
Je amyloid podstatný pro senilní demenci?
Is the role of amyloid in senile dementia substantial?
Extensive literature from the past 40 years accuses brain amyloid as one of the causal factors of senile dementia. Amyloid is considered, together with intracellular inclusions of τ-protein, as the originator for a specific Alzheimer‘s disease. After being dependent for many decades on amyloid detection only in autopsy specimens, the application of radioactive ligands using positron emission tomography (PET) currently achieves highly informative in vivo findings. The recognition of relations between mental acuity and the intensity of amyloid deposits has thus obtained a new dimension. We are providing a review of methods and their conclusions which shed light onto the mechanisms of brain ageing: histological properties of amyloid, techniques and the yield of its detection in PET, relation of amyloid density to that of neurofibrillary tangles, its relation to tissue metabolism and atrophy, dynamics of amyloid presence according to age and correlative studies between amyloid and mental effectivity. The given interpretation puts doubt on the causal role of amyloid in the psychic deterioration of seniors and thus also on one of the pillars of the so-called Alzheimer‘s disease.
Key words:
brain amyloid – Alzheimer‘s disease – dementia – senile atrophy – neurofibrillary tangles – hypometabolism – brain ageing – PET in dementia
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
P. Kalvach 1; K. Kupka 2; M. Vogner 1
Authors‘ workplace:
Neurologická klinika 3. LF UK a FN Královské Vinohrady, Praha
1; Ústav nukleární medicíny, 1. LF UK a VFN v Praze
2
Published in:
Cesk Slov Neurol N 2018; 81(2): 164-170
Category:
Review Article
doi:
https://doi.org/10.14735/amcsnn2018csnn.eu1
Overview
Rozsáhlá literatura posledních 40 let obviňuje mozkový amyloid jako jeden z kauzálních faktorů stařecké demence. Amyloid je tak tradován spolu s fibrilárními intracelulárními inkluzemi τ-proteinu jako původce specifické Alzheimerovy nemoci. Poté, co jsme byli dlouhá desetiletí závislí na amyloidní detekci v pitevních vzorcích, dosahuje dnes použití radioaktivních ligandů cestou pozitronové emisní tomografie (PET) vysoce instruktivních nálezů in vivo. Poznání souvislostí mezi duševním výkonem a intenzitou amyloidních depozit dostalo tedy nový rozměr. Poskytujeme přehled metod a jejich závěrů, které přinášejí nové světlo do mechanizmů stárnoucího mozku: histologické vlastnosti amyloidu, techniky a výtěžnost jeho detekce v PET, vztah denzity amyloidu k denzitě neurofibrilárních klubíček, vztah k intenzitě tkáňového metabolizmu a atrofii, dynamiku výskytu vzhledem k věku probanda a korelační studie mezi amyloidem a duševním výkonem. Podaný výklad zpochybňuje kauzální roli amyloidu v duševní deterioraci seniorů, a tím i jeden z pilířů tzv. Alzheimerovy nemoci.
Klíčová slova:
mozkový amyloid – Alzheimerova choroba – demence – senilní atrofie – neurofibrilární klubka – hypometabolizmus – stárnutí mozku – PET u demence
Úvod
Po první dekádě výzkumu průkazu mozkového amyloidu in vivo se dostává i do ČR vyšetření distribuce ligandů β-amyloidu (Aβ) značených radioaktivním fluorem pomocí pozitronové emisní tomografie (PET). Pracoviště PET v Praze, Plzni, v Brně a ve Zlíně zahájila vyšetřování flutemetamolem (Vizamyl) a florbetabenem (NeuraCeq) a shromáždila zkušenosti s prvními desítkami pacientů. Co má být cílem těchto vyšetření? Poznání amyloidních agregací ve stárnoucím mozku? Prověření potenciálu detekce mozkového amyloidu novou metodou? Etiologická diagnostika neurodegenerativních demencí?
Ke kritickému zhodnocení reálného významu této metody je vhodné se připravit rozborem podstaty tohoto nového diagnostického instrumentu a shrnutím zkušeností, které nastřádala již na tisícovkách vyšetřených renomovaná světová pracoviště.
Historie poznání mozkových amyloidních plak
Histologické studie mozku se rozvinuly v poslední čtvrtině 19. století, hlavně po Golgiho objevu argyrofilní povahy neuronů. P. Blocq a G. Marinesco popsali v roce 1892 v Paříži skvrnité nepravidelnosti v mozkovém parenchymu a nazvali je „amas ronds“. O 6 let později studoval ve Vídni podobnou patologii u dvou senilních demencí moravský rodák E. Redlich. Ten popsal své případy jako miliární sklerózu a jako první použil slova „plaky“ [1]. Tyto histologické nálezy již vysoce převyšovaly znalosti makroskopické, s nimiž popsali v roce 1892 A. Pick s H. Chiarim svůj historický případ frontální atrofie. Mikroskopická technika v začátku 20. století poutala při pitvách starých lidí pozornost k agregátům „svérázné“ bílkoviny, které dnes nazýváme neuritickými plakami. Zasloužili se o to prvními publikacemi toho druhu A. Alzheimer v Mnichově [2] a O. Fischer v Praze [3], kteří v roce 1907 poprvé upozornili na depozita fibrilárního materiálu v mozkové kůře v souvislosti s demencí. Alzheimer na jednom případu pacientky (54 let), Fischer na 12 případech ze skupiny 16 dementních osob. Fischer zavedl pro tato ložiska granulární nekrózy nový termín „drusige Wucherung“ (voštinovité bujení), jinak též jako „Sphaerotrichia multiplex cerebri“ u presbyofrenní demence. V následujících letech upřesnili Alzheimer i Fischer dále svá pozorování, když první z nich popsal spolu s G. Perusinim 4 případy a Fischer již 58 u dementních osob, porovnaných v souboru 257 případů s pitvami osob normálních, psychotických a lueticko-paralytických. O. Fischer v tomto článku o 100 stranách rozvedl stadia maturace neuritických plak do osmi stupňů [4], podobně jako o 4 roky později Bielschowski [5,6]. Zatímco pražské pojetí (A. Pick s O. Fischerem) připouštělo plaky jako projev stárnutí všeobecně, mnichovská škola Kraepelina s Alzheimerem razila jejich výskyt u presenilní demence jako samostatné onemocnění [7].
Co je amyloid?
Amyloid je protein o β struktuře, skládající se z amyloidových fibril a neurofibrilární amyloidové hmoty. Ukládá se extracelulárně ve tkáních a orgánech, kde vede k jejich strukturální a funkční destrukci. Vzniká transformací bílkovin, které jsou přítomny v plazmě buď fyziologicky nebo za určitých patologických stavů ve zvýšené koncentraci nebo v modifikované formě. Těchto bílkovin je v současné době známo 23 (např. lehké řetězce imunoglobulinů, transthyretin, sérový amyloidový protein A [SAA], apolipoprotein, β2-mikroglobulin a další). Ty se pak mohou stát za spoluúčasti dosud ne zcela jednoznačně definovaných faktorů prekurzory amyloidových fibril, a tím příčinou tvorby a ukládání amyloidu ve tkáních (amyloidózy). Amyloid není jednotná látka, liší se podle prekurzorového proteinu, ale i typem, počtem a sekvencí aminokyselin. Amyloidózy dělíme na systémové, k nimž např. patří AL (z lehkých řetězů immunoglobulinů), AA (z SAA), ATTR (z transthyretinu), Aβ2MG (z β2 mikroglobulinu) a lokalizované, např. atriální srdeční amyloidóza (z atriálního natriuretického peptidu). Mezi tyto lokalizované patří i β-amyloidóza mozku u některých neurodegenerativních onemocnění, ale i u starších osob bez klinické symptomatiky.
Jako patologickou substanci popsal amyloid poprvé roku 1842 K. Rokitansky v játrech; název pochází od R. Virchowa z roku 1854, který jej však považoval za látku škrobové povahy (latinsky amylum) [8].
Detekce amyloidu v histologických řezech se daří nejspolehlivěji pomocí Kongo červeně se zeleným dichromizmem v polarizovaném světle (Benholdova reakce). Druhou možností je fluorescence používající barviva thioflavinu S nebo T. Nověji se uplatňuje metodika imunohistochemická na principu vazby specifických protilátek. Průkaz amyloidu v orgánech žijících osob je však o mnoho složitější. Ideální marker dosud k dispozici není, jsou však popsány pozitivní zkušenosti se semispecifickým 123I-SAP u AA a AL amyloidózy, či nespecifickými 99mTc-DPDF a 99mTc-Aprotininem pro průkaz amyloidu v srdci.
V centru našeho zájmu je průkaz fibrilárního Aβ v mozku. Rozlišují se dvě hlavní izoformy: 1. Aβ40, která převažuje, je rozpustná a z těla je vylučována, a 2. Aβ42, která agreguje rychleji a je dominující formou Aβ plak. Amyloidní β-peptid je normální součástí séra i mozkomíšního moku. Jeho prekurzor je trvale přítomen v buněčné membráně, hraje fyziologickou roli v transmembránových procesech, celulární adhezi a zřejmě se účastní synaptické plasticity [9].
Plaky jsou extracelulární agregáty fibrilárního Aβ, kolem jejichž jádra jsou patrné dystrofické neurity, mikroglie a reaktivní astrocyty. Nejvíce se hromadí ve II. a III. vrstvě šedé hmoty mozkové. Rozeznáváme „méně škodlivé“ plaky difuzní a „škodlivější“ neuritické. První z nich jsou vločkovitá depozita bílkoviny bez denzního centrálního jádra; extracelulární ložiska, která nedeformují neurony ani neurity ložiskem procházející, ani v bezprostředním okolí. Hovoří se o nich také jako o „non-fibrilized Aβ deposits“, „diffuse deposits“ nebo „benign plaques“. Plaky neuritické jsou pokročilejší a vyznačují se denzním jádrem. Neurity, které jimi procházejí, jsou ztluštělé, pokroucené a histologicky se silně barví. Mají-li denzní jádro obklopené prstencem bez imunoreaktivity, jsou označovány jako „cored plaques“, mají-li pouhé denzní jádro bez lemu celulárních procesů, označují se jako „burned-out plaques“ nebo „compact plaques“ [10].
Když se nahromadila pozorování, že mikrovaskulární léze nejsou podmínkou pro extravazaci neurotoxických amyloidních oligomérů, nastal od teorie amyloidové kaskády určitý odklon. Mezi dalšími teoriemi našla důvěru také představa, že primární závadou je neschopnost mozku udržet Aβ peptidy v solubilní formě. Podle ní by degenerující neuron ztratil schopnost jakýmsi hypotetickým faktorem odvrátit agregaci [11]. V literatuře se diskutuje i o kalciové hypotéze. Agregovaná forma Aβ podle ní vytváří arteficiální kalciové kanály, kterými se do buňky dostává vápník ve vysokém množství, jež nedovede buňka pufrovat a hyne toxickou koncentrací intracelulárního kalcia.
Využití ligandů β-amyloidu
První ligand pro diagnostiku mozkového amyloidu in vivo připravil tým pracovníků Pittsburgské university pod vedením gerontopsychiatra W. E. Klunka a radiochemika Ch. A. Mathise. Těm se podařilo syntetizovat z T thioflavinu neutrální benzothiazolový derivát 2 – 4’-methylaminoprophenyl-6-hydroxybenzothiazol, zvaný Pittsburgská sloučenina B. Ta, po označení radioaktivním uhlíkem 11C (11CPIB) je vhodná pro detekci pomocí PET. V prvních klinických studiích s tímto preparátem ve švédské Uppsale v únoru 2002 Engler se spolupracovníky dokázali, že retence 11CPIB v korových partiích u nemocných s AD je přibližně 2× vyšší než u kontrolní skupiny a reflektuje nálezy známé ze sekčních studií [12]. Ligand se preferenčně vázal téměř specificky na amyloidové fibrily Aβ40 a Aβ42. Většímu rozšíření tohoto ligandu brání skutečnost, že radiofarmaka značená 11C uhlíkem s poločasem přeměny 25 min je nutno připravovat cyklotronem přímo v diagnostickém centru. První preparát značený izotopem 18F s delším poločasem rozpadu byl v USA uvolněn pro humánní použití jako preparát firmy Eli Lilly 18F-florbetapir (firemní název Amyvid). Následovala EU, kde jsou nyní registrovány preparáty 18F-florbetaben (NeuraCeq) a 18F-flutemetamol (Vizamyl). S posledními dvěma jsou od druhé poloviny roku 2015 určité, i když zatím nevelké, zkušenosti i v ČR. Zobrazení Aβ těmito ligandy velmi dobře koreluje se sekčním průkazem amyloidových plak (98% specificita, 89% senzitivita), absence zobrazení tedy s přijatelně vysokou spolehlivostí vylučuje patologii Aβ [13].
Oba preparáty se aplikují nitrožilně. Hodnocení se provádí vizuálně, s doplněním semikvantitativním, srovnávajícím aktivitu klíčových oblastí s referenční aktivitou mozečku, případně pontu. Vhodné je provést automatickou, poloautomatickou či manuální fúzi s individuálním obrazem MR pro spolehlivou separaci šedé a bílé hmoty mozkové.
Interpretace nálezů
Za normální se považuje aktivita pouze v bílé hmotě. Ta se významně neliší u osob s přítomností korových depozit Aβ a bez nich, je považována za nespecifickou. Není jednoznačně vysvětlena, rozhodně se však nejedná o vazbu na amyloid, jak bylo histochemicky prokázáno [14]. Vysvětlením nebude zřejmě ani pomalejší clearence ligandů z méně prokrvené bílé hmoty [15]. Uvažuje se o vazbě na některé látky bílkovinné povahy, např. myelin [16], o vazbě lipofilních ligandů na bílou hmotu s vyšším obsahem lipidů [17], případně o vazbě ligandů na cévy (CAA). K prohlášení nálezu za patologický stačí průkaz abnormní aktivity v jedné z klíčových korových oblastí (frontální laloky a přední cingulum, laterální temporální laloky, precuneus se zadním cingulem a temporoparietální kůra) či ve striatu (obr. 1).

Zkušenosti posledních let však vedou k určité skepsi při interpretací nálezů mozkového Aβ. Frey klade výstižně otázku: „Zobrazení amyloidu – Přínos nebo zmatek?“ [18]. Pochybnosti vznikají z toho, že „korelace amyloid vs. demence“, „amyloid vs. neurofibrilární klubka“, „amyloid vs. metabolizmus glukózy“, nebo „amyloid vs. atrofie“ nedávají obraz dostatečně konzistentní psychometrické či lokální závislosti. Studovat tyto elementární závislosti je samozřejmě mnohem spolehlivější než studovat závislost amyloid vs. Alzheimerova demence (AD), když víme, že spolehlivost stanovení této diagnózy je za života malá.
Literární nejednoznačné názory na význam stanovení patologie Aβ u seniorů reflektují i přísná indikační kritéria Americké Alzheimerovské asociace, Americké společnosti nukleární medicíny a molekulárního zobrazení („appropriate use criteria“), ale i našeho Státního úřadu pro kontrolu léčiv pro odesílání pacientů k těmto finančně náročným vyšetřením PET.
Souvislost amyloidu s mentální výkonností?
Předpokládaná neurotoxicita mozkových amyloidních agregátů je předmětem intenzivní diskuze. Ani na myším modelu [19], ani v klinicko-neuropatologických studiích [20] však nebyly přesvědčivé vazby mezi amyloidem a demencí nalezeny.
Mnozí autoři pokládají vztah demence a depozit mozkového amyloidu za přímý a kauzální. Z běžné klinické praxe však víme, že demenci vyvolává dlouhá řada příčin, které nemají s výsevem amyloidu nic společného. Posttraumatická demence je způsobena mechanizmy kontuzními s přerušením synaptických dotyků; posthemoragická mikro - nebo makrovaskulárním krvácením do zraněné tkáně a dystrofií parenchymu po narušení hematoencefalické bariéry na kapilární stěně. V postischemickou demenci vyústí mozkový iktus, který totálně nebo částečně deprivoval důležitá centra paměti, řeči či asociací o kyslíkovou dodávku. Také u demencí demyelinizačních není žádné řeči o amyloidu. Též spongiformní vakuolizace u prionových onemocnění dovede zmařit kogitační a kognitivní funkce rychleji a významněji než pár amyloidních plak, které se u nich někdy, spíše v mozečku, naleznou [21]. Celá skupina „rapidly progressive dementias“ nabízí řadu rozmanitých příčin, které potlačí mentální kapacitu bez jakýchkoli thesaurací v parenchymu (alkohol, nedostatek thyreohormonu, epilepsie, uremie, limbické imunitní encefalopatie apod.) [22].
Četné studie Aβ in vivo dovolují nyní posoudit korelaci této tkáňové zátěže se stupněm stařeckého mentálního oploštění – presbyofrenie. Stává se zřejmým, že mnoho kognitivně normálních lidí má významná depozita amyloidu v mozku. S použitím PIB-PET zjistili Rowe et al ve velké kohortě 177 osob, že 33 % kognitivně intaktních jedinců patřilo ke skupině Aβ pozitivních případů [23]. Ještě vyšší byla proporce těchto nálezů (47 %) u Jagusta et al [24]. V několika dalších studiích [25–27] se pozitivita amyloidu u mentálně výkonných lidí pohybovala mezi 15 a 30 %. Nálezy PIB-PET ve tkáni principiálně souhlasí s patologickými nálezy Aβ42 v likvoru, avšak opět nekorelují s kognitivní insuficiencí. Nejen studie s PIB, ale také s použitím Florbetapiru nalezly pozitivní denzity amyloidu ve 21 – 28 % [28], 20 % [29], či ve 14 – 23 % [30] kognitivně zachovalých osob. Tyto výsledky korespondují také se staršími neuropatologickými studiemi dle Katzmana 1988, Braaka 1997 i Priceové 1999 [31,32].
Roli Aβ zpochybňují i nálezy opačné, totiž absence amyloidu u osob diagnostikovaných s AD. Takováto nezávislost byla nalezena např. ve 32 % ze 31 osob s AD ve studii Doraiswamyho et al [33].
Nálož Aβ v mozku vzrůstá s časem a traduje se jako vyšší u populací v riziku AD. Také osoby s jeho vyšším obsahem mají mít horší prognózu. Přesto, pokusy rozdělit stárnoucí lidi dichotomickým způsobem na ty, kteří amyloid akumulují a kteří ne, selhaly. Podle Chételata intermediární případy nejsou nijak zanedbatelnou částí množiny stárnoucích osob [34]. Tato pozorování vedou k závěrům, že skupiny kognitivně normální s amyloidními depozity a bez nich nelze separovat při adici dalších markerů, rizikových faktorů a klinických projevů. Francouzští autoři tudíž rekapitulují, že úvodní markery mozkového poškození (hypometabolizmus a hipokampální atrofie) u kognitivně normálních lidí nezávisejí na β-amyloidóze [35].
Odborníci stavějící na etiologické kauzalitě amyloidu pro AD rozlišují tři stadia vývoje choroby: nejprve abnormální biomarkery Aβ, v druhém stadiu se přidávají neurodegenerativní biomarkery (snížení metabolizmu glukózy, NFT [neurofibrillary tangles], atrofie) a ve třetím se přidává kognitivní porucha. Níže uvedené studie jim však nedávají za pravdu.
Progresivní nárůst amyloidu v neokortikálních, později v alokortikálních oblastech po dosažení určité hladiny ustrne. Ve velké pitevní populační studii bylo zjevné, že do 75 let se poměr neuritických plak a postupující mentální poruchy vyvíjel v poměrné korelaci. Další stárnutí však již očekávané zahuštění amyloidu nepřinášelo. Ze 456 osob ve věku 69 – 103 let bylo 243 dementních. Zatímco v 75 letech byl v kohortě poměr amyloidu, neurofibrilárních klubíček a demence ještě v relativní korelaci, vyšší věkové kategorie se již z této korelace vymykaly. Autoři upozornili, že i vzdor dalšímu nárůstu histologických projevů AD (plak a NFT) vydrží některé velmi staré osoby bez demence [36]. Predikce demence na základě histologického nálezu amyloidu tedy ve vysokém věku selhává.
Koincidence s neurofibrilárními klubky?
Kromě amyloidu jsou druhou historickou jednotkou degenerativního zmaru (již z Fischerových a Alzheimerových popisů před 100 lety) deformované intracelulární fibrily – NFT. V polovině 80. let minulého století bylo zjištěno, že základem těchto fibril je hyperfosforylovaný τ-protein. Ten se v iniciálním stadiu shromažďuje v cytoplazmatu kolem buněčného jádra a z těchto „pretangle“ formací se postupně vyvine fibrilární struktura v neuronálním těle a z něho vycházejících dendritech. Tyto fibrilární smotky jsou argyrofilní, při barvení hematoxylinem-eozinem pak bazofilní intracelulárně, zatímco méně časté τ-fibrilární formace extracelulární jsou spíše eozinofilní [37].
Nabízí se otázka, nakolik amyloidní plaky a neurofibrilární klubka koincidují ve shodných tkáňových lokalizacích. Nové patologicko-anatomické studie nenalézají mezi těmito dvěma úkazy úzkou závislost. Intracelulární fibrilární formace τ-proteinu se objevují v mozku mnohem dříve, někdy dokonce již před 30. rokem věku a s věkem pak jejich denzita narůstá. Místem jejich časného vzniku je hlavně limbický systém. Amyloidní plaky jsou na nich nezávislé, dostavují se později a patologové je nacházejí hlavně v neokortexu. Na základě Braakových kritérií předpokládají Duyckaerts et al, že časná stadia amyloidu se vyskytují v 50 % všech osob ve věku 73 let [37]. Pro neurofibrilární klubka, rodící se již od časného dětství, se přítomnost u 50 % osob odhaduje již na stáří 48 let. V dalších letech jich přibývá a od 65 let je vzrůst jejich denzity i u non-dementních osob exponenciální. Lze proto usuzovat, že amyloid a neurofibrilární degenerace jsou dva nezávislé procesy, separované jak časově, tak lokalizací [11]. Kromě volného vztahu neurofibrilárních klubek s plakami je i vztah hyperfosforylovaných fibril k demenci značně nezávislý. U svých 39 non-dementních osob ve věku 51 – 93 let je Price se spolupracovníky nalezl ve všech případech. V těchto benigních stadiích se nacházejí hlavně v hipokampu a ento - a perirhinálních kortexech [38].
V imunocytochemické studii 2 332 neselektovaných mozků osob od 1 do 100 let nalezli Braak et al intraneurální depozita τ-proteinu v narůstající míře již od 10 let věku, zatímco vzrůstající depozita amyloidu byla shledána až od věku mezi 40 a 50 lety [39]. Vazba demence na neurofibrilární τ-inkluze je pevnější než vazba na intenzitu amyloidu [40].
Koincidence se sníženým metabolickým obratem?
Pro posouzení škodlivosti amyloidu ve tkáni je rozhodující, zda jeho depozita vyvolávají snížení tkáňového metabolického obratu – tedy mozkové lokální aktivity; zda akcelerují atrofii a zda jsou příčinou demence. Kauzalita amyloidu ve vývoji demence byla opakovaně zpochybněna [41].
Platí-li představa, že amyloid způsobuje zánik synapsí, dendritů a tím také lokálního metabolizmu, měly by se oblasti nahromaděného Aβ shodovat s oblastmi sníženého obratu glukózy (metabolic rate of glucose; MRGI) na pozitronové tomografii (PET) 18FDG. Do jisté míry tato zjištění byla učiněna. Snížený metabolizmus ruku v ruce s plakami amyloidu byl potvrzen i u mírné kognitivní poruchy - v předním cingulu a v prekuneu [42]. V téže metaanalýze bylo však zjištěno, že mnohé oblasti mozku v těchto dvou parametrech nekoincidují: v zadním cingulu, pravé inzule, nc. lentiformis i putamen byl shledán pouze zvýšený amyloid, zatímco naopak – snížený metabolizmus FDG byl nalezen pouze ve frontálním a angulárním gyru. Zóny snížené synaptické aktivity a zvýšeného Aβ se tedy nemusí překrývat (obr. 2).
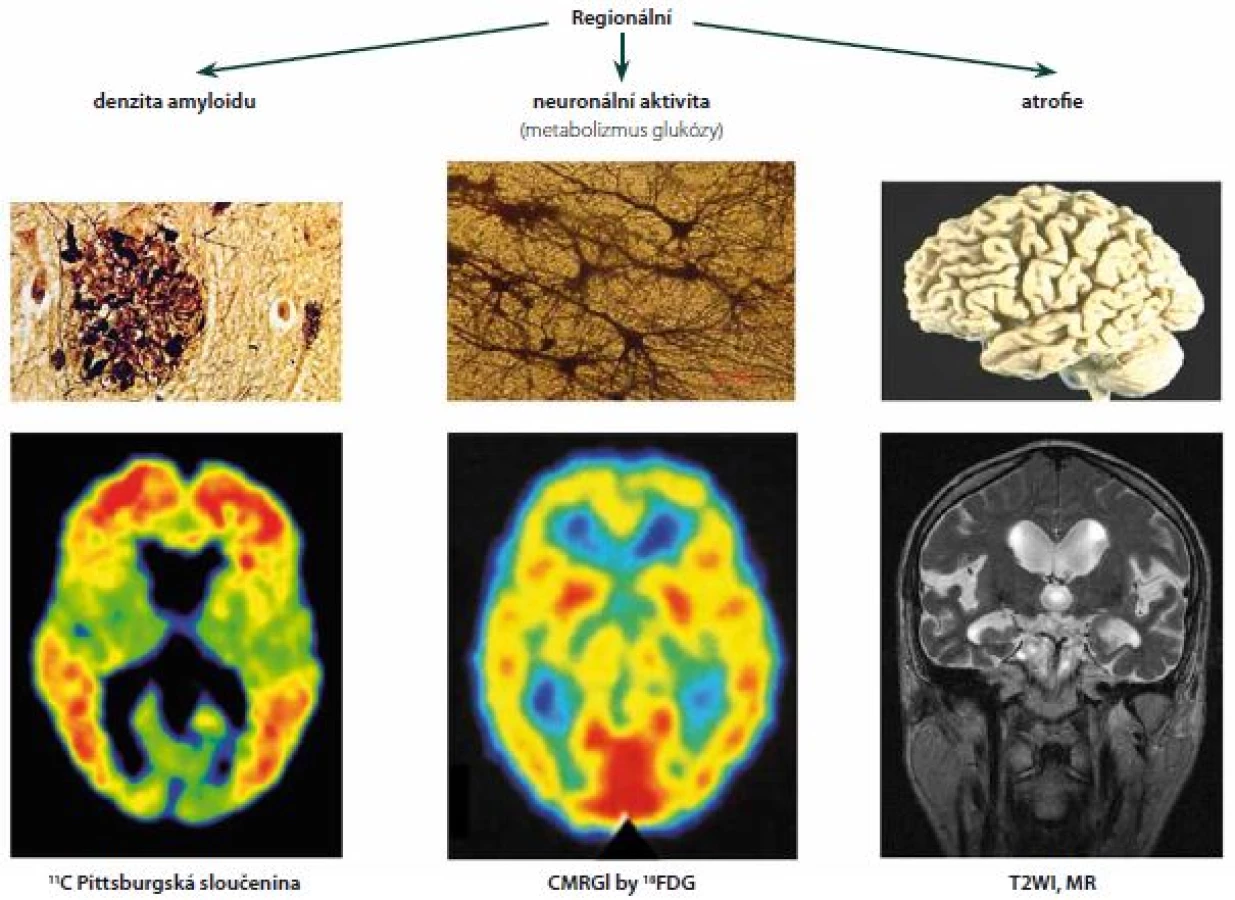
Rozsáhlá studie Mayo kliniky vyšetřila kohortu 430 kognitivně normálních lidí (věkový medián 78 let) jednak 3T MR, jednak PET s amyloidním ligandem PIB a 18FDG (dokonce téhož dne). Jejich mimořádně hodnotné výsledky dovolují srovnání amyloidu s hypometabolizmem, objemem hipokampu a navíc ještě s rozsahem leukoaraiózy. Vysoce kvalitní integrace s použitím statistických parametrických map k unifikaci tvaru a velikosti mozků vynesla velmi instruktivní výsledky – 32 % probandů mělo zvýšený kortikální amyloid nad úroveň 1,5 SUV (standardized uptake value) a z nich polovina současně i hipokampální atrofii a snížený metabolizmus FDG. Naopak, skupina s amyloidem pod 1,5 SUV obsahovala 35 % jednotlivců s hipokampální atrofií a hypometabolizmem FDG. Autoři tuto skupinu postrádající zvýšený amyloid označili za tzv. „suspected non-Alzheimer pathway“ variantu stárnutí [43].
Má se tedy brát idea o toxicitě amyloidu vážně? Mají případně Aβ oligomery jako nejranější fáze agregátů působit toxicky na buňky? Stěží. V neuropatologických studiích běžné populace se opakovaně nalézá překryv mezi tzv. AD typy a vaskulárními typy histologických senilních změn. V britské analýze 209 osob ve věku 70 – 103 let v okamžiku smrti (medián 86) bylo nalezeno při pitvě 78 % cerebrovaskulárních defektů a 70 % příznaků AD. Pouze 100 osob, tedy méně než polovina, byla „dementních“. Při členění na dementní a non-dementní se našly AD změny v poměru 64 vs. 33 % a cerebrovaskulární změny 46 vs. 33 % [20].
Non-amyloidní procesy stárnutí mozku
Zrekapitulovali jsme studie, které objasňují roli mozkového amyloidu při stárnutí CNS. Jsou pouhým fragmentem ze široké literatury, která toto téma prověřuje z nejrůznějších stran [44]. Za 110 let od popisů, jimiž průkopníci tématu mozkového amyloidu, totiž Oskar Fischer v Praze a Alois Alzheimer s Gaetanem Perusinim a Francesco Bonfigliem ve Frankfurtu a Mnichově obdivuhodně rozebrali své histologické nálezy, je vědecký svět 21. století bohatší o několik nových metod studujících senilní změny in vivo. Triumfální poznatky posledních 10 let ukazují chřadnutí mozkového parenchymu v netušených nových podobách a Alzheimerovskou trias demence na základě amyloidu a hyperfosforylovaných fibril zanořují do historického šerosvitu.
Které přesvědčivé poznatky dokládají odlišné způsoby všeobecného stárnutí mozku u široké populace? Jsou to deteriorace vaskulární a další jevy mikrostrukturální, které objevuje hlavně pokročilá metodika magneticko-rezonanční. Uvádíme jen heslovitě tyto nové kategorie:
- Ischemické inzulty tkáně.
- Kromě známých makrovaskulárních iktů jsou to tiché mozkové infarkty o velikosti lakun a menší (silent brain infarctions); vzniku takovéto léze si není v daném okamžiku daná osoba vědoma [45].
- Leukoaraióza jako důsledek plošné perfuzní, opakující se, nouze v bílé hmotě [46–48].
- Mikroinfarkty kortexu zjistitelné při použití 7 Tesla MR [49–52]. Nalézají se ve velkém počtu u běžné populace; jde o miniaturní ischemie o průměru např. 0,5 mm, které buď kolikvují za plného rozpadu neuronů i glie, nebo při ischemii parciální ztratí pouze neurony a zachovalá glie udrží konzistenci tkáně.
- Mikrokrvácení pod úrovní několika milimetrů, subjektivně nepozorovaná. Detekuje je MR sekvence na hemosiderin [53,54].
- Postupný vzrůst střední difuzivity mozku dle MR; svědčí o rozvolňujících se celulárních membránách ve tkáni [55].
- Postupný pokles anizotropie mozkových drah svědčící o rozvolňujících se membránách myelinových pochev [56].
- Poruchy hematoencefalické bariéry s extravazací albuminu; akcentují se hyperglykemií.
- Studie normotenzního hydrocefalu dodávají pohledy na harmonické a dysharmonické pulzace mozku s příznivou či narušenou expulzí likvoru z komorového systému.
- τ-proteiny a jejich narušená fosforylace jsou zjišťovány při experimentální mozkové ischemii.
- MR spektroskopie odhaluje změny tkáňových metabolitů, jako jsou snížený glutamát, aspartát, askorbát, nebo zvýšený cholin, glutamin, myo-inositol nebo N-acetylaspartylglutamát u stárnoucí populace.
Konzistence konceptu Alzheimerovy choroby
Obecná lidská zkušenost pozorující progresivní pokles mentální kapacity starých osob se shoduje s výkladem B. Reisberga. Ten sestavil v roce 1999 přehled dějů, které probíhají v analogickém recipročním trendu k vývojovým stadiím dítěte. Nazývá je retrogenezí; míní tím zrcadlový obraz k maturaci při dospívání, a to v 16 stupních tzv. „functional assessment staging“. Vzestupy kognitivní, např. podle Brief Cognitive Rating Scale, úkazy behaviorální v náboru dovedností, reciproční proti deterioraci stařecké, vývojové reflexy dítěte vs. axiální ve stáří, se kryjí se stadii myelinizace v dětství proti stadiím narušených myelinových drah ve stáří [57]. U řady „specifických“ neurodegenerativních chorob také detailní histologické rozbory nacházejí ve stoupající frekvenci překryv jejich patologických proteinů a inkluzí [58].
Po 110 letech od Alzheimerových popisů zůstává již pohled na výhradní etiologickou závislost stařecké demence na amyloidu a neurofibrilárních τ-inkluzích v neuronech entitou historickou. Je zřejmé, že stárneme několika dalšími cestami mozkové tkáňové deteriorace, které nemohly být Aloisi Alzheimerovi ani jeho vrstevníkům známy. To však neznamená, že by další výzkum mozkového amyloidu ztrácel na své přitažlivosti.
Závěr
Dostupností pozitronového sledování mozkových amyloidních depot zaživa získaly teorie o účincích mozkového amyloidu na demenci nesmírně cennou novou platformu. Usnadněné korelace mezi histologickými a behaviorálními charakteristikami jednotlivců rozptylují konzistenci dosud uznávaných nozologických jednotek. Praktické vyšetřování amyloidu pomocí PET nemůže dnes tudíž sloužit k diagnostické verifikaci předpokládaných chorob, ale osvětluje mechanizmy stárnutí v kontextu s dalšími histologickými abnormitami.
Práce byla podpořena granty Progres PQ 35 a 260388/SVV/2017.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 19. 5. 2017
Přijato do tisku: 11. 1. 2018
prof. MUDr. Pavel Kalvach, CSc., FEAN
Neurologická klinika
3. LF UK a FN Královské Vinohrady
Ruská 87
100 00 Praha 10
e-mail: pavel.kalvach@fnkv.cz
Sources
1. Goedert M. Oskar Fischer and the study of dementia. Brain 2009; 132(4): 1002–1011. doi: 10.1093/brain/awn256.
2. Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift Psychiat Psychisch-Gerichtlich Med 1907; 64 : 146–148.
3. Fischer O. Miliäre Nekrosen mit drusigen Wucherungen der Neurofibrillen, eine regelmässige Veränderung der Hirnrinde bei seniler Demenz. Mschr Psychiat Neurol 1907; 22 : 361–372. doi:10.1159/000211873.
4. Fischer O. Die presbyophrene Demenz, deren anatomische Grundlage und klinische Abgrenzung. Zeitschr Ges Neurol Psychiatr 1910; 3(1): 371–471.
5. Kalvach P, Kalvach Z. History of dementia research in Bohemia and middle Europe. Neurodegener Dis 2010; 7(1–3): 6–9. doi: 10.1159/000283474.
6. Peng F. Alzheimer‘s disease: what is it after all? Taipei: Ho-Chi Book Publishing Company 2012.
7. Braun B, Stadlober-Degwerth M, Klünemann HH. La malattia di Alzheimer-Perusini. Zum 100. Jahrestag der Publikation Gaetano Perusinis. Nervenarzt 2011; 82(3): 363–369. doi: 10.1007/s00115-010-2984-x.
8. Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol 2000; 130 : 88–98.
9. Hardy JA, Higgins GA. Alzheimer‘s disease: the amyloid cascade hypothesis. Science 1992; 256(5054): 184–185.
10. Price JL, Morris JC. Tangles and plaques in nondemented aging and „preclinical“ Alzheimer´s disease. Ann Neurol 1999; 45(3): 358–368.
11. Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer‘s disease. Acta Neuropathol Commun 2014; 2 : 135. doi: 10.1186/s40478-014-0135-5.
12. Klunk WE, Engler H, Nordberg A et al. Imaging brain amyloid in Alzheimer‘s disease with Pittsburgh Compound-B. Ann Neurol 2004; 55(3): 306–319.
13. Seibyl J, Stephens A, Barthel H et al. A negative florbetaben PET scan reliably excludes amyloid pathology – a histopathology confirmation multi-centre study. Eur J Nucl Med Mol Imaging 2014; 41 (Suppl 2): S259.
14. Fodero-Tavoletti MT, Rowe ChC, McLean CA et al. Characterization of PiB binding to white matter in Alzheimer disease and other dementias. J Nucl Med 2009; 50(2): 198–204. doi: 10.2967/jnumed.108.057984.
15. Kepe V, Moghbel MC, Långström B et al. Amyloid beta - positron emission tomography imaging probes: a critical review. J Alzheimers Dis 2013; 36(4): 613–631. doi: 10.3233/JAD-130485.
16. Stankoff B, Freeman L, Aigrot MS et al. Imaging central nervous system myelin by positron emission tomography in multiple sclerosis using [methyl-11C]-2-(4´-methylaminophenyl)-6-hydroxybenzylthiazole. Ann Neurol 2011; 69(4): 673–680. doi: 10.1002/ana.22320.
17. Glodzik L, Rusinek H, Li J et al. Reduced retention of Pittsburg compound B in white matter lesions. Eur J Nucl Med Mol Imaging 2015; 42(1): 97–102. doi: 10.1007/s00259-014-2897-1.
18. Frey KA. Amyloid imaging in dementia: contribution or confusion? J Nucl Med 2015; 56(3): 331–332. doi: 10.2967/jnumed.114.151571.
19. Westerman M, Cooper-Blacketer D, Mariash A et al. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer‘s disease. J Neurosci 2002; 22(5): 1858–1867.
20. Neuropathology Group. Medical research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre community-based population in England and Wales. Lancet 2001; 357(9251): 169–175.
21. Love S, Louis D, Ellison DW et al. Greenfield‘s Neuropathology. 8th ed. London: CRC Press. 2008.
22. Geschwind MD. Rapidly progessive dementia. Continuum (Minneap Minn) 2016; 22 (2 Dementia): 510–537. doi: 10.1212/CON.0000000000000319.
23. Rowe CC, Ellis KA, Rimajova M et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010; 31(8): 1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007.
24. Jagust WJ, Landau SM, Shaw LM et al. Relationships between biomarkers in aging and dementia. Neurology 2009; 73(15): 1193–1199. doi: 10.1212/WNL.0b013e3181bc010c.
25. Lowe WJ, Kemp BJ, Jack CR Jr et al. Comparison of 18F-FDG and PIB PET in cognitive impairment. J Nucl Med 2009; 50(6): 878–886. doi: 10.2967/jnumed.108.058529.
26. Jack CR Jr, Lowe WJ, Senjem ML et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer‘s disease and amnestic mild cognitive impairment. Brain 2008; 131(3): 665–680. doi: 10.1093/brain/awm336.
27. Mormino EC, Brandel MG, Madison CM et al. Not quite PIB-positive, not quite PIB-negative: slight PIB elevations in elderly normal control subjects are biologically relevant. Neuroimage 2012; 59(2): 1152–1160. doi: 10.1016/j.neuroimage.2011.07.098.
28. Fleischer AS, Chen K, Liu X et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol 2011; 68(11): 1404–1411. doi: 10.1001/archneurol.2011.150.
29. Rodrigue KM, Kennedy KM, Devous MD Sr et al. β-amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology 2012; 78(6): 387–395. doi: 10.1212/WNL.0b013e318245d295.
30. Sperling RA, Johnson KA, Doraiswamy PM et al. Amyloid deposition detected with florbetapir F18 ((18)F-AV-45) is related to lower episodic memory performance in clinically normal older individuals. Neurobiol Aging 2013; 34(3): 822–831. doi: 10.1016/j.neurobiolaging.2012.06.014.
31. Katzman R. Alzheimer´s disease as an age-dependent disorder. In: Research and the ageing population. CIBA Foundation Symposium 134. Chichester: John Wiley and sons 1988 : 69–85. Available from URL: http://onlinelibrary.wiley.com/doi/10.1002/9780470513583.fmatter/pdf.
32. Price DL, Walker LC, Martin LJ et al. Amyloidosis in aging and Alzheimer´s disease. Am J Pathol 1992; 141(4): 762–772.
33. Doraiswamy PM, Sperling RA, Coleman RE et al. Amyloid-β assessed by florbetapir F18 PET and 18 months cognitive decline: a multicenter study. Neurology 2012; 79(16): 1636–1644. doi: 10.1212/WNL.0b013e3182661f74.
34. Chételat G, La Joie R, Villain N et al. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer´s disease. Neuroimage Clin 2013; 2 : 356–365. doi: 10.1016/j.nicl.2013.02.006.
35. Chételat G. Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nat Rev Neurol 2013; 9(3): 123–124. doi: 10.1038/nrneurol.2013.21.
36. Savva GM, Wharton SB, Ince PG et al. Age, neuropathology and dementia. N Engl J Med 2009; 360(22): 2302–2309. doi: 10.1056/NEJMoa0806142.
37. Duyckaerts C, Dickson D. Neuropathology of Alzheimer‘s disease and its variants. In: Neurodegeneration: The molecular pathology of dementia and movement disorders. Oxford: Wiley-Blackwell 2011 : 477.
38. Price JL, Morris JC. Tangles and plaques in nondemented aging and „preclinical“ Alzheimer‘s disease. Ann Neurol 1999; 45(3): 358–368.
39. Braak H, Thal DR, Ghebremedhin E et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011; 70(11): 960–969. doi: 10.1097/NEN.0b013e318232a379.
40. Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 2009; 68(1): 1–14. doi: 10.1097/NEN.0b013e3181919a48.
41. Korczyn AD. The amyloid cascade hypothesis. Alzheimers Dement 2008; 4(3): 176–178. doi: 10.1016/j.jalz.2007.11.008.
42. He W, Liu D, Radua J et al. Meta-analytic comparison between PIB-PET and FDG-PET results in Alzheimer‘s disease and MCI. Cell Biochem Biophys 2015; 71(1): 17–26. doi: 10.1007/s12013-014-0138-7.
43. Knopman DS, Jack CR Jr, Wiste HJ et al. Brain injury biomarkers are not dependent on β-amyloid in normal elderly. Ann Neurol 2013; 73(4): 472–480. doi: 10.1002/ana.23816.
44. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer‘s disease at 25 years. EMBO Mol Med 2016; 8(6): 595–608. doi: 10.15252/emmm.201606210.
45. de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke 2002; 33(4): 1152–1162.
46. Iadecola C. The pathobiology of vascular dementia. Neuron 2013; 80(4): 844–866. doi: 10.1016/j.neuron.2013.10.008.
47. Xiong YY, Mok V. Age-related white matter changes. J Aging Res 2011; 2011 : 617927. doi: 10.4061/2011/617927.
48. Schmidt R, Petrovic K, Ropele S et al. Progression of leukoaraiosis and cognition. Stroke 2007; 38(9): 2619–2625.
49. Brundel M, de Bresser J, van Dillen JJ et al. Cerebral microinfarcts: a systematic review of neuropathological studies. J Cereb Blood Flow Metab 2012; 32(3): 425–436. doi: 10.1038/jcbfm.2011.200.
50. Van Rooden S, Goos JDC, van Opstal AM et al. Increased number of microinfarcts in Alzheimer disease at 7-T MR imaging. Radiology 2014; 270(1): 205–211. doi: 10.1148/radiol.13130743.
51. Kalaria RN, Kenny RA, Ballard CG et al. Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci 2004; 226(1–2): 75–80.
52. McAleese KE, Alafuzoff I, Charidimou A et al. Post-mortem assessment in vascular dementia: advances and aspirations. BMC Med 2016; 14(1): 129–140. doi.10.1186/s12916-016-0676-5.
53. Yates PA, Villemagne VL, Ellis KA et al. Cerebral microbleeds: a review of clinical, genetic, and neuroimaging associations. Front Neurol 2013; 4 : 205–218. doi: 10.3389/fneur.2013.00205.
54. Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited. Recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 2012; 83(2): 124–137. doi: 10.1136/jnnp-2011-301308.
55. Giorgio A, Santelli L, Tomassini V et al. Age-related changes in gray and white matter structure throughout adulthood. Neuroimage 2010; 51(3): 943–951. doi: 10.1016/j.neuroimage.2010.03.004.
56. Sexton CE, Walhovd KB, Storsve AB et al. Accelerated changes in white matter microstructure during aging: a longitudinal diffusion tensor imaging study. J Neurosci 2014; 34(46): 15425–15436. doi: 10.1523/JNEUROSCI.0203-14.2014.
57. Reisberg B, Franssen EH, Hasan SM et al. Retrogenesis: clinical, physiologic and pathologic mechanisms in brain aging, Alzheimer‘s and other dementing processes. Eur Arch Psychiatry Clin Neurosci 1999; 249 (Suppl 3):28–36.
58. Rohan Z, Matěj R, Rusina R. Překrývání neurodegenerativních demencí. Cesk Slov Neurol N 2015; 78/111(6): 641–648. doi: 10.14735/amcsnn2015641.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2018 Issue 2
Most read in this issue
- Ataxie
- Biopsie mozku v deseti bodech – co může neurolog očekávat od neurochirurga a neuropatologa?
- Fabryho choroba, přehled problematiky a nejčastější neurologické projevy
- Syndrom GLUT-1 deficience – expandující klinické spektrum léčitelného onemocnění