
Myotonická dystrofie – jednota v různosti
Myotonic Dystrophy – Unity in Diversity
Myotonic dystrophy is the most frequent muscular dystrophy of adult age characterised by muscle weakness, myotonia, cataracts, and autosomal dominant inheritance. The disease is caused by trinucleotide expansion in the DMPK gene in case of myotonic dystrophy type 1 (MD1) and tetranucleotide expansion in the CNBP/ZNF9 gene in myotonic dystrophy type 2 (MD2). The accumulation of RNA transcripts and its toxicity leads to dysregulation of many other genes, providing a clue for understanding of the broad clinical spectrum of the disease. MD1 manifests from birth (congenital) to adulthood. Severity and time of onset correlate with the number of repeats. On the contrary, MD2 is a disease with an onset during adult age only. Localisation of muscle weakness is also diff erent; the facial muscles, paraspinal, distal muscles of upper and lower limbs are aff ected in case of MD1 and the proximal muscles, esp. of lower limbs are involved in patients suff ering from MD2 – this localisation determined the former name: proximal myotonic myopathy. In contrast to MD1 that has worldwide prevalence, MD2 is predominantly restricted to middle and northern Europe. The heart conduction system (arrhythmias) is aff ected in patients with either type of the disease. In general, the impact of the disease is more severe in patients with MD1 than in MD2. Due to involvement of many systems, a multidisciplinary approach and team should be involved in the management of these patients.
Key words:
myotonic dystrophy type 1 – myotonic dystrophy type 2 – proximal myotonic myopathy
The authors declare they have no potential confl icts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
:
S. Voháňka
:
Neurologická klinika LF MU a FN Brno
:
Cesk Slov Neurol N 2017; 80/113(3): 255-265
:
Minimonography
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amcsnn2017255
Myotonická dystrofie je nejčastější svalová dystrofie dospělého věku. Jde o autozomálně dominantní onemocnění charakterizované klíčovou triádou příznaků: svalová slabost, myotonie a katarakta. Podstatou je expanze trinukleotidových sekvencí CTG v genu DMPK u myotonické dystrofie 1. typu (MD1) a expanze tetranukleotidových sekvencí v genu CNBP/ ZNF9 u myotonické dystrofie 2. typu (MD2). Akumulace RNA potom vede k ovlivnění celé řady dalších genů, což je podkladem rozsáhlého postižení mnoha systémů. MD1 se manifestuje od narození do dospělosti a tíže a začátek koreluje s počtem opakování. MD2 se rozvíjí vždy pouze v dospělosti. Důležitý je také rozdíl v distribuci svalové slabosti. V případě MD1 jde především o mimické svalstvo, šíjové svalstvo a distální svaly horních a dolních končetin. U MD2 se jedná dominantně o kořenové svaly, především DK. Tato lokalizace také dala chorobě dříve používané synonymum: proximální myotonická myopatie. Na rozdíl od MD1, která má celosvětový výskyt, se MD2 dominantně vyskytuje ve střední Evropě a Skandinávii. U obou forem je postižen i převodní systém srdeční, kognitivní a exekutivní funkce, zažívací trakt a řada dalších orgánů. Onemocnění je u MD1 zpravidla závažnější než u MD2. Vzhledem k postižení řady systému, nejen svalů, je v péči o nemocné důležitý multidisciplinární přístup.
Klíčová slova:
myotonická dystrofie typ 1 – myotonická dystrofie typ 2 – proximální myotonická myopatie
Úvod
Myotonická dystrofie (MD) je nejčastější svalová dystrofie dospělých. Je charakterizována známou triádou příznaků: svalová slabost, myotonie, katarakta. Dále je přítomna celá řada systémových příznaků a autozomální dědičnost. První popisy pocházejí od Curschmanna, Battena a Steinerta [1– 3]. Původ choroby byl objeven v roce 1992, kdy byla popsána nestabilní expanze trinukleotidů (CTG)n na dlouhém raménku 19. chromozomu [4]. Gen později dostal jméno DMPK (Dystrophic Myotonia Protein Kinase). Dva roky nato publikovali američtí autoři Thornton et al [5] a Ricker et al [6] popis pacientů s projevy MD, u kterých tuto nestabilní expanzi na 19q13.3 nenalezli. Také klinický obraz byl poněkud odlišný od dosud známého fenotypu. Hlavním rozdílem byla akcentace svalové slabosti v oblasti kořenového svalstva dolních končetin. Nazvali tedy tuto MD proximální myotonickou myopatií (PROMM). Později se začal používat termín myotonická dystrofie 2. typu (MD2) (jako kontrast k původní myotonické dystrofii 1. typu; MD1). Ranum et al [7] a Ricker et al [8] nalezli kauzální vztah nemoci k dlouhému raménku 3. chromozomu a Liquori et al [9] zjistili, že podkladem je nestabilní expanze tetranukleotidů v intronu 1 genu ZNF9 (Zinc Finger Protein 9). Tato oblast obsahuje motiv (TG)n(TCTG)n(CCTG)n, který v případě standardní DNA má velikost 104– 196 pb. Expanze spojená s MD2 se týká tetranukleotidové repetice (CCTG)n, kdy v případě onemocnění její počet vzroste na 75– 11 000. V současné době se místo ZNF9 používá název CNBP (Cellular Nucleic acid-binding Protein) nebo kombinace ZNF9/ CNBP. Jde tedy o dvě různé nemoci, které sdílejí řadu znaků a patogenetických mechanizmů, s podobnými diagnostickými přístupy a terapeutickými konsekvencemi.
Epidemiologie
Kombinovaná prevalence byla pro MD (diagnostikované klinicky) udávána 12,5/ 100 tis. obyvatel [10]. MD1 se vyskytuje celosvětově, v některých regionech je však výskyt zřetelně vyšší (Quebec, Baskicko), MD2 je naproti tomu vázána na středoevropský region a severní Evropu (Finsko) [11]. Ve Finsku byla provedena populační anonymní studie u dárců krve a prevalence pro MD2 byla 1/ 1 830 a pro MD1 1/ 2 760 [11]. Vzhledem k tomu, že se předpokládá 100% penetrance, lze kalkulovat souhrnnou prevalenci na 20 případů na 100 tis. obyvatel. V Německu je výskyt MD2 stejný jako MD1 [12], v České republice je prevalence MD2 podle údajů z registru vyšší než MD1 [13,14]. Můžeme se důvodně domnívat, že nejen v uvedených regionech je skutečný výskyt MD2 podstatně vyšší, než je udáván [10]. Jde často o lehčí postižení v pozdějším věku, což vede v řadě případů k chybné diagnóze nebo se potíže skrývají mezi jinými chorobami.
Molekulárně biologický podklad
Podkladem MD1 je mutace v genu DMPK. Gen je lokalizován na dlouhém raménku 19. chromozomu, má 14 exonů a 13 kb genomické DNA. U zdravých osob je počet opakování CTG na jedné alele v 3’nepřekládané oblasti genu DMPK mezi 5 a 37 (obr. 1). Počet 38– 50 je považován za premutaci, kdežto 51– 100 za tzv. protomutaci. Obě jsou nestabilní a mají tendenci ke zvyšování počtu opakování. Nosiči těchto pre- a protomutací jsou často asymptomatičtí nebo oligosymptomatičtí (např. pouze katarakta). Pacienti s klasickým adultním fenotypem MD1 mají zpravidla více než 100 opakování, nemocní s kongenitální formou více než 1 000 opakování. U MD1 je počet opakování úměrný tíži postižení a věku manifestace. Některé další korelace mezi genotypem a fenotypickými aspekty jsou nejasné: patrně pro nekonzistentní klinická data a různý stupeň nestability mutovaných alel v různých tkáních [15– 18]. V rodinách existuje zřetelná anticipace, tedy zhoršování klinického obrazu v dalších generacích. Příčinou je nestabilita tripletů v zárodečných buňkách. Ta je podstatně větší v mateřských alelách (děti s kongenitální formou MD1 se téměř výlučně rodí postiženým matkám), naopak nestabilita v mužských alelách může vést kromě expanze až v 6 % ke zkrácení expandovaného segmentu, dokonce až do normálních hodnot [17,19,20].
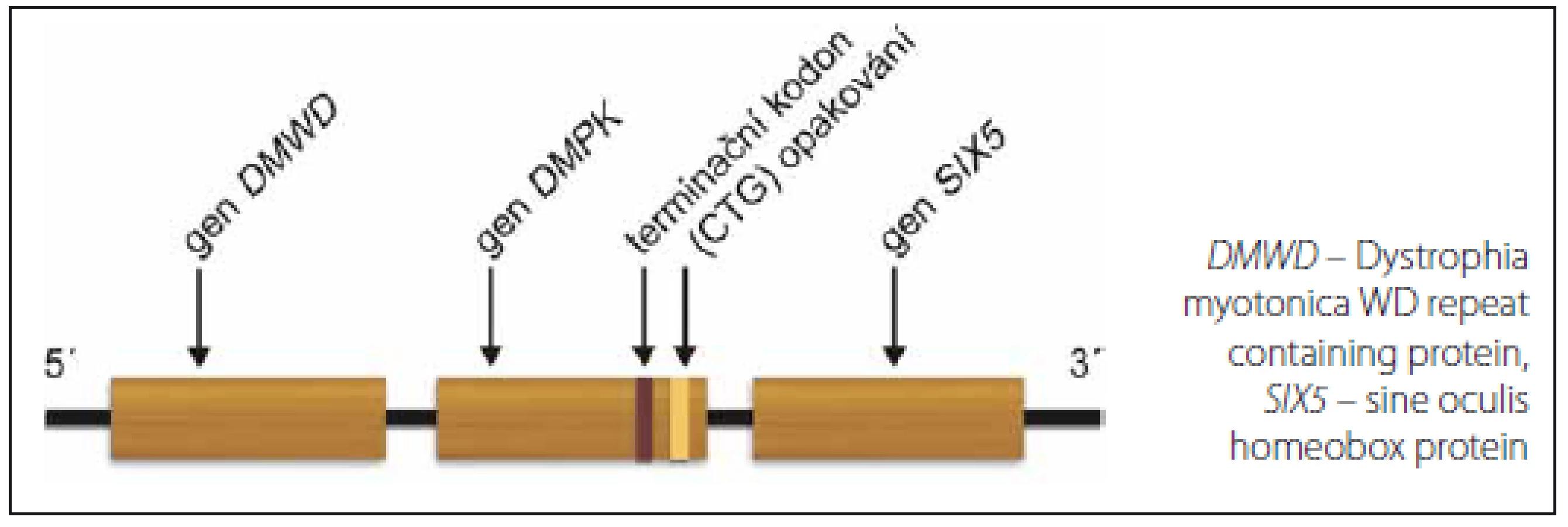
U MD2 je opakování (CCTG)n částí komplexního motivu (TG)n(TCTG)n(CCTG)n(NCTG)n(CCTG)n [9,21–23] v intronu 1 genu CNBP (dříve Zinc Finger Protein 9). Alely zdravých osob obsahují méně než 30 kopií tetrapletu (CCTG)n. Nejmenší zaznamenaná alela, která se u nositele manifestovala klinickým obrazem MD2, měla 55– 75 opakování (není ale jisté, zda v některých tkáních nebyl podstatně větší stupeň opakování díky mozaicizmu), obvykle se však nachází kolem 5 000 opakování a byla nalezena alela s 11 000 opakováními [9,21– 24]. Tak jako u MD1 můžeme najít pre- a protomutační rozsah opakování: počty mezi 23– 33 vykazují v evropské populaci významnou nestabilitu [11,23].
Trvalá somatická nestabilita je charakteristická pro oba typy MD a vytváří mezitkáňovou a mezibuněčnou variabilitu počtu opakování, tedy somatickou mozaiku. Počet opakování v leukocytech (obvyklý zdroj DNA pro diagnostické účely) a počet opakování v tkáních je rozdílný, což platí pro oba typy, u MD1 je však v postižených tkáních (svaly, mozek) výraznější [25,26]. Mezigenerační přenos je však mezi oběma typy odlišný. Zatímco u MD1 je jasná tendence k mezigenerační anticipaci, u MD2 nacházíme v mezigeneračním srovnání nejen expanze, ale i kontrakce [21,27]. Není tedy zřetelná korelace mezi rozsahem expanze a klinickým postižením či věkem začátku. Stejně tak není přítomna anticipace či kongenitální formy.
Obě MD jsou monoalelické dominantní choroby – stačí tedy k plnému rozvoji jedna mutovaná alela. Obecně platí, že homozygotní stav u expanzivních autozomálně dominantních genových mutací nemění fenotyp nemoci ve srovnání s heterozygotním stavem. U MD je velmi vzácný a byl doposud popsán jen u malého počtu pacientů [28– 30]. Byl popsán jak u pacientů s MD1, tak MD2. Klinický charakter a průběh se nelišil od nositelů heterozygotních monoalelických mutací [29,30].
Patogeneze
Předchozí kapitola nabízí otázku, proč mají tyto choroby navzdory různé lokalizaci genového defektu natolik podobné rysy: kromě svalové slabost jde o myotonii, kataraktu, systémové projevy a dominantní dědičnost. Klíčem pro pochopení původu řady klinických projevů je akumulace expandované transkribované ribonukleové kyseliny (RNA), která poškozuje buněčné procesy (schéma 1) [10,31,32]. Je v zásadě jedno, zda se jedná o triplety (CTG)n nebo tetraplety (CCTG)n, [9,10,27,33,34]. Mutované transkripty RNA (CUG, resp. CCUG) vytvářejí shluky a prostřednictvím proteinu CELF1 (CUGBP/ Elav-like family member 1) inhibují vazebné jaderné regulační proteiny MBLN1-3 (muscleblind-like protein 1-3) [35– 40]. Ty potom dalekosáhle ovlivňují sestřih RNA dalších genů. Hovoří se o „sestřihopatiích“ (spliceopathy) [41] a RNA toxicitě [10]. Bylo identifikováno více než 30 genů, jejichž sestřih je ovlivněn CELF1 a MBLN1 [10]. Dále se na patogenezi podílí poruchy v regulaci transkripce některých genů, dysregulace mikroRNA a RNA interference [10]. Exprese je změněna až u 2 000 genů, v řadě případů asi jde i o kompenzatorní mechanizmy [14]. U obou typů se tyto patogenetické vlivy v detailech liší, což vede k různé manifestaci.

Myopatie
V patogenezi svalové slabosti hraje roli patologický sestřih exonu 11 amfifyzinového genu (BIN1) [42], je více prokazatelný ve svalech nemocných MD1 než u nemocných MD2. Dále byl popsán abnormální sestřih řady sarkomerických protenů jako např. LDB3, MYOM1 a MYH14 [36,43– 45]. Na patogenezi svalové slabosti se také podílí alternativní sestřih (ztráta exonu 29) podjednotky vápníkového kanálu CAV1.1 (CACNA1S) [46].
Myotonie
Myotonie vzniká při poruchách iontových kanálu na sarkolemě, dominantně napětím řízeného chloridového kanálu. Je popsáno více než 60 mutací v genu ClCN1/ ClC1, které jsou příčinou kongenitální myotonie [47,48]. Tyto mutace vedou k redukci konduktance sarkolemy, elektrické hyperexcitabilitě a repetitivní aktivitě svalových vláken. U MD naproti tomu nejde o primární mutaci v genu ClCN1, ale o alternativní sestřih [49]. Dochází k retenci exonu 2, který obsahuje předčasný stop kodon, nebo přidání dvou nových exonů mezi exony 6 a 7 [50] a řadě dalších možností jako přeskakování exonů apod. [49].
Inzulinová rezistence
U pacientů s MD je zvýšená hladina inzulinu jako reakce beta buněk na tkáňovou inzulinovou rezistenci. Příčinou je alternativní sestřih v genu inzulinového receptoru (INSR), který kóduje tetramerický protein složený ze dvou alfa a dvou beta podjednotek. V případě MD1 i MD2 dochází k abnormální inzerci exonu 11. Tato verze se potom exprimuje ve svalové a tukové tkáni a v játrech s vlivem na metabolizmus glukózy [51].
Katarakta
Expanze tripletů CTG u MD1 redukuje transkripci sousedního genu SIX5 [52]. Jestliže je katarakta u MD1 způsobena haploinsuficiencí SIX5, u MD2 je příčinou „vzdálená“ toxicita CCTG transkriptů [53,54].
Klinické obrazy
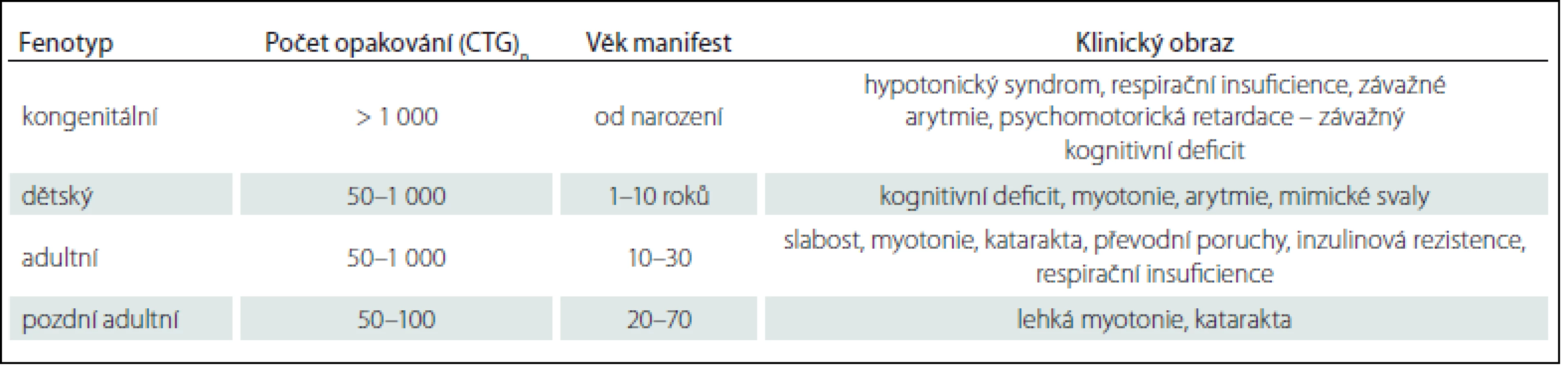
MD1 se manifestuje čtyřmi formami: kongenitální forma, dětská forma, adultní forma a pozdní adultní/ oligosymptomatická forma. Formu a věk manifestace určuje počet opakování (tab. 1) [31]. Naproti tomu má MD2 pouze adultní formu s různým věkem začátku. Vrozená manifestace nebo rozvoj v dětství dosud nebyl popsán. Rozdíl mezi oběma adultními formami zobrazuje tab. 2 [10].

Adultní forma MD1
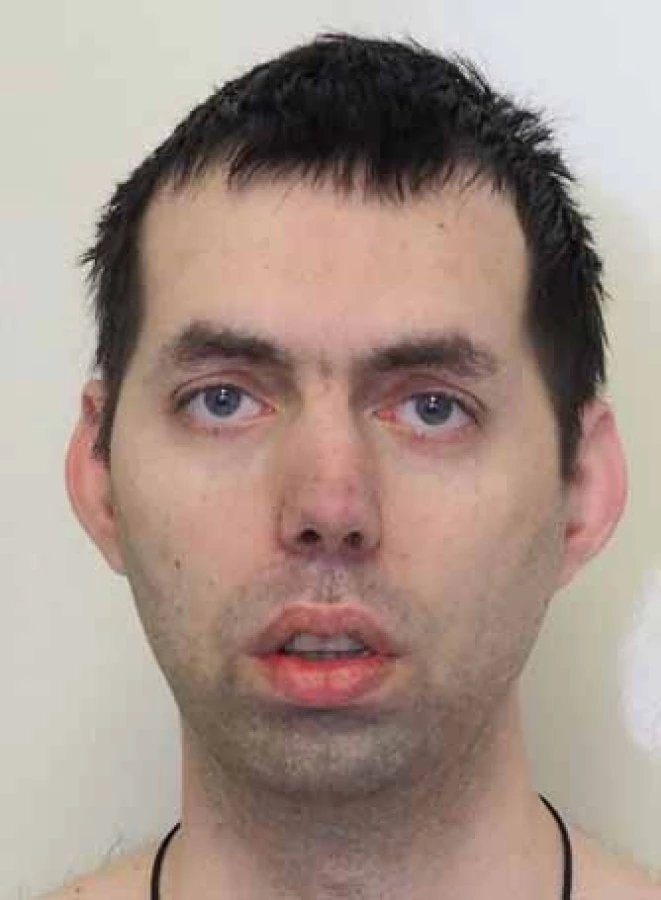
Pacienti zpravidla přicházejí s některým ze základní trojice příznaků: myotonie, svalová slabost nebo katarakta. Myotonii, kterou definujeme jako poruchu relaxace kosterního svalstva po volní kontrakci, často udávají jako tuhost nebo ztuhlost. Je přítomna už od školního věku a bývá zřetelná jak klinicky, tak elektrofyziologicky. Zjevně se zmírňuje opakovanými pohyby (warm-up fenomén). Často dlouho předchází svalovou slabost a svalová síla může být v časné dospělosti zcela normální. Katarakta, zvláště pokud se objeví až v pozdějším věku a svalové příznaky jsou malé, tak často nevyvolá podezření na MD. Svalová slabost a atrofie postihují především mimické, šíjové svalstvo a distální svaly končetin. Kombinace atrofie temporálních svalů, katarakty a frontální plešatosti vytváří charakteristický obraz (obr. 2). V pozdějších stadiích dochází k rozvoji imobility, respirační insuficienci (slabost bránice), dysartrii a dysfagii [55]. Postižení myokardu se manifestuje kardiálními kondukčními defekty vedoucími k tachyarytmiím, synkopám a riziku náhle srdeční smrti [56]. Podkladem je fibróza kondukčního systému a sinoatriálního uzlu. Dilatační kardiomyopatie není obvyklá, byť nálezy subklinických změn jsou časté [57]. Zadní subkapsulární katarakta je téměř patognomická a její výskyt před 50. rokem věku by měl lékaře vždy vést k úvaze o MD [58]. Výskyt stoupá s délkou trvání nemoci a celoživotní prevalence se blíží 100 %. Dále můžeme z oftalmologického hlediska najít retinální degeneraci a nízký intraokulární tlak [59]. Postižení CNS má strukturální i funkční charakter. Typickými projevy jsou vedle lehkého kognitivního deficitu (20– 80 % podle různých studií, korelují s počtem opakování [60]) i vyhýbavé osobní rysy (avoidance personality) [60], excesivní denní spavost a apatie. Excesivní denní spavost, která postihuje 70– 80 % pacientů [61– 63], není obvykle spojena se známkami obstrukční spánkové apnoe [64,65] či respirační insuficiencí. Obě zmíněné situace vedou k fragmentaci spánku a musíme je vždy vyloučit polysomnografickým a spirometrickým vyšetřením. Příčinou je ztráta serotoninových 5-HT neuronů v oblasti dorzální raphe a kmenových jader. Tyto změny vedou k řadě socioekonomických problémů a snižují zřetelně kvalitu života pacientů [66]. MR vyšetření prokazuje difuzní změny bílé hmoty, které jsou zřetelnější než atrofie [67]. Mezi časté potíže patří také postižení gastrointestinálního traktu [68– 70]. Dysfagie (faryngální nebo ezofageální) vede kromě váhového úbytku k riziku aspirační pneumonie, pacienty také sužuje obstipace nebo naopak průjmy. Příčinou je postižení hladkých svalů i Meissnerova a Auerbachova plexu. Různá míra inkontinence stolice postihuje až 56 % nemocných [71]. Časté jsou potíže s močením: určitá forma inkontinence postihuje až 42 % nemocných [71]. Mezi endokrinními dysfunkcemi je nejčastější inzulinová rezistence (manifestní diabetes ale není častý), hypotyreóza, která může zhoršovat příznaky MD, a poruchy reprodukčních funkcí: mužská infertilita a zvýšená frekvence spontánních potratů. Frontální plešatost, která byla již uvedena, jako prominentní příznak, není jedinou kožní změnou. Nacházíme častý výskyt papilomatrikomů, často i mnohočetných [72,73] (pozn.: papilomatrikom = kalcifikovaný epiteliom = nádor z vlasového folikulu). Je zvýšené riziko dalších nádorových onemocnění, především seminomu [74– 76], endometriálního karcinomu, nádorů mozku a non-hodgkinského lymfomu.
Kongenitální forma MD1
Tato nejzávažnější forma se projevuje již prenatálně sníženou pohyblivostí plodu, vzniká polyhydramnion a řada deformit detekovatelných ultrazvukovým vyšetřením [77]. Při narození jsou děti hypotonické s tendencí k respirační insuficienci a potížemi s přijímáním potravy. Jde o vývojovou centrální poruchu, ne o nervosvalové postižení. Je zřetelná mentální retardace a děti se opožďují v dosahování vývojových milníků. Myotonie se vyvíjí později a v 2. dekádě se objevuje svalová slabost.
MD1 se začátkem v dětství
Dlouho opomíjená forma, protože není přítomna svalová slabost, atrofie ani myotonie. Většinou se vyskytují školní problémy a děti jsou vyšetřovány pro podezření na mentální retardaci. Svalové příznaky se vyvíjejí až v dospělosti a klíčem ke správné diagnóze v dětství je diagnóza MD jednoho z rodičů (zpravidla matky) [78– 80].
Pozdní oligosymptomatická forma MD1
Nemocní mají pouze kataraktu a lehké svalové obtíže v pozdní dospělosti. Jde o osoby, u jejichž potomků se zpravidla ve třetí generaci objevuje díky anticipaci klasická forma MD1 [10].
MD2
Nejčastější manifestací je slabost pánevního pletence, která se rozvíjí od 4. dekády (nebo později) [66,81]. Dala této variantě i původní název (proximální myotonická myopatie). Pacienti přicházejí k lékaři s potížemi při chůzi do schodů, při nastupování do dopravních prostředků nebo při zvedání se z nízké židle či toalety. Slabost je často mírného stupně. Vzhledem k rozvoji v dospělosti je mnohdy zvažována některá ze získaných myopatií. Ve fázi rozvoje svalové slabosti nemusí být klinicky přítomna myotonie (ani elektrofyziologicky) a většinou nenalezneme ani kataraktu. Na druhou stranu je poměrně častá situace, kdy je pacient diagnostikován na základě náhodného nálezu při EMG vyšetření z jiné indikace a teprve při cíleném dotazování zjistíme kořenovou slabost. Myotonie většinou nebývá v popředí a je méně výrazná než u typu 1. Je-li výrazná a dominující, může jít i o koincidenci s primární mutací v chloridovém kanálu (kongenitální myotonie), jak bylo opakovaně popsáno [82], vč. tuzemského písemnictví [83].
Uvádí se, že choroba má lehčí průběh než MD1, délka života prakticky není zkrácena a vliv na kvalitu života je podstatně menší. Také je menší procento závažných kardiálních postižení a nemocní nemají ani v pozdních fázích respirační či polykací potíže [14]. Na rozdíl od typu 1 bývají přítomny hypertrofie lýtek [14]. Pacienti si stěžují na chronickou únavu, mají poruchy spánku a bdění, které jsou nejčastěji přičítány zhoršenému usínání při bolestech, ale není charakteristická excesivní denní spavost [14]. Uvádí se, že kognitivní poruchy jsou mírnější než u MD1, není přítomna mentální retardace. Vliv na kvalitu života je však významný a v jedné nizozemské studii (porovnání srovnatelných kohort dospělých pacientů, MD1 vs. MD2 a věk, trvání nemoci atd.) byl vliv kognitivní poruchy na kvalitu života prakticky srovnatelný [84]. Při podrobnějším vyšetření nalézáme zhoršení pozornosti a exekutivních funkcí [85], pokles vizuospaciálních a konstrukčních schopností, vyhýbavou osobnost [60], únavu a depresivní syndrom. To také vede často k horšímu společenskému a pracovnímu uplatnění, než bychom očekávali podle stupně pohybového deficitu. Častým příznakem je nadměrné pocení – hyperhidróza (u nemocných s MD1 se vyskytuje v podstatně menší míře) [86].
Bolest
U MD, zvláště u MD2, je bolest velmi dobře známým příznakem. Výskyt se udává od 50 do 75 % a je četnější než jakákoli chronická bolest v populaci a vyskytuje se pravděpodobně častěji než bolest u jiných nezánětlivých svalových chorob [87– 90].
Traduje se, že bolest je častější problém u nemocných s MD2 než MD1. Proto je více studií zaměřeno na zkoumání bolesti u pacientů s MD2. Při cílené exploraci a porovnání se však ukázalo, že se frekvence, intenzita a charakter bolesti u obou forem MD v zásadě neliší [91]. Fakt, že je bolest častěji popisována u pacientů s MD2, se někdy vysvětluje rozdílným vnímáním nemoci u obou skupin. U pacientů trpících MD1 jsou bolesti zastíněny závažnějším svalovým deficitem a celkově těžším průběhem. Bolest může být také iniciálním a dlouho jediným symptomem projevů MD [92,93].
Bolest má dlouhodobý charakter [89] a často kolísavý průběh [88]. Nepříliš hojně také udávají nemocní stabilní bolesti. Lokalizována bývá do oblastí, které jsou při svalových chorobách nejvíce postiženy. Nejčastěji jde o dolní oblast zad, stehna, lýtka, paže, ramena, krk [89,94]. Běžný je výskyt bolestí na více místech [95]. Nemocní popisují bolesti jako trhavé, bodavé, tíživé, křečovité, vyčerpávající či pobolívání, popř. má bolest charakter nepříjemných pocitů ve svalech, jako jsou napětí, pocit tíhy, citlivost, pálení či brnění [88]. Častá je zkušenost s více typy bolesti. Charakter bolestí by mohl poukazovat na neuropatický původ [91], ale přesvědčivé důkazy zatím chybí. Němečtí autoři [96] nedávno popsali potlačení nebo naopak zvýšenou expresi některých genů v myocytech u pacientů s MD2 a bolestmi ve srovnání s pacienty bez bolestí.
Postižení srdce
Kardiální patologie je významnou příčinou mortality u obou typů MD [97– 100]. Poruchy převodního systému (blokády, arytmie) jsou u MD1 běžné a vyžadují pravidelné sledování. Při vyšetření (EKG, Holterovo monitorování) je nacházíme až v 90 %, klinicky významné jsou asi v polovině případů [101,102] a až v 30 % jsou příčinou úmrtí [55]. Významná kardiomyopatie je vzácná [103], nepředstavuje více než 6 % případů. Meola et al však nalezli určitý stupeň hypertrofie levé komory u 24 % vyšetřovaných [102]. U MD2 je frekvence a tíže kardiálních abnormalit menší (asi 20 %). Vzhledem k vyššímu věku manifestace vzniká překryv s ischemickým postižením myokardu. O tom, zda rozsah CTG expanze koreluje se stupněm kardiálního postižení, panují kontroverzní názory. Velikost CTG fragmentu je podle některých starších studií nejsilnějším prediktorem kardiálního postižení [104– 106] a pozitivně koreluje s incidencí ventrikulárních tachyarytmií [104]. Některé novější práce ale tuto korelaci nepotvrzují [57]. V poslední době byla prokázána asociace obou typů MD a Brugada syndromu [100,107]. Podkladem je alternativní sestřih exonu 6 napětím řízeného natriového kanálu SCN5A [107,108].
Riziko rakoviny
Nemocní s oběma typy MD mají obecně zvýšené riziko nádorového onemocnění [109]. Kromě již zmíněného papilomatrikomu [110] jde podle americké studie zvláště o choroidální melanom a karcinom štítné žlázy, menší riziko je potom pro karcinom varlete a prostaty [111]. Jiné práce zdůrazňují u MD1, kromě výše uvedeného i zvýšený výskyt endometriálního karcinomu, nádorů mozku a non-hodgkinského lymfomu [74– 76].
Klinická diagnostika
Laboratorní vyšetření
Kreatinkináza (CK) je obvykle lehce zvýšena, maximum je 10násobek, asi u 1/ 5 může být i normální [112,113]. Často se také nalézá IgG, event. IgM hypogamaglobulinemie a u MD2 elevace jaterních testů (zvl. GGT) neznámé etiopatogeneze [112]. Nemocní MD2 mají zvýšenou řadu autoimunitních markerů a mají také častěji různá autoimunitní onemocnění [114]. U MD1 tato asociace chybí.
Elektromyografie
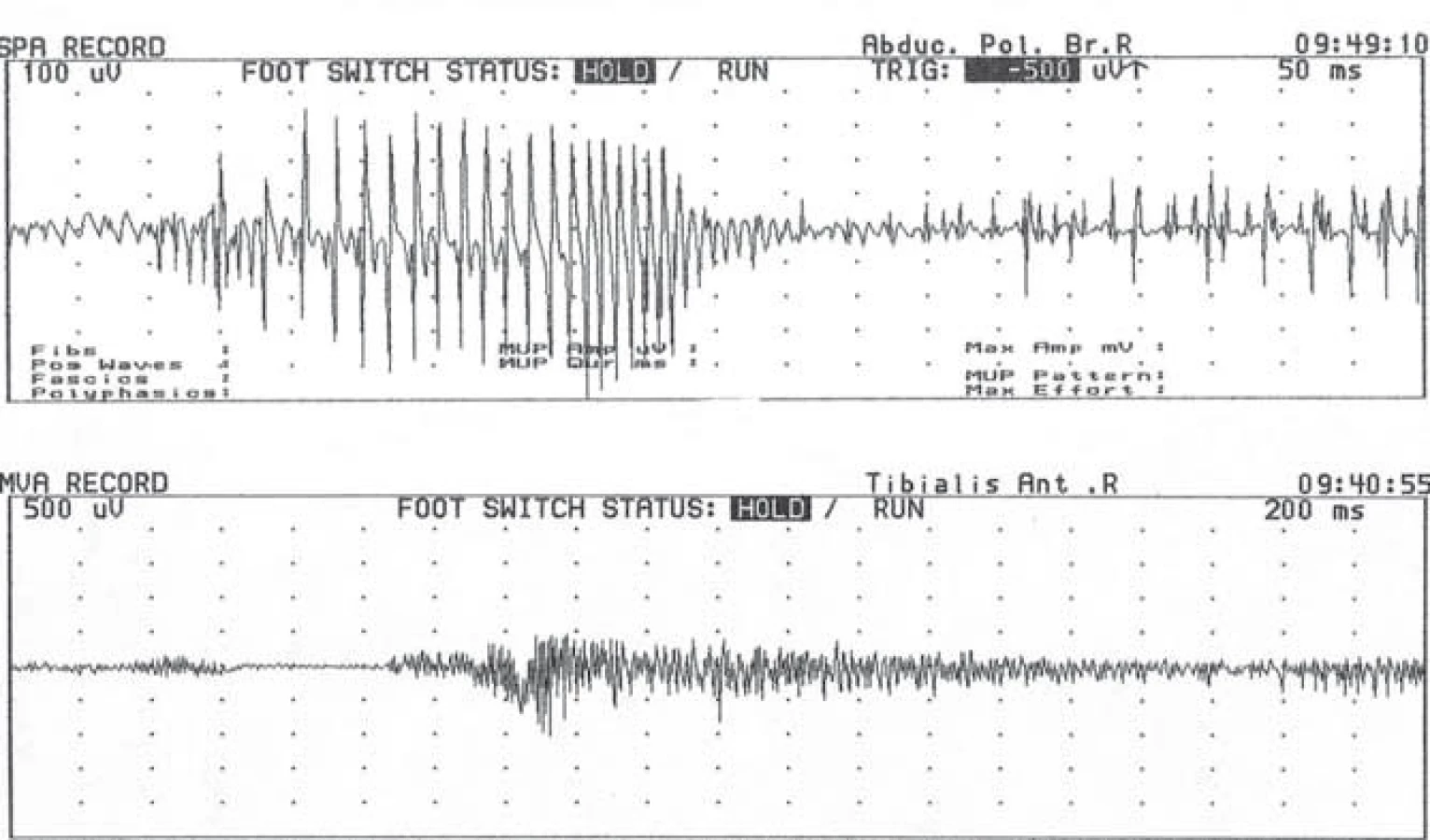
Kombinace myotonických výbojů a myopatické přestavby potenciálů motorických jednotek je pro tyto choroby charakteristická. Myotonické výboje jsou prakticky patognomické pro svalové kanálopatie a MD – představují tedy velmi cenný diagnostický nástroj (obr. 3). Nicméně u mladších pacientů s MD1 nebo u nemocných s MD2 v časnější fázi nemusí být myopatický vzorec přítomen a u MD2 lze někdy zachytit myotonické výboje až při velmi extenzívním jehlovém vyšetření [14,112].
Histologické vyšetření
Histopatologický nález ve svalech pacientů s MD1 je více vyjádřen v distálních svalech než v proximálních. Dominuje atrofie vláken typu I a obecné myopatické změny. V případě MD2 se naopak jedná o charakteristickou atrofii subpopulace vláken 2A. Další myopatické znaky jako centrální jádra jsou také dominantně vyjádřeny ve vláknech II. typu [115]. Atrofická vlákna lze nalézt v proximálních svalech ještě před klinickou manifestací [116]. V tkáních pacientů s MD1 i MD2 se dá také u obou forem pozorovat pomocí metody FISH inkluze tvořené expandovanou RNA a MBNL proteiny [117]. V současné době již histologické vyšetření nepatří do základního diagnostického schématu MD. Prokáže pouze myopatii, ale etiologii ve smyslu MD1 či MD2 určit nedokáže.
Vyšetření DNA
Jde o klíčovou metodu k potvrzení choroby. U MD1 můžeme díky rodokmenu a poměrně charakteristickému fenotypu diagnostikovat chorobu s velkou pravděpodobností již z klinického obrazu. U MD2 však jde často o nespecifickou kořenovou slabost bez dalších rozlišovacích znaků. Vzhledem k relativně pozdnímu výskytu jsou často informace z rodokmenu zavádějící. Rodiče zemřeli ještě před manifestací choroby nebo byly potíže překryty jinými chorobami dospělého věku.
U MD je metodou volby triplet-repeat primed PCR (TP PCR), která spolehlivě prokáže heterozygotní expanzi. Metoda však není schopna prokázat exaktně rozsah počtu opakování nad určitý práh. K tomu se používá Southernova analýza [118]. TP PCR má asi 3– 5 % falešně negativních výsledků. K jejich odhalení lze použít modifikaci TP PCR s oboustranným (3a 5’) značením [119]. Pro diagnostiku MD2 se dominantně používá metoda repeat-primed PCR (RP PCR). Sallinen et al [24] ve své práci provedli srovnání s jinými diagnostickými přístupy a ve všech případech potvrdili shodu výsledků použitých technik jak v případě pacientů s MD2, tak v případě kontrolních vzorků. Senzitivita je 99 % [31].
Zobrazovací vyšetření
U obou typů nalézáme náhradu svalové tkáně tukem. V případě MD1 jsou výrazné změny v m. triceps surae (především gastrocnemius medialis a soleus), méně je postižen přední kompartment (m. tibialis anterior) a zřetelně bývá ušetřen m. tibialis posterior. V oblasti stehna je postižen více čtyřhlavý sval (s ušetřením m. rectus femoris) než zadní skupina [120,121].
U MD2 dominují změny v m. quadriceps, často s relativně ušetřeným m. rectus femoris a m. gracilis, na rozdíl od MD1 bývá mnohdy nález v oblasti lýtek prakticky normální [120]. Dále nacházíme postižení m. semitendinosus a m. semimembranosus, trupových svalů (m. rectus a obliquus abdominis) a především svalů paravertebrálních a m. gluteus maximus [121].
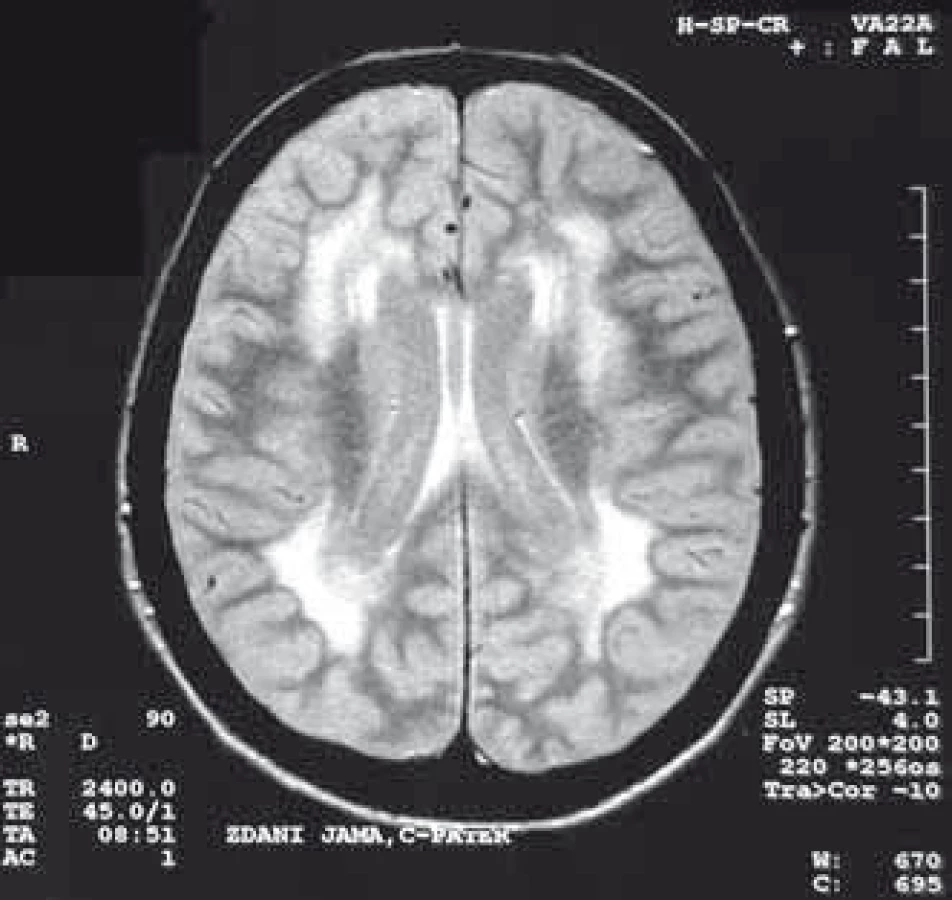
U pacientů s MD nalézáme také významné změny při MR vyšetření mozku, kde dominuje především postižení bílé hmoty (obr. 4), ale pokročilými metodami lze nalézt i postižení šedé hmoty. Metoda VBM (Voxel-Based Morphometry) prokázala postižení některých kortikálních regionů, thalamu a hipokampu [121] a modalita DTI (Diffusion Tensor Imaging) zobrazila u některých pacientů atrofii corpus callosum [121]. Vyšetření pomocí PET MR prokázalo hypoperfuzi částí frontálních laloků a parietookcipitálních oblastí [60]. Změny v mozku u MD2 jsou podobné jako u MD1, jen méně vyjádřeny, zvláště v šedé hmotě.
Prognóza onemocnění
Před koncem druhého tisíciletí byla délka života nemocných s MD1 významně zkrácena, v současné době se situace zlepšuje, ale je stále signifikantně kratší než u zdravé populace, hlavní příčinou je arytmie, respirační insuficience a pneumonie [122]. Asi polovina nemocných je před smrtí upoutána na invalidní vozík. Délka života u MD2 není obecně zkrácena, v riziku je jen malá skupina nemocných se závažnou arytmií [120].
Klinický management a terapie
Pokles svalové síly nelze v současné době ovlivnit a pacienti jsou odkázáni na ortopedické pomůcky a doplňky do domácnosti k usnadnění života. Pravidelné cvičení je prospěšné [120]. Je nutné v pravidelných intervalech sledovat spirometrické funkce, pátrat po známkách noční respirační insuficience a případně indikovat použití přístrojové podpory (CPAP).
U nemocných s MD1 také sledujeme nutriční status (sledování hmotnosti, BMI, laboratorních markerů výživy), hodnotíme polykání a riziko aspirace.
Pro ovlivnění myotonie se používá nejčastěji mexiletin a procainamid, ale na rozdíl od vrozené myotonie zde musíme vždy zvažovat kardiální riziko.
Vzhledem k zvýšenému riziku nádorových onemocnění je na místě pravidelná onkologická prevence.
Kognitivní změny vyžadují v řadě případů psychologickou péči a sociální poradenství.
U nemocných trpících MD1 je třeba cíleného dotazování na denní spavost a indikovat vyšetření ve spánkové laboratoři. Léčebnou modalitou je podávání modafinilu.
Neexistuje jednoznačné doporučení pro léčbu bolestí a myalgií, řídíme se obecnými doporučeními.
Léčba katarakty je chirurgická a neliší se nijak od obvyklých postupů. Ptózu charakteristickou pro MD1 lze upravit plastickým zákrokem.
Pečlivá kardiologická anamnéza s dotazem na výskyt synkop, palpitací, presynkop či epizod dušnosti je nezbytnou součástí vyšetření. Za minimální frekvenci EKG kontrol lze považovat interval jedenkrát ročně [102]. 24hodinový Holterův EKG monitoring by měl být ordinován nejlépe u všech pacientů, rozhodně však vždy při výskytu EKG abnormit či kardiálních symptomů [123].
Použití statinů je kontroverzní. Vzhledem k častým svalovým bolestem a elevaci CK chybí obvyklé markery pro hodnocení nežádoucích účinků. Vždy musíme pečlivě zvážit riziko vs. prospěch v každém individuálním případě [120].
Patogeneticky orientovaná terapie: Jádrem celého řetězce je akumulace RNA a její toxicita. Experimentální postupy se zaměřují právě k tomuto cíli pomocí antisense oligonukleotidů či dalších látek [124] vč. vnášení do buněk pomocí virových vektorů [125].
Gravidita
Problémy se týkají podstatně častěji žen s MD1, protože rozvinutá MD2 postihuje většinou ženy v období po narození dětí. U žen s MD1 je zvýšené riziko potratů a předčasných porodů. Při významné slabosti je riziko prolongace porodu, není ale indikace k rutinnímu provádění císařských řezů. Během gravidity je indikován ultrazvukový skríning (placenta previa, polyhydramnion) [126,127]. Je možná preimplantační genetická diagnostika. U obou typů, pokud je to aktuální, je při plánování gravidity indikována genetická konzultace a spolupráce mezi porodníkem gynekologem a specialistou z neuromuskulárního centra.
Anestezie
Vždy jde o nemocné s arytmogenním potenciálem a rizikem rabdomyolýzy. Zásadně se tedy vyhýbáme depolarizujícím myorelaxanciím (sukcinylcholin) a volatilním plynům (halothan, isofluran, desfluran). Nedepolarizující myorelaxancia jsou bezpečná. Není riziko maligní hypertermie. Riziko arytmie také zvyšuje event. iontová dysbalance. Důležitá je rovněž prevence podchlazení (chlad zhoršuje myotonii a navozuje třes = zvýšení rizika rabdomyolýzy). Před zákrokem v celkové anestezii bychom měli znát aktuální respirační funkce a schopnost polykání a toalety dýchacích cest [128– 130].
Vzhledem k postižení řady systémů jde při péči o nemocné s MD o komplexní multidisciplinární přístup, na kterém se podílí řada odborníků a odborností. Pacienti by měli být vedeni na pracovištích, která jsou schopna tuto komplexní péči zajistit a mají s ní dostatek zkušeností (neuromuskulární centra). Pacienti by měli být dispenzarizováni a sledováni v ročních intervalech. Nezastupitelnou roli v kultivaci péče a informacích o zdravotním stavu hrají také registry. V případě České republiky jde o Národní registr myotonických poruch ReaDy (www.ready.registry.cz).
Seznam použitých zkratek
CK – kreatinkináza
CNBP – Cellular Nucleic acid-binding Protein
DMPK – Dystrophic Myotonia Protein Kinase
DMWD – Dystrophia myotonica WD repeat containing protein
FISH – fluorescent in situ hybridisation
MD1 – myotonická dystrofie typu 1
MD2 – myotonická dystrofie typu 2
PCR – polymerázová řetězová reakce
PROMM – proximální myotonická myopatie
RNA – ribonukleová kyselina
RP PCR – repeat-primed PCR
SIX5 – sine oculis homeobox protein
TP PCR – triplet-repeat primed PCR
ZNF9 – Zinc Finger Protein 9
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Stanislav Voháňka, CSc., MBA
Neurologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: vohanka.stanislav@fnbrno.cz
Přijato k recenzi: 20. 2. 2017
Přijato do tisku: 10. 4. 2017
MUDr. Stanislav Voháňka, CSc., MBA

Doktor Voháňka promoval na Masarykově univerzitě v roce 1982. Neurologickou erudici zahájil na Klinice dětské neurologie tehdejší Fakultní dětské nemocnice v Brně. Od roku 1989 pracuje na Neurologické klinice LF MU a FN Brno, kde je od roku 1993 zástupcem přednosty pro LPP. Dlouhodobě se věnuje problematice nervosvalových onemocnění a chorobám bederní páteře. Vhled do vertebrogenní problematiky získal při pobytu na spinální jednotce Schulthessovy kliniky v Zurichu pod vedením prof. Dvořáka (1991–1992). V 90. letech minulého století se intenzivně věnoval elektrofyziologii a tyto zkušenosti využil při budování elektromyografi cké laboratoře a pracoviště evokovaných potenciálů v nově vzniklé Fakultní nemocnici Brno v Bohunicích. V roce 1993 obhájil kandidátskou dizertační práci, jejímž těžištěm byly elektrofyziologické nálezy u vybraných vertebrogenních onemocnění. Je zakládajícím členem Neuromuskulární sekce České neurologické společnosti a od roku 2005 jejím předsedou. Prezentoval více než 350 sdělení a je autorem nebo spoluautorem 250 publikací převážně z oblasti neuromuskulárních chorob. V roce 2016 se pod jeho vedením Neuromuskulární centrum FN Brno stalo součástí evropské sítě neuromuskulárních center (European Reference Network).
Sources
1. Curschmann H. Uber partielle Myotonie unter dem Bilde einer Beschaftigungsneurose und Lahmung. Berl Klin Wochenschr 1905;42:1175– 85.
2. Batten FE, Gibb HP. Myotonia atrophica. Brain 1909;32:187– 205.
3. Steinert HHW. Myopathologische Beiträge. I. Über das klinische und anatomische Bilde des Muskelschwunds des Myotoniker. Dtsch Z Nervenheilkd 1909;37:58– 104.
4. Fu YH, Pizzuti A, Fenwick RG, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992;255(5049):1256– 8.
5. Thornton CA, Griggs RC, Moxley RT. Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol 1994;35(3):269– 72.
6. Ricker K, Koch MC, Lehmann-Horn F, et al. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology 1994;44(8):1448– 52.
7. Ranum LP, Rasmussen PF, Benzow KA, et al. Genetic mapping of a second myotonic dystrophy locus. Nat Genet 1998;19(2):196– 8.
8. Ricker K, Grimm T, Koch MC, et al. Linkage of proximal myotonic myopathy to chromosome 3q. Neurology 1999;52(1):170– 1.
9. Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001;293(5531):864– 7.
10. Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 2012;11(10):891– 905. doi: 10.1016/ S1474-4422(12)70204-1.
11. Suominen T, Bachinski LL, Auvinen S, et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet 2011;19(7):776– 82. doi: 10.1038/ ejhg.2011.23.
12. Ricker K. Myotonic dystrophy and proximal myotonic myophathy. J Neurol 1999;246(5):334– 8.
13. Vohanka S, Parmova O, Mazanec R, et al. Myotonic dystrophy in Czech Republic: data from the national registry. J Neurol Sci 2015;357:e347– 8. doi: 10.1016/ j.jns.2015.08.1232.
14. Udd B, Meola G, Krahe R, et al. Myotonic dystrophy type 2 (DM2) and related disorders report of the 180th ENMC workshop including guidelines on diagnostics and management 3–5 December 2010, Naarden, The Netherlands. Neuromuscul Disord NMD 2011;21(6):443– 50. doi: 10.1016/ j.nmd.2011.03.013.
15. Morales F, Couto JM, Higham CF, et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum Mol Genet 2012;21(16):3558– 67. doi: 10.1093/ hmg/ dds185.
16. Wong LJ, Ashizawa T, Monckton DG, et al. Somatic heterogeneity of the CTG repeat in myotonic dystrophy is age and size dependent. Am J Hum Genet 1995;56(1):114– 22.
17. Monckton DG, Wong LJ, Ashizawa T, et al. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum Mol Genet 1995;4(1):1– 8.
18. Monckton DG, Caskey CT. Unstable triplet repeat diseases. Circulation 1995;91(2):513– 20.
19. Ashizawa T, Dunne PW, Ward PA, et al. Effects of the sex of myotonic dystrophy patients on the unstable triplet repeat in their affected offspring. Neurology 1994;44(1):120– 2.
20. Martorell L, Gamez J, Cayuela ML, et al. Germline mutational dynamics in myotonic dystrophy type 1 males: allele length and age effects. Neurology 2004;62(2):269– 74.
21. Bachinski LL, Udd B, Meola G, et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/ proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet 2003;73(4):835– 48.
22. Liquori CL, Ikeda Y, Weatherspoon M, et al. Myotonic dystrophy type 2: human founder haplotype and evolutionary conservation of the repeat tract. Am J Hum Genet 2003;73(4):849– 62.
23. Bachinski LL, Czernuszewicz T, Ramagli LS, et al. Premutation allele pool in myotonic dystrophy type 2. Neurology 2009;72(6):490– 7. doi: 10.1212/ 01.wnl.0000333665.01888.33.
24. Sallinen R, Vihola A, Bachinski LL, et al. New methods for molecular diagnosis and demonstration of the (CCTG)n mutation in myotonic dystrophy type 2 (DM2). Neuromuscul Disord 2004;14(4):274– 83.
25. Thornton CA, Johnson K, Moxley RT. Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann Neurol 1994;35(1):104– 7.
26. Jansen G, Willems P, Coerwinkel M, et al. Gonosomal mosaicism in myotonic dystrophy patients: involvement of mitotic events in (CTG)n repeat variation and selection against extreme expansion in sperm. Am J Hum Genet 1994;54(4):575– 85.
27. Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 2003;60(4):657– 64.
28. Cerghet M, Tapos D, Serajee FJ, et al. Homozygous myotonic dystrophy with craniosynostosis. J Child Neurol 2008;23(8):930– 3. doi: 10.1177/ 0883073808314965.
29. Martorell L, Illa I, Rosell J, et al. Homozygous myotonic dystrophy: clinical and molecular studies of three unrelated cases. J Med Genet 1996;33(9):783– 5.
30. Schoser BG, Kress W, Walter MC, et al. Homozygosity for CCTG mutation in myotonic dystrophy type 2. Brain 2004;127(8):1868– 77.
31. Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta 2015;1852(4):594– 606. doi: 10.1016/ j.bbadis.2014.05.019.
32. Meola G, Cardani R. Myotonic dystrophy type 2 and modifier genes: an update on clinical and pathomolecular aspects. Neurol Sci 2017;38(4):535– 46. doi: 10.1007/ s10072-016-2805-5.
33. Osborne RJ, Thornton CA. RNA-dominant dis-eases. Hum Mol Genet 2006;15 Spec No 2:R162– 9. 34. Cho DH, Tapscott SJ. Myotonic dystrophy: emerging mechanisms for DM1 and DM2. Biochim Biophys Acta 2007;1772(2):195– 204. doi: 10.1016/ j.bbadis.2006.05.013.
35. Mankodi A, Teng-Umnuay P, Krym M, et al. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann Neurol 2003;54(6):760– 8.
36. Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 2006;15(13):2087– 97. doi: 10.1093/ hmg/ ddl132.
37. Jiang H, Mankodi A, Swanson MS, et al. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet 2004;13(24):3079– 88.
38. Fardaei M, Rogers MT, Thorpe HM, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet 2002;11(7):805– 14.
39. Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science 2003;302(5652):1978– 80.
40. Mankodi A, Urbinati CR, Yuan QP, et al. Muscle-blind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet 2001;10(19):2165– 70.
41. Meola G, Cardani R. Myotonic Dystrophy Type 2: an Update on Clinical Aspects, Genetic and Pathomolecular Mechanism. J Neuromuscul Dis 2015;2:S59– 71. doi: 10.3233/ JND-150088.
42. Fugier C, Klein AF, Hammer C, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 2011;17(6):720– 5. doi:1 0.1038/ nm.2374.
43. Vihola A, Bachinski LL, Sirito M, et al. Differences in aberrant expression and splicing of sarcomeric proteins in the myotonic dystrophies DM1 and DM2. Acta Neuropathol 2010;119(4):465– 79. doi: 10.1007/ s00401-010-0637-6.
44. Koebis M, Ohsawa N, Kino Y, et al. Alternative splicing of myomesin 1 gene is aberrantly regulated in myotonic dystrophy type 1. Genes Cells Devoted Mol Cell Mech 2011;16(9):961– 72. doi: 10.1111/ j.1365-2443.2011.01542.x.
45. Rinaldi F, Terracciano C, Pisani V, et al. Aberrant splicing and expression of the non muscle myosin heavy-chain gene MYH14 in DM1 muscle tissues. Neurobiol Dis 2012;45(1):264– 71. doi: 10.1016/ j.nbd.2011.08.010.
46. Tang ZZ, Yarotskyy V, Wei L, et al. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum Mol Genet 2012;21(6):1312– 24. doi: 10.1093/ hmg/ ddr568.
47. Jurkat-Rott K, Lehmann-Horn F. State of the art in hereditary muscle channelopathies. Acta Myol 2010;29(2):343– 50.
48. Jurkat-Rott K, Lehmann-Horn F. Muscle channelopathies and critical points in functional and genetic studies. J Clin Invest 2005;115(8):2000– 9.
49. Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 2002;10(1):35– 44.
50. Charlet B, Savkur RS, Singh G, et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell 2002;10(1):45– 53.
51. Machuca-Tzili L, Brook D, Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: a review. Muscle Nerve 2005;32(1):1– 18. doi: 10.1002/ mus.20301.
52. Ranum LP, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet 2004;74(5):793– 804.
53. Day JW, Ranum LP. RNA pathogenesis of the myotonic dystrophies. Neuromuscul Disord 2005;15(1):5– 16.
54. Day JW, Ranum LP. Genetics and molecular pathogenesis of the myotonic dystrophies. Curr Neurol Neurosci Rep 2005;5(1):55– 9.
55. Mathieu J, Allard P, Potvin L, et al. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology 1999;52(8):1658– 62.
56. Groh WJ, Groh MR, Saha C, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med 2008;358(25):2688– 97. doi: 10.1056/ NEJMoa062800.
57. Hermans MC, Faber CG, Bekkers SCAM, et al. Structural and functional cardiac changes in myotonic dystrophy type 1: a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson 2012;14:48. doi: 10.1186/ 1532-429X-14-48.
58. Garrott HM, Walland MJ, O’Day J. Recurrent posterior capsular opacification and capsulorhexis contracture after cataract surgery in myotonic dystrophy. Clin Experiment Ophthalmol 2004;32(6):653– 5.
59. Rosa N, Lanza M, Borrelli M, et al. Low intraocular pressure resulting from ciliary body detachment in patients with myotonic dystrophy. Ophthalmology 2011;118(2):260– 4. doi: 10.1016/ j.ophtha.2010.06.020.
60. Meola G, Sansone V, Perani D, et al. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/ DM-2). Neuromuscul Disord 2003;13(10):813– 21.
61. Yu H, Laberge L, Jaussent I, et al. Daytime sleepiness and REM sleep characteristics in myotonic dystrophy: a case-control study. Sleep 2011;34(2):165– 70.
62. Dauvilliers YA, Laberge L. Myotonic dystrophy type 1, daytime sleepiness and REM sleep dysregulation. Sleep Med Rev 2012;16(6):539– 45. doi: 10.1016/ j.smrv.2012.01.001.
63. Laberge L, Gagnon C, Dauvilliers Y. Daytime sleepiness and myotonic dystrophy. Curr Neurol Neurosci Rep 2013;13(4):340. doi: 10.1007/ s11910-013-0340-9.
64. Winblad S, Lindberg C, Hansen S. Temperament and character in patients with classical myotonic dystrophy type 1 (DM-1). Neuromuscul Disord 2005;15(4):287– 92.
65. van der Werf S, Kalkman J, Bleijenberg G, et al. The relation between daytime sleepiness, fatigue, and reduced motivation in patients with adult onset myotonic dystrophy. J Neurol Neurosurg Psychiatry 2003;74(1):138– 9.
66. van Engelen BG, Eymard B, Wilcox D. 123rd ENMC International Workshop: Management and Therapy in Myotonic Dystrophy, 6– 8 February 2004, Naarden, The Netherlands. Neuromuscul Disord 2005;15(5):389– 94.
67. Minnerop M, Weber B, Schoene-Bake JC, et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 2011;134(12):3527– 43. doi: 10.1093/ brain/ awr299.
68. Ronnblom A, Andersson S, Danielsson A. Mechanisms of diarrhoea in myotonic dystrophy. Eur J Gastroenterol Hepatol 1998;10(7):607– 10.
69. Ronnblom A, Danielsson A, el Salhy M. Intestinal endocrine cells in myotonic dystrophy: an immunocy-tochemical and computed image analytical study. J Intern Med 1999;245(4):91– 7.
70. Ronnblom A, Forsberg H, Danielsson A. Gastrointestinal symptoms in myotonic dystrophy. Scand J Gastroenterol 1996;31(7):654– 7.
71. Vohanka S, Parmova O, Strenkova J. Lower Urinary Tract and Bowel Dysfunction in Patients with Myotonic Dystrophy. J Neuromuscul Dis 2014;1:200– 1. doi: 10.3233/ JND-149002.
72. Sherrod QJ, Chiu MW, Gutierrez M. Multiple pilomatricomas: cutaneous marker for myotonic dystrophy. Dermatol Online J 2008;14(7):22.
73. Mueller CM, Hilbert JE, Martens W, et al. Hypothesis: neoplasms in myotonic dystrophy. Cancer Causes Control 2009;20:2009– 20. doi: 10.1007/ s10552-009-9395-y.
74. Win AK, Perattur PG, Pulido JS, et al. Increased cancer risks in myotonic dystrophy. Mayo Clin Proc 2012;87(2):130– 5. doi: 10.1016/ j.mayocp.2011.09.005.
75. Gadalla SM, Lund M, Pfeiffer RM, et al. Cancer risk among patients with myotonic muscular dystrophy. JAMA 2011;306(22):2480– 6. doi: 10.1001/ jama.2011.1796.
76. Gadalla SM, Pfeiffer RM, Kristinsson SY, et al. Quantifying cancer absolute risk and cancer mortality in the presence of competing events after a myotonic dystrophy diagnosis. PLoS One 2013;8(11):e79851. doi: 10.1371/ journal.pone.0079851.
77. Ashizawa T, Sarkar PS. Myotonic dystrophy types 1and 2. Handb Clin Neurol 2011;101:193– 237. doi: 10.1016/ B978-0-08-045031-5.00015-3.
78. Angeard N, Gargiulo M, Jacquette A, et al. Cognitive profile in childhood myotonic dystrophy type 1: is there a global impairment? Neuromuscul Disord 2007;17(6):451– 8. doi: 10.1016/ j.nmd.2007.02.012.
79. Angeard N, Jacquette A, Gargiulo M, et al. A new window on neurocognitive dysfunction in the childhood form of myotonic dystrophy type 1 (DM1). Neuromuscul Disord 2011;21(7):468– 76. doi: 10.1016/ j.nmd.2011.04.009.
80. Echenne B, Rideau A, Roubertie A, et al. Myotonic dystrophy type I in childhood. Long-term evolution in patients surviving the neonatal period. Eur J Paediatr Neurol 2008;12(3):210– 23. doi: 10.1016/ j.ejpn.2007.07.014.
81. Meola G, Moxley RT. Myotonic dystrophy type 2 and related myotonic disorders. J Neurol 2004;251(10):1173– 82.
82. Sun C, Van Ghelue M, Tranebjærg L, et al. Myotonia congenita and myotonic dystrophy in the same family: coexistence of a CLCN1 mutation and expansion in the CNBP (ZNF9) gene. Clin Genet 2011;80(6):574– 80. doi: 10.1111/ j.1399-0004.2010.01616.x.
83. Parmová O, Voháňka S, Fajkusová L, et al. Souběžný výskyt mutace v genu ZNF9 (myotonická dystrofie typu 2) a v genu CLCN1 (myotonia congenita) v jedné rodině – kazuistika. Čes Slov Neurol N 2013;76/ 109(6):648– 51.
84. Tieleman AA, Jenks KM, Kalkman JS, et al. High disease impact of myotonic dystrophy type 2 on physical and mental functioning. J Neurol 2011;258(10):1820– 6. doi: 10.1007/ s00415-011-6027-8.
85. Sansone V, Gandossini S, Cotelli M, et al. Cognitive impairment in adult myotonic dystrophies: a longitudinal study. Neurol Sci 2007;28(1):9– 15.
86. Aminoff MJ, Beckley DJ, McIlroy MB. Autonomic function in myotonic dystrophy. ArchNeurol 1985;42(1):16.
87. Suokas KI, Haanpää M, Kautiainen H, et al. Pain in patients with myotonic dystrophy type 2: a postal survey in Finland. Muscle Nerve 2012;45(1):70– 4. doi: 10.1002/ mus.22249.
88. George A, Schneider-Gold C, Zier S, et al. Musculoskeletal pain in patients with myotonic dystrophy type 2. Arch Neurol 2004;61(12):1938– 42.
89. Jensen MP, Hoffman AJ, Stoelb BL, et al. Chronic pain in persons with myotonic dystrophy and facioscapulohumeral dystrophy. Arch Phys Med Rehabil 2008;89(2):320– 8. doi: 10.1016/ j.apmr.2007.08.153.
90. Mäntyselkä PT, Turunen JHO, Ahonen RS, et al. Chronic pain and poor self-rated health. JAMA 2003;290(18):2435– 42.
91. Parmova O, Vohanka S, Strenkova J. The Character and Frequency of Muscular Pain in Myotonic Dystrophy and Their Relationship to Myotonia. Int J Neurol Neurother 2014;1:2. doi: 10.23937/ 2378-3001/ 1/ 1/ 1009.
92. Papadimas GK, Kekou K, Papadopoulos C, et al. Phenotypic variability and molecular genetics in proximal myotonic myopathy. Muscle Nerve 2015;51(5):686– 91. doi: 10.1002/ mus.24440.
93. Hilbert JE, Ashizawa T, Day JW, et al. Diagnostic odyssey of patients with myotonic dystrophy. J Neurol 2013;260(10):2497– 504. doi: 10.1007/ s00415-013-6993-0.
94. Schneider-Gold C, Beer M, Kostler H, et al. Cardiac and skeletal muscle involvement in myotonic dystrophy type 2 (DM2): a quantitative 31P-MRS and MRI study. Muscle Nerve 2004;30(5):636– 44.
95. Parmová O, Voháňka S. Výskyt bolesti u myotonické dystrofie. Neurol Praxi 2016;17(4):240– 3.
96. Moshourab R, Palada V, Grunwald S, et al. A Molecular Signature of Myalgia in Myotonic Dystrophy 2. EBioMedicine 2016;7:205– 11. doi: 10.1016/ j.ebiom.2016.03.017.
97. Sansone VA, Brigonzi E, Schoser B, et al. The frequency and severity of cardiac involvement in myotonic dystrophy type 2 (DM2): long-term outcomes. Int J Cardiol 2013;168(2):1147– 53. doi: 10.1016/ j.ijcard.2012.11.076.
98. Schoser BG, Ricker K, Schneider-Gold C, et al. Sudden cardiac death in myotonic dystrophy type 2. Neurology 2004;63(12):2402– 4.
99. Groh WJ, Groh MR, Saha C, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med 2008;358(25):2688– 97. doi: 10.1056/ NEJMoa062800.
100. Rudnik-Schöneborn S, Schaupp M, Lindner A, et al. Brugada-like cardiac disease in myotonic dystrophy type 2: report of two unrelated patients. Eur J Neurol 2011;18(1):191– 4. doi: 10.1111/ j.1468-1331.2010.03077.x.
101. Motta J, Guilleminault C, Billingham M, et al. Cardiac abnormalities in myotonic dystrophy. Electrophysiologic and histopathologic studies. Am J Med 1979;67(3):467– 73.
102. Meola G, Sansone V, Marinou K, et al. Proximal myotonic myopathy: a syndrome with a favourable prognosis? J Neurol Sci 2002;193(2):89– 96.
103. Bushby K, Muntoni F, Bourke JP. 107th ENMC international workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th-9th June 2002, Naarden, the Netherlands. Neuromuscul Disord 2003;13(2):166– 72.
104. Melacini P, Villanova C, Menegazzo E, et al. Correlation between cardiac involvement and CTG trinucleotide repeat length in myotonic dystrophy. J Am Coll Cardiol 1995;25(1):239– 45.
105. Tokgozoglu LS, Ashizawa T, Pacifico A, et al. Cardiac involvement in a large kindred with myotonic dystrophy. Quantitative assessment and relation to size of CTG repeat expansion. JAMA 1995;274(10):813– 9.
106. Redman JB, Fenwick RG, Fu YH, et al. Relationship between parental trinucleotide GCT repeat length and severity of myotonic dystrophy in offspring. JAMA 1993;269(15):1960– 5.
107. Wahbi K, Algalarrondo V, Bécane HM, et al. Brugada syndrome and abnormal splicing of SCN5A in myotonic dystrophy type 1. Arch Cardiovasc Dis 2013;106(12):635– 43. doi: 10.1016/ j.acvd.2013.08.003.
108. Freyermuth F, Rau F, Kokunai Y, et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat Commun 2016;7:11067. doi: 10.1038/ ncomms11067.
109. Gadalla SM, Lund M, Pfeiffer RM, et al. Cancer risk among patients with myotonic muscular dystrophy. JAMA 2011;306(22):2480– 6. doi: 10.1001/ jama.2011.1796.
110. Mueller CM, Hilbert JE, Martens W, et al. Hypothesis: neoplasms in myotonic dystrophy. Cancer Causes Control 2009;20(10):2009– 20. doi: 10.1007/ s10552-009-9395-y.
111. Win AK, Perattur PG, Pulido JS, et al. Increased cancer risks in myotonic dystrophy. Mayo Clin Proc 2012;87(2):130– 5. doi: 10.1016/ j.mayocp.2011.09.005.
112. Harper PS. Myotonic dystrophy. 3rd ed. London: Harcourt Publishers Ltd 2001.
113. Voháňka S. Zvýšená hladina kreatinkinázy. Interní Med 2012;14(2):62– 5.
114. Tieleman AA, den Broeder AA, van de Logt AE, et al. Strong association between myotonic dystrophy type 2 and autoimmune diseases. J Neurol Neurosurg Psychiatry 2009;80(11):1293– 5. doi: 10.1136/ jnnp.2008.156562.
115. Bassez G, Chapoy E, Bastuji-Garin S, et al. Type 2 myotonic dystrophy can be predicted by the combination of type 2 muscle fiber central nucleation and scattered atrophy. J Neuropathol Exp Neurol 2008;67(4):319– 25. doi: 10.1097/ NEN.0b013e31816b4acc.
116. Vihola A, Bassez G, Meola G, et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/ DM2. Neurology 2003;60(11):1854– 7.
117. Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta 2015;1852(4):594– 606. doi: 10.1016/ j.bbadis.2014.05.019.
118. Kakourou G, Dhanjal S, Mamas T, et al. Modification of the triplet repeat primed polymerase chain reaction method for detection of the CTG repeat expansion in myotonic dystrophy type 1: application in preimplantation genetic diagnosis. Fertil Steril 2010;94(5):1674– 9. doi: 10.1016/ j.fertnstert.2009.10.050.
119. Radvansky J, Ficek A, Kadasi L. Upgrading molecular diagnostics of myotonic dystrophies: Multiplexing for simultaneous characterization of the DMPK and ZNF9 repeat motifs. Mol Cell Probes 2011;25(4):182– 5. doi: 10.1016/ j.mcp.2011.04.006.
120. Udd B, Meola G, Krahe R, et al. Myotonic dystrophy type 2 (DM2) and related disorders. Neuromuscul Disord 2011;21(6):443– 50. doi: 10.1016/ j.nmd.2011.03.013.
121. Kornblum C. Myotonic Dystrophies. In: Neuromuscular Imaging. New York: Springer Science & Business Media 2013:279– 93.
122. Die-Smulders CE, Howeler CJ, Thijs C, et al. Age and causes of death in adult-onset myotonic dystrophy. Brain 1998;121(8):1557– 63.
123. Vytopil M, Voháňka S, Šišáková M. Postižení srdce u hereditárních svalových onemocnění. Část II. Myotonická dystrofie, sarkoglykanopatie a Emeryho-Dreifussova svalová dystrofie. Čes Slov Neurol Neurochir 2001;64/ 97(2):144– 51.
124. Magaña JJ, Cisneros B. Perspectives on gene therapy in myotonic dystrophy type 1. J Neurosci Res 2011;89(3):275– 85. doi: 10.1002/ jnr.22551.
125. Bisset DR, Stepniak-Konieczna EA, Zavaljevski M, et al. Therapeutic impact of systemic AAV-mediated RNA interference in a mouse model of myotonic dystrophy. Hum Mol Genet 2015;24(17):4971– 83. doi: 10.1093/ hmg/ ddv219
126. Argov Z, de Visser M. What we do not know about pregnancy in hereditary neuromuscular disorders. Neuromuscul Disord 2009;19(10):675– 9. doi: 10.1016/ j.nmd.2009.07.004.
127. Awater C, Zerres K, Rudnik-Schöneborn S. Pregnancy course and outcome in women with hereditary neuromuscular disorders: comparison of obstetric risks in 178 patients. Eur J Obstet Gynecol Reprod Biol 2012;162(2):153– 9. doi: 10.1016/ j.ejogrb.2012.02.020.
128. Adams DC, Heyer EJ. Problems of anesthesia in patients with neuromuscular disease. Anesthesiol Clin N Am 1997;15:673– 89.
129. Brambrink AM, Kirsch JR. Perioperative care of patients with neuromuscular disease and dysfunction. Anesthesiol Clin 2007;25(3):483– 509. doi: 10. 1016/ j.anclin.2007.05.005.
130. Racca F, Mongini T, Wolfler A, et al. Recommendations for anesthesia and perioperative management of patients with neuromuscular disorders. Minerva Anestesiol 2013;79(4):419– 33.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2017 Issue 3
Most read in this issue
- Myotonic Dystrophy – Unity in Diversity
- Fetal Radiation Risk Due to X-ray Procedures Performed on Pregnant Women
- Febrile Seizures – Sometimes Less is More
- Low Back Pain – Evidence-based Medicine and Current Clinical Practice. Is there Any Reason to Change Anything?