
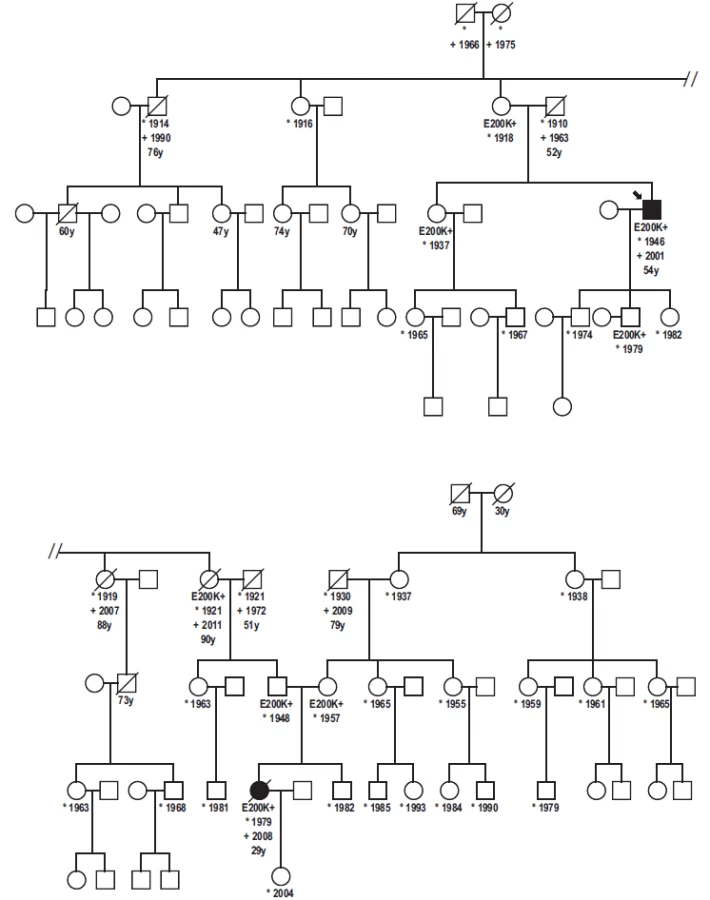
A Patient Homozygous for the E200K Mutation from a Family of the Slovak Cluster of Genetic Creutzfeldt-Jakob Disease
Pacient s homozygozitou mutácie E200K v rodine slovenského klastru genetickej formy Creutzfeldtovej-Jakobovej choroby
Práca informuje o prvom prípade homozygozity mutácie E200K na géne prionového proteínu v slovenskom súbore 227 pacientov postihnutých genetickou formou Creutzfeldtovej-Jakobovej choroby. Klinický priebeh, epidemiologické údaje, rodinná anamnéza ako aj výsledky imunoblotovania, nálezy histologické a imunohistologické u homozygotnej pacientky sú porovnávané s nálezmi u pacientov heterozygotných s rovnakou mutáciou. Kým neurologická a psychiatrická symptomatológia, ani dĺžka klinicky manifestnej fázy nevykazovali žiadne výrazné rozdiely, pri porovnaní dožitého veku bol zistený nápadný rozdiel. Pacientka s homozygozitou mutácie E200K mala 29 rokov a je najmladšou v súbore Creutzfeldtovej-Jakobovej choroby na Slovensku. Významný rozdiel dožitého veku vo vzťahu k postihnutému probandovi z predchádzajúcej generácie (25 rokov) je v súlade s nálezom anticipácie, ktorá charakterizuje familiárne prípady genetickej formy Creutzfeldtovej-Jakobovej choroby na Slovensku. Táto upozorňuje na klesajúci vek pri manifestácii ochorenia u nosičov mutácie E200K v postupne pribúdajúcich, následných generáciách, čo je významné tak z hľadiska včasnej diagnostiky, ako aj prevencie iatrogénnej nákazy. Neobvyklý výskyt nosičov mutácie E200K vo veku nad 90 rokov bez príznakov Creutzfeldtovej-Jakobovej choroby 1. poskytuje ďalší dôkaz nekompletnej penetrancie mutácie E200K stredoeuropského typu, 2. poukazuje na možný vplyv exogénnych ko-faktorov, prípadne spúšťacích podnetov pri manifestácii ochorenia. Ich podrobnejší výskum a objasnenie môže prispieť k prevencii klinickej manifestácie Creutzfeldtovej-Jakobovej choroby u asymptomatických nosičov najčastejšej a najrozšírenejšej mutácie E200K.
Kľúčové slová:
Creutzfeldtova-Jakobova choroba – mutácia génu prionového proteínu E200K – homozygozita
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Authors:
E. Mitrová 1; D. Slivarichova-Zakova 1; D. Kosorinova 1; M. Stelzer 1; S. Koscova 1; B. Berndtova 2; R. Junas 2
Authors‘ workplace:
Department of Prion Diseases, Slovak Medical University in Bratislava, Slovakia
1; Neurological Department, General Hospital in Ziar nad Hronom, Slovakia
2
Published in:
Cesk Slov Neurol N 2016; 79/112(3): 336-342
Category:
Case Report
doi:
https://doi.org/10.14735/amcsnn2016336
Overview
The first report of a patient homozygous for the E200K mutation of the prion protein gene in the Slovak cluster of 227 genetic Creutzfeldt-Jakob disease cases is presented. Consanguinity between parents or in the family was not reported. Clinical course, epidemiological data, family history, results obtained by immunoblotting, histopathology and immunohistochemistry are described and compared with patients heterozygous for the same mutation. While no striking difference was observed in neurological and psychiatric symptomatology or duration of symptomatic course, a significant age difference was found. The patient homozygous for the E200K mutation is the youngest case (29 years old) of Creutzfeldt-Jakob disease in Slovakia. The significant difference in age at the time of death between the patient and his affected relative from the previous generation (25 years) is in agreement with the concept of anticipation that characterizes familial cases of the Creutzfeldt-Jakob disease in Slovakia. This emphasises the decreasing age of onset of the disease in carriers of the E200K mutation in successive generations. This is important with respect to early diagnosis of the disease and prevention of iatrogenic transmission. Unexpected survival of E200K carriers aged 90 years or over without any signs of Creutzfeldt-Jakob disease, provides 1. further evidence of the incomplete penetrance of the Central European type of the E200K mutation and 2. emphasises the possible role of co-factors and triggering stimuli. Their more detailed study and elucidation may contribute to prevention of clinical manifestation of the genetic Creutzfeldt-Jakob disease in asymptomatic carriers of the most frequent, worldwide spread E200K mutation.
Key words:
Creutzfeldt-Jakob disease – prion protein gene mutation E200K – homozygosity
Introduction
Creutzfeldt-Jakob disease (CJD) is a rare, neurodegenerative, transmissible disorder of the central nervous system (CNS), clinically characterized by progressive dementia and death. It is the most frequent and worldwide spread human prion disease also called transmissible spongiform encephalopathy (TSE). Aggregates of a pathologically misfolded isoform of the cellular prion protein (PrPc), encoded by the host prion protein gene (PRNP) are detected in the brain of an affected person. The pathological prion protein – prion – is partially protease resistant (PrPres) [1]. Western blot analysis reveals two distinct, differently sized non-glycosylated PrPres fragments: type 1 (21 kD) and type 2 (19 kD). These two types, in combination with the PRNP codon 129 polymorphism, have been proposed to define six phenotypic variants of sporadic CJD [2]. The main difference between TSEs and other conformational neurodegenerative disorders, such as Alzheimer’s or Parkinson’s diseases, is their transmissibility. Quite unique feature of PDs is that they are both transmissible and genetic.
CJD occurs in sporadic (sCJD), genetic (gCJD) or iatrogenic (iCJD) forms [3] and as a new variant (vCJD) [4]. While origin of the most common sCJD is still unknown, iCJD is caused by accidental transmission of the disease in the course of diagnostic or therapeutic interventions, and vCJD – the first human prion zoonosis – is related to bovine spongiform encephalopathy.
Genetic CJD is associated with disease-specific mutations (insertions, deletions) of the PRNP. They represent about 10– 15% of all CJD cases [3,5,6]. Preventive genetic testing in gCJD-affected families revealed ”healthy”, asymptomatic carriers of the disease-specific mutations. They represent a “genetic CJD risk group” in the general population. Proportion of carriers developing the disease depends on the penetrance of individual mutations. It is unknown whether and when asymptomatic carriers could be infectious. These data are, therefore, important for prevention of possible iatrogenic transmission as well as for prophylactic treatment of carriers, when effective therapy is available.
The most frequent CJD-specific mutation is at codon 200 (E200K). The range of penetrance in individual mutations varies between 12–97/100% [6–10], and their clinical course is indistinguishable from sporadic CJD (sCJD) [11,12]. CJD patients with this mutation (gCJDE200K) have occurred worldwide, mainly in Chile [13], France [14], Italy [15], Hungary [6], Japan [16] and the Czech Republic [12]. Two large clusters have been confirmed in Slovakia [10,17] and in Israel [18].
At least five patients homozygous for the E200K mutation have been identified in an ethnic CJD cluster of Libyan Jews in Israel [19]. The authors ascribed its origin to consanguinity. Despite the high number (227) of confirmed gCJDE200K cases with numerous marriages between the gCJDE200K-affected families [20] and between relatives, patients homozygous for E200K have not been detected yet, and the presented patient is the first one in the Slovak CJD cluster.
Patients and methods
Patient 1
A proband in the gCJDE200K-affected family was a 54-years-old civil engineer, working as a deputy director. He had a personal history of cholelithiasis, steatosis hepatis and hemangioma hepatis. No surgical or pharmacological treatment was reported. Since July 2000, his family noticed that he had become nervous and irritated, ascribing his behaviour to the unwanted but forthcoming position of a director. Since October 2000, he observed posture disturbances, unstable gait, weakness and memory impairment. In December 2000, he was hospitalized due to rapid progression of neurological symptoms, including epileptic seizures and evident cognitive deficit. During his hospitalisation, he developed rapid progressive dementia, repeated grand mal seizures and left gaze-evoked, horizontal nystagmoid involuntary bulbar movements. CT showed diffuse cortical atrophy. His initial EEG revealed diffuse abnormal graph with epileptiform discharges on the right side. His second EEG showed typical bilateral synchronous periodic discharges (SW complexes 2.5–3 Hz). Genetic testing confirmed the E200K mutation with methionine homozygosity at codon 129. The patient died four months after the onset of clinical symptoms, on February 10, 2001. Definite CJD was confirmed by histopathology and immuhistochemistry.
His father died at the age of 54 (of silicosis) while his mother, born in 1918, is still alive. According to the family members, no memory loss or disorientation was evident so far. His sister, born in 1937 (a physician), a carrier of the E200K mutation, is alive, taking care of her 90-year-old mother. The patient has three children, two married sons (41 and 35 years old) and a daughter. The only tested 35-year-old son is a carrier of the E200K mutation. The patient’s daughter is a 33 years old single woman who was suffering from repeated depressions after her father fell ill. At present, she is well, working at a foreign embassy. Except for his mother, sister and one of his sons, all relatives refused genetic testing.
Patient 2
Patient 2 was a 29-year-old woman, a niece of the proband. She was working as a chambermaid in a hotel. She was married and had a four-year-old daughter. Her 60-year-old father and 51-year-old mother are still alive. Consanguinity between parents was not confirmed. She had a brother, a 33-year-old healthy joiner.
In September 2008, she complained aboutvisual impairment, tinnitus, and occasional disorientation. Her family observed behavioural abnormalities (apathy, withdrawal, fatigue, forgetfulness) and gradual onset of gait imbalance, uncoordinated walking and dizziness. She became untidy and was not able to do her usual housework. Both, her husband and her employer, reproached her with her laziness.
In November 2008, she was hospitalized with the diagnosis of vestibulocerebellar syndrome. During hospitalisation, she developed mild tremor in upper extremities, vertigo and occasional falls to the left side. Myoclonus was not observed. There was obvious intellectual decline and rapid memory loss. CT revealed supra- and infratentorial atrophy not corresponding to her age. EEG showed diffuse slow activity with delta and theta waves. Magnetic resonance imaging (MRI) (T1- and T2-weighted) confirmed profound diffuse infra- and supratentorial atrophy, not corresponding to the patient’s age. Focal abnormalities in the gray and white matter were absent. CSF findings were normal or negative, except for positive P 14-3-3. Genetic testing revealed homozygosity of the E200K mutation and of the methionine at codon 129. She died on December 16, 2008, following a rapid progression of the neurological symptoms and mental functions.
Histopathological and immunohisto-chemical findings confirmed definite CJD.
Besides the two gCJS patients, molecular genetic testing was performed on the mother, sister and one of the proband’s sons, and on the paternal grandmother and parents (repeatedly) of the second affected family member. The rest of the family have thus far refused the testing.
Neurohistopathology and immunohistochemistry: Serial sections (6 um) were cut from formalin-fixed and paraffin-embedded brain excisions, slides were stained with H & E as well as according to Masson, Kluver and Barrera and impregnated according to Cajal and Holmes. Immunohistochemical detection of PrPres was performed on formic ACID pre-treated, auto-claved and proteinase K (PK) treated slides with anti-PrPres monoclonal antibodies (MoAbs) 3F4 (DAKO) and 6H4 (Prionics).
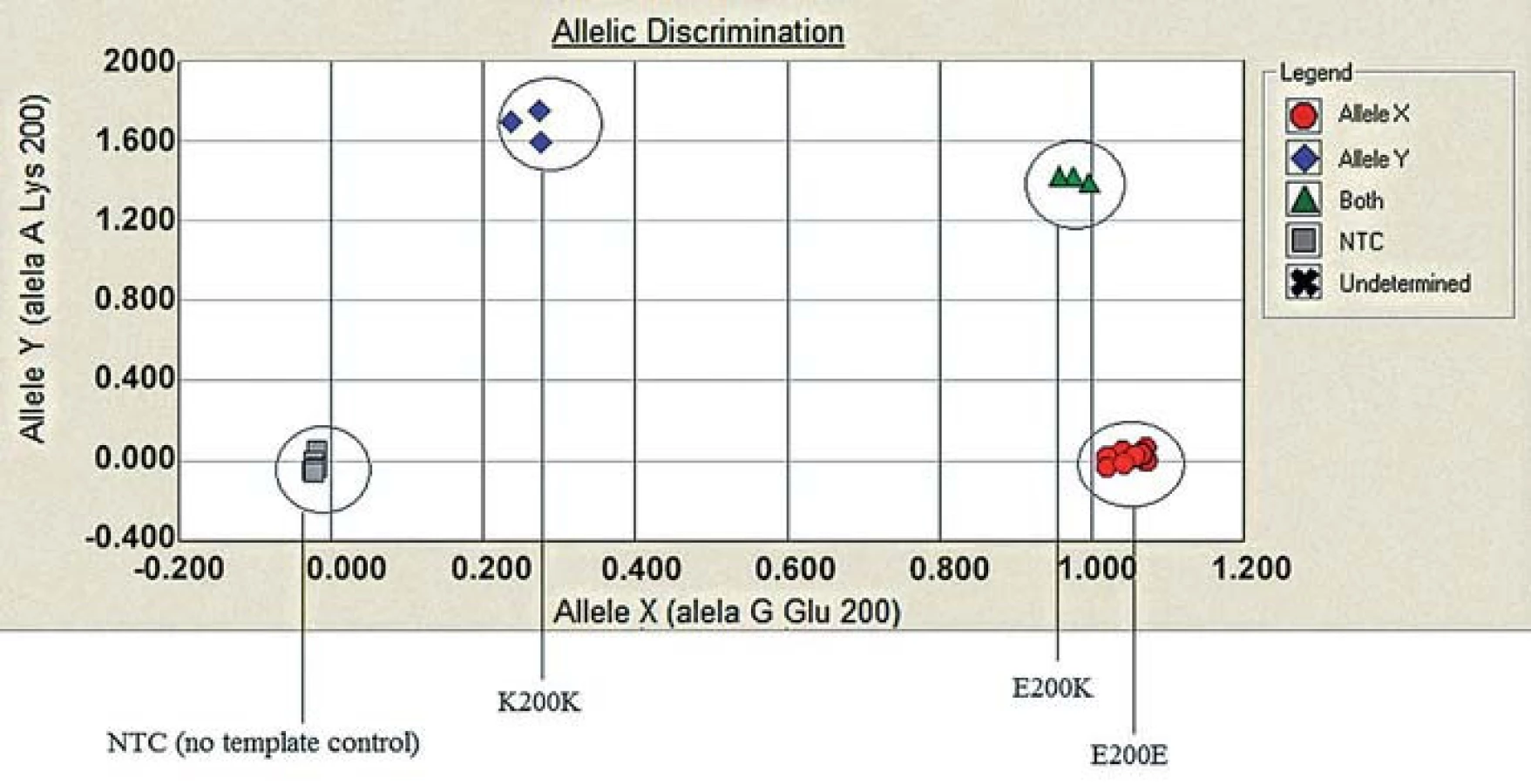
Molecular genetic testing: DNA was isolated from peripheral blood and amplified by PCR reaction performed with thermal cycler PTC-200 from MJ research. Detection of the point mutation (E200K) and the M129V polymorphism were carried out either using restriction enzymes BsmA1 (E200K) and MaeII/HpyCHIV (M129V) or a special AB 7500 real time PCR system with TaqMan SNP Genotyping Assay E200K (Applied Biosystem). Products/ results of the real time PCR were used for allelic discrimination assay plot. Genetic testing was performed on two patients and on 6 healthy relatives who agreed with the investigation and signed an informative consent.
Immunoblotting: Brain homogenates (10%) were prepared in PBS from the frozen brain with or without proteinase K digestion, denaturated with sodium dodecyl sulphate and separated by polyacrylamide gel electrophoresis; separated proteins were then transferred to nitrocellulose membranes. PrPres was detected using MoAbs 3F4 (DACO).
Results
Patient 1 (proband)
Histopathological examination showed cortical atrophy, neuronal loss, diffuse, moderate spongiform changes in all layers, hypertrophy, proliferation and clasmatodendrosis of astrocytes. Mild loss of granular and incipient spongiosis in the molecular layer was observed in the cerebellum. Immunostaining (3F4, 6H4) showed synaptic and fine granular positivity in the molecular layer and granular positivity in the granular layer.
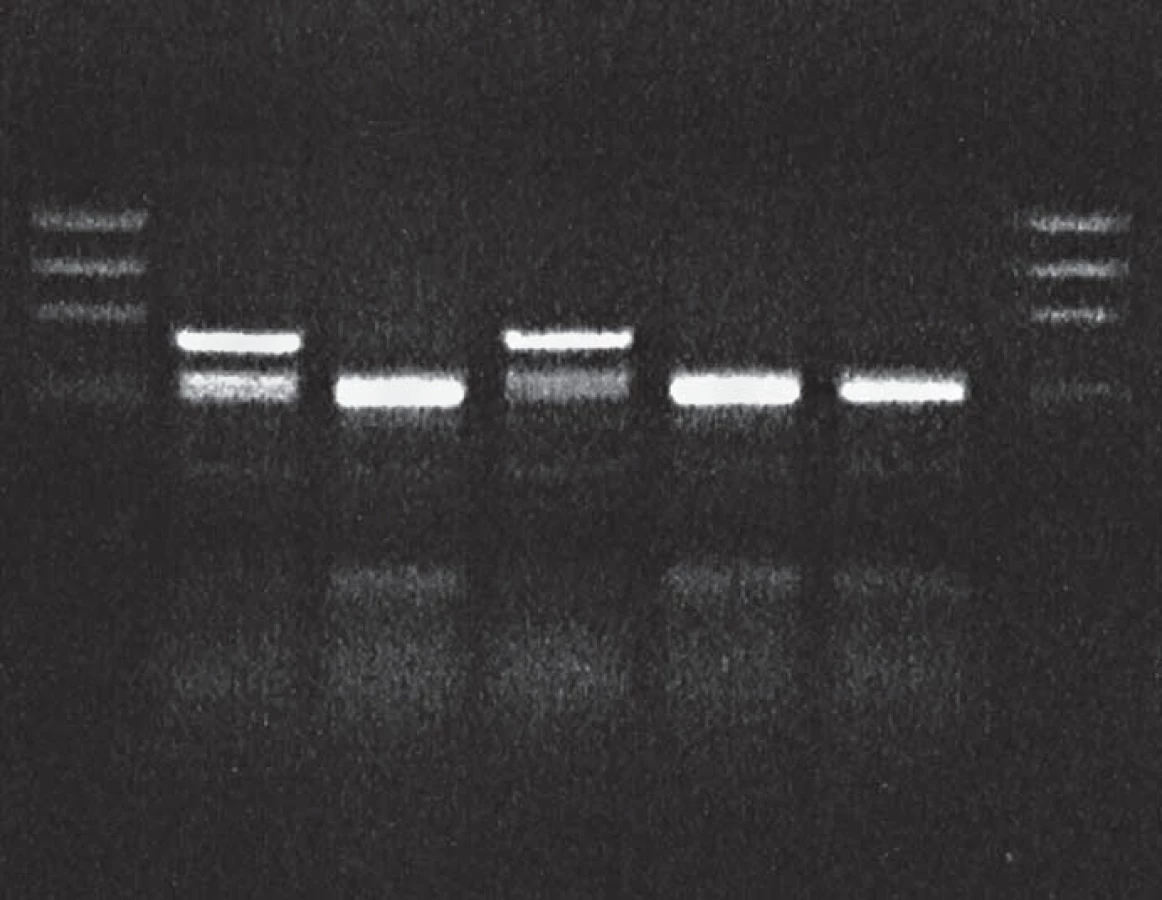
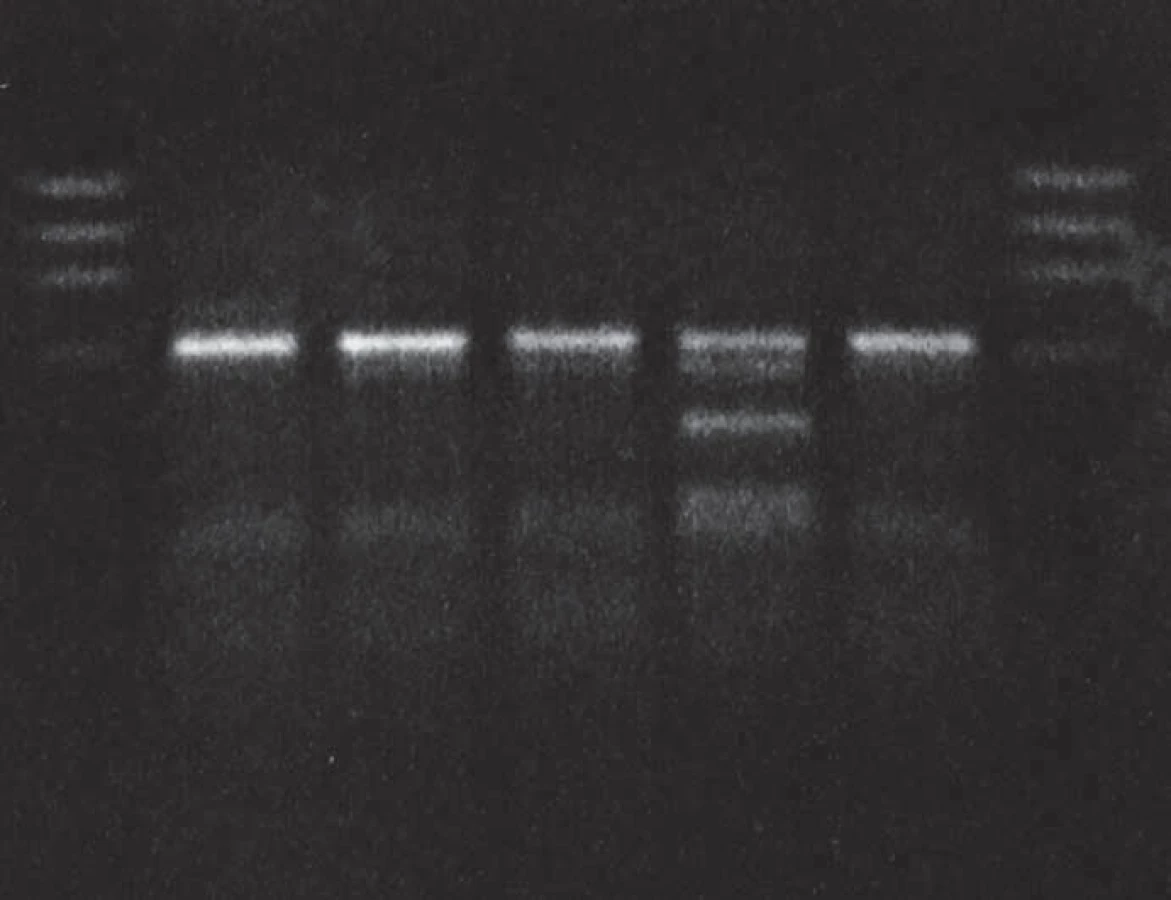
Molecular genetic analysis revealed PRNP gene mutation at codon 200 (E200K) and homozygosity Met/Met at codon 129 in the patient (Fig. 1, 2) and the E200K mutation in his mother, sister and son.
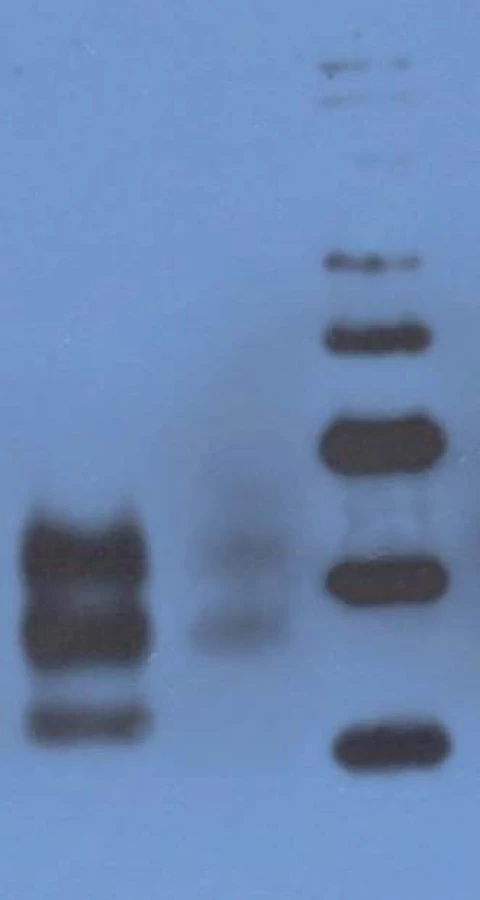
Western blot analysis (immunoblotting) showed prominent mono- and di-glycosylated bands and significantly thinner non-glycolysated band.
Patient 2 (homozygous for the E200K mutation)
Histopathological examination showed profound atrophy, severe neuronal loss, increased number of lipofuscin-loaded neurons not corresponding to the patient’s age, diffuse spongiosis with proliferation and hypertrophy of astrocytes in all cortical regions, less in basal ganglia. Immunostaining revealed synaptic and granular pattern of anti-prion immunopositivity in the molecular layer of the cerebellum.
Molecular genetic analysis (using restriction enzymes as well as allelic discrimination plot) showed PRNP gene mutation at codon 200 on both alleles (homozygosity of the E200K mutation) and homozygosity Met/Met at codon 129 (Fig. 3, 4). The E200K mutation was present in the mother, sister and son of the proband and in the grandmother and both parents (tested twice) of the patient homozygous for the E200K mutation (Fig. 5).
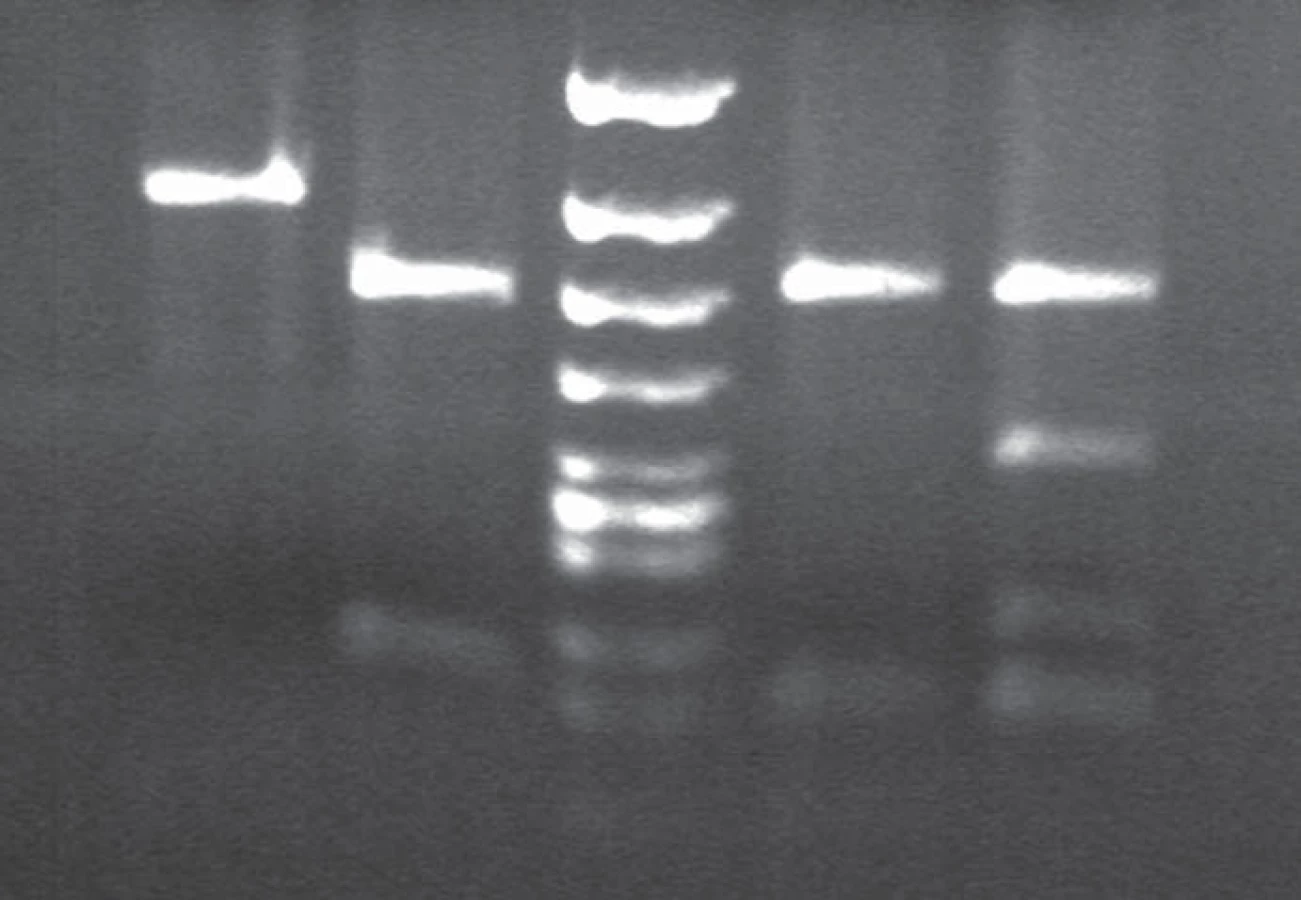
Immunoblotting revealed preserved mono- and di-glycosylated bands and absence of the non-glycolysated band (Fig. 6).
Discussion and Summary
More than 30 disease-specific point mutations of the PRNP have been described in human genetic TSEs. In the genetic TSE, even single-nucleotide mutations in the gene encoding the prion protein (PRNP) can increase likelihood of aggregation and neurodegeneration [19]. According to Van der Kamp, these mutations undoubtadly influence stability, processing and/or cellular interactions of the prion protein but the mechanism involved in the development of the mutation and the disease onset is not yet fully understood [20]. There is neither any explanation for why homozygosity of the mutation is so rare in such a large cluster of gCJDE200K (227 cases) with rather frequent consanguinity.
To explore the possible effect of the E200K homozygosity, we compared a patient homozygous for the E200K mutation with an affected relative as well as with other gCJDE200K patients. No difference in the clinical course and neurological symptomatology was observed. Prodromal stage, characterized by visual problems lasting three months, was followed by behavioural changes, progressive mental deterioration, motoric dysfunction and death within four months, corresponding to the course of gCJDs.
There was no triggering stimulus (divorce, death in the family, accident, surgery, loss of job, chronic occupational stress) reported in the proband, frequently seen in gCJDE200K patients and most evident in our four twin pairs with one CJD-affected twin.
The only histopathological abnormality observed was the unexpectedly high number of lipofuscin-loaded neurons in such a young patient; and while immunoblotting of the heterozygous patients had shown prominent mono- and di-glycosylated bands with only thinner non-glycosylated band, there was no non-glycosylated band in the E200K homozygous patient. Age at onset of the disease (29 years) also was a noteworthy feature, as she was the youngest patient in the Slovak genetic CJD cluster. The age difference at the onset of the disease between her and the proband was 25 years, and this was in agreement with the anticipation characteristic for the analysed CJD cluster [21]. Notable here is the decreasing age at the onset of gCJD in successive generations, important with respect to early diagnosis of the disease and prevention of iatrogenic CJD.
There is an obvious difference between our patient and five E200K homozygous patients in Israel relating to the age of the disease onset. All homozygous cases had lower age of onset (29 years our patient, 50.4 ± 6.2 years Israel homozygotes) compared to heterozygous cases (58.07 ± 8.3 years Slovak heterozygotes, 59.1 ± 9.0 years Israel heterozygotes). However, while our patient was only 29 years old, the range of age of onset in homozygous cases from Israel was as high as 41–58 years. Since the prion protein gene polymorphism M129V was not tested in the compared homozygous group, possible influence of methionine homozygosity cannot be reviewed. No difference was observed in disease duration; it was 4 months in our patient and it ranged between 2–24 months in the compared homozygous cases (19).
Detailed family history of our patients revealed some unexpected observations. In addition to the two CJD patients, there were six asymptomatic E200K mutation carriers in the presented family. Five were from the paternal side, since, except from the patient’s mother, there was no cooperation on the mother’s side.
The maternal grandmother is 78 years old, immobile due to stroke, deaf with good mental health. Grandfather was a miner and alcohol abuser who lived away from the family for years and had no contact with his three daughters. He died at the age of 79 in 2009. Maternal great-grandmother died at the age of 30 during delivery of her third child. Great-grandfather died at the age of 69 (prostatic cancer).
The paternal side is positive for both CJD patients and asymptomatic carriers of the E200K mutation. Within the first genetically tested generation, the 97-years-old mother of the proband (and great-aunt of the E200K homozygous patient) is still alive. She is almost imobile but her memory and mental capacities are preserved. She is taken care of by her 78-years-old daughter (a physician), a carrier. Her siblings (not genetically tested) died without any signs of mental deterioration at the age of 76 (pneumonia), 88 and 91 (stroke). Her sister, who was positive for the mutation (grandmother of the E200K homozygous patient), died at the age of 90 (peritonitis) without any signs of mental deterioration.
Our data suggest that the pathogenic effect of the mutation may depend on some additional (exo- and endogenous) factors. The advanced age E200K carriers not de-veloping CJD and dying of other diagnoses provide further evidence on incomplete penetrance of the E200K mutation of the Central European type, confirmed in our group [8,10] and also reported in other gCJDE200K groups [9]. This corresponds with the observation that while all CJD patients in families with more than one affected family member (familial cases) have point mutation of the PRNP gene, i.e. they form the genetic CJD (gCJD) group, genealogical studies of gCJD cases demonstrated that not all gCJD patients have a positive family history and familial CJD represent less than 60% of all genetic CJD cases [5,6,10]. This also is in accordance with the observation that there are families where gCJD is “skipped” and there might be no patients between two affected generations.
Described findings, i.e. carriers of the pathogenic mutation at unusually high age without any signs of CJD, cannot be explained with the available data. They draw attention to additional co-factors, either endo- or exogenous, as well as the need for further, more detailed epidemiological and molecular genetic studies. Elucidation of the possible role of exogenous co-factors, including triggering stimuli, may contribute to prevention of clinical manifestation of the genetic CJD in identified asymptomatic carriers of the most frequent and worldwide spread E200K mutation.
Acknowledgement
The project was supported by a grant from the EC ”Protecting the food chain from prions: shaping European priorities through basic and applied research, PRIORITY, No222887 and by a grant from the EU Joint Programme ”Neurodegenerative Disease Research Biomarker-based diagnosis of rapid progressive dementias, JPND-DEMTEST, 01ED1201A.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Eva Mitrova, MD, PhD, DSc.
Department of Prion Diseases
Slovak Medical University in Bratislava
Limbová 14
833 03 Bratislava
Slovakia
e-mail: eva.mitrova@szu.sk
Accepted for review: 9. 11. 2015
Accepted for print: 7. 1. 2015
Sources
1. Prusiner SB. Some speculations about prions, amyloid and Alzheimer’s disease. N Engl J Med 1984;310:661–3.
2. Gambetti PL, Cali I, Notari S, et al. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol 2011;121(1):79–90. doi: 10.1007/s00401-010-0761-3.
3. WHO manual for surveillance of human transmissible spongiform ecephalopathies including variant Creutzfeldz-Jakob disease. Geneva, WHO 2003:1–75.
4. Will R, Ironside J, Zeidler JW. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996;374:921–5.
5. Alperovitch A, Zerr I, Windl O, et al. Genetic epidemiology of Creutzfeldt-Jakob disease in Europe. Rev Neurol 2002;157(1–7):633–7.
6. Kovacs G, Puopulo M, Ladogana A, et al. Genetic prion disease: the EUROCJD experience. Hum Genet 2005;118(2):166–74.
7. Goldfarb L, Brown P, Mitrová E, et al. Creutzfeldt-Jakob disease associated with the PRNP codon 200Lys mutation: an analysis of 45 families. Eur J Epidemiol 1991;7(5):477–86.
8. Spudich S, Mastrianni JA, Wrench M, et al. Complete penetrance of Creutzfeldt-Jakob disease in Libyan Jews carrying the E200K mutation of the prion protein gene. Molecular Medicine 1995;1(6):607–13.
9. D’Alessandro M, Petraroli R, Ladogana A, et al. High incidence of Creutzfeldt-Jakob disease in rural Calabria, Italy. Lancet 1998;352(9145):1989–90.
10. Mitrová E, Belay G. Creutzfeldt-Jakob disease with E200K mutation in Slovakia: characterization and development. Acta Virol 2002;46(1):31–9.
11. Gdovinová Z. Creutzfeldtova-jakobova choroba.Cesk Slov Neurol N 2013;76/ 109(2):138–54.
12. Rohan Z, Parobková E, Johanidesová S, et al. Lidské prionové nemoci v České republice – 10 let zkušenosti s diagnostikou. Cesk Slov Neurol N 2013;76/109(3):300–6.
13. Galvez S, Masters C, Gajdusek CD. Descriptive epididemiology of Creutzfeldt-Jakob disease in Chile. Arch Neurology 1980;37(1):11–4.
14. Laplanche J, Delasnerie-Laupretre N, Brandel J, et al. Molecular genetic s of prion diseases in France. Neurology 1994;44(12):2347-51.
15. Ladogana A, Puopulo M, Poleggi D, et al. High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology 2005;64(11):1592–97.
16. Miyakawa T, Inoue K, Iseki E. Japanese Creutzfeldt-Jakob disease patients exhibiting high incidence of the E200K PRNP mutation and located in the basin of river. Neurol Res 1998;20(8):684–8.
17. Goldfarb L, Mitrová E, Brown P, et al. Mutation in codon 200 of scrapie amyloid protein in two clusters of Creutzfeldt-Jakob disease in Slovakia. Lancet 1990;336(8713):514.
18. Hsiao K, Meiner Z, Kahana E, et al. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N Eng J Med 1991;324(16):1091–7.
19. Simon ES, Kahana E, Chapman J, et al. Creutzfeldt-Jakob disease profile in patients homozygous for the PRNP E200K mutation. Ann Neurol 2000;47(2):257–60.
20. Mitrová E. Some new aspects of Creutzfeldt-Jakob disease epidemiology in Slovakia. Eur J Epid 1991;7(5):439–49.
21. Mead S. Prion disease genetics. Europ J Human Genet 2006;14(3):273-81.
22. van der Kamp MV, Dagget V. The consequences of pathogenic mutations to the human prion protein. Protein Eng Des Sel 2009;22(8):461–8. doi: 10.1093/protein/gzp039.
23. Mitrová E. Increasing incidence, anticipation and prophylaxis of genetic Creutzfeldt-Jakob disease. J Neurol 2010;257(Suppl):2002–3.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2016 Issue 3
Most read in this issue
- Sympathetic Chain Schwannoma – a Case Report
- Clinical Guideline for the Diagnostics and Treatment of Patients with Ischemic Stroke and Transitory Ischemic Attack – Version 2016
- Validity Study of the Boston Naming Test Czech Version
- Pre-motor and Non-motor Symptoms of Parkinson’s Disease – Taxonomy, Clinical Manifestation and Neuropathological Correlates