow progression and prolonged survival: a diagnostic catch. Mov Disord 2012; 27(9): 1186– 1190. doi: 10.1002/ mds.25115.
63. Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med 2015; 372(3): 249– 263. doi: 10.1056/ NEJMra1311488.
64. Chelban V, Bocchetta M, Hassanein S et al. An update on advances in magnetic resonance imaging of multiple system atrophy. J Neurol 2019; 266(4): 1036– 1045. doi: 10.1007/ s00415-018-9121-3.
65. Burk K, Skalej M, Dichgans J. Pontine MRI hyperintensities („the cross sign“) are not pathognomonic for multiple system atrophy (MSA). Mov Disord 2001; 16(3): 535.
66. Brooks DJ, Seppi K, Neuroimaging Working Group on MSA. Proposed neuroimaging criteria for the diagnosis of multiple system atrophy. Mov Disord 2009; 24(7): 949– 964. doi: 10.1002/ mds.22413.
67. Bajaj S, Krismer F, Palma JA et al. Diffusion-weighted MRI distinguishes Parkinson disease from the parkinsonian variant of multiple system atrophy: a systematic review and meta-analysis. PLoS One 2017; 12(12): e0189897. doi: 10.1371/ journal.pone.0189897.
68. Paviour DC, Thornton JS, Lees AJ et al. Diffusion-weighted magnetic resonance imaging differentiates Parkinsonian variant of multiple-system atrophy from progressive supranuclear palsy. Mov Disord 2007; 22(1): 68– 74. doi: 10.1002/ mds.21204.
69. Tang CC, Poston KL, Eckert T et al. Differential diagnosis of parkinsonism: a metabolic imaging study using pattern analysis. Lancet Neurol 2010; 9(2): 149– 158. doi: 10.1016/ S1474-4422(10)70002-8.
70. Walter U, Dressler D, Probst T et al. Transcranial brain sonography findings in discriminating between parkinsonism and idiopathic Parkinson disease. Arch Neurol 2007; 64(11): 1635– 1640. doi: 10.1001/ archneur.64.11.1635.
71. Bouwmans AE, Vlaar AM, Srulijes K et al. Transcranial sonography for the discrimination of idiopathic Parkinson‘s disease from the atypical parkinsonian syndromes. Int Rev Neurobiol 2010; 90: 121– 146. doi: 10.1016/ S0074-7742(10)90009-3.
72. Hellwig S, Reinhard M, Amtage F et al. Transcranial sonography and [18F]fluorodeoxyglucose positron emission tomography for the differential diagnosis of parkinsonism: a head-to-head comparison. Eur J Neurol 2014; 21(6): 860– 866. doi: 10.1111/ ene.12394.
73. Deguchi K, Ikeda K, Sasaki I et al. Effects of daily water drinking on orthostatic and postprandial hypotension in patients with multiple system atrophy. J Neurol 2007; 254(6): 735– 740. doi: 10.1007/ s00415-006-0425-3.
74. Mathias CJ, Young TM. Water drinking in the management of orthostatic intolerance due to orthostatic hypotension, vasovagal syncope and the postural tachycardia syndrome. Eur J Neurol 2004; 11(9): 613– 619. doi: 10.1111/ j.1468-1331.2004.00840.x.
75. Palma JA. Autonomic dysfunction in Parkinson‘s disease and other synucleinopathies: Introduction to the series. Mov Disord 2018; 33(3): 347– 348. doi: 10.1002/ mds.27347.
76. European Reference Network on Rare Neurological Diseases. [online]. Available from URL: http: / / www.ern-rnd.eu/ .
77. European Multisystem Atrophy Study Group. [online]. Available from URL: http: / / www.emsa-sg.org/ .
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2019 Issue 4
Most read in this issue
- Multiple system atrophy
- Superior semicircular canal dehiscence
- Spina bifida in the Czech Republic – incidence and prenatal diagnostics
- Hearing loss after spinal anesthesia