
Diastematomyelie u dospělých – kazuistika
Diastematomyelia in Adults – a Case Report
Diastematomyelia is a rare congenital defect, a type of spinal dysraphisms, often associated with the tethered cord syndrome. Diastematomyelia is formed by the two halves of the split spinal cord that each has own dural sac separated by a rigid or fibrotic septum. This defect is mainly diagnosed in children. Pain is the dominant symptom in adult patients. The authors present a case of a woman aged 44 years with diastematomyelia who complained of chronic back pain and progressive weakness in her legs. In this patient, laminectomy was performed, the spinal cord released and the dural sac restored. After the surgery, the patient had no neurological deficits and reported significant pain relief.
Key words:
diastematomyelia in adults – tethered cord syndrom – split cord malformation
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
V. Novák; L. Hrabálek; M. Vaverka; M. Halaj
Authors‘ workplace:
Neurochirurgická klinika LF UP a FN Olomouc
Published in:
Cesk Slov Neurol N 2015; 78/111(4): 486-489
Category:
Case Report
Overview
Diastematomyelie je vzácná vrozená vada patřící mezi spinální dysrafizmy a obvykle je asociována se syndromem míšního ukotvení. Diastematomyelie je tvořena dvěma polovinami rozštěpené míchy s vlastními durálními vaky, které jsou rozděleny osteokartilaginózním septem. Tato vada bývá diagnostikována především v dětském věku. U dospělých je vzácná a dominujícím příznakem je bolest. Autoři prezentují kazuistiku ženy ve věku 44 let s diastematomyelií, která si stěžovala na chronické bolesti zad a progredující slabost dolních končetin. U pacientky jsme provedli laminektomii, deliberaci míchy a plastiku durálního vaku. Po operaci pacientka zůstala bez neurologického deficitu a udávala výraznou úlevu od bolestí.
Klíčová slova:
diastematomyelie u dospělých – syndrom míšního ukotvení – rozštěpové malformace míchy
Úvod
Diastematomyelie je vzácná vrozená vada patřící mezi spinální dysrafizmy a obvykle je asociována se syndromem míšního ukotvení. Je charakterizována dvěma polovinami rozštěpené míchy s vlastními durálními vaky, které jsou rozděleny rigidním nebo fibrózním septem. Pang et al v roce 1992 publikovali práci, ve které diastematomyelii řadí do skupiny rozštěpových vad míchy nazývaných „split cord malformation“ (dále jen SCM) [1]. SCM rozdělují na typ I a typ II. Typ I, původně označovaný jako diastematomyelie, je tvořen dvěma polovinami rozštěpené míchy s vlastními durálními vaky, které jsou rozděleny osteokartilaginózním septem. Typ II, označovaný také jako diplomyelie, je tvořen dvěma kompletními částmi míchy v jednom durálním vaku. Jako kompozitní typ je nazýván výskyt obou předchozích typů tandemově za sebou. SCM bývá diagnostikována především v dětském věku a bývá doprovázena celou škálou anomálií jak spinálních, tak extraspinálních. Mezi spinální anomálie patří myelomeningokéla, zkrácené filum terminale, lipom, teratom, syringohydromyelie, neuroenterické a dermoidní cysty. Z extraspinálních anomálií se nejčastěji jedná o kožní stigmata, deformity nohou, hydrocefalus a vývojové vady urogenitálního traktu. Mezi průvodní příznaky syndromu míšního ukotvení patří bolest, slabost dolních končetin, sfinkterové dysfunkce, skolióza. U dospělých pacientů se mohou objevit všechny výše popsané příznaky, ale dominujícím příznakem je bolest. Popisovány jsou i asymptomatické případy [2– 6]. V české a slovenské literatuře se články věnující diastematomyelii vyskytují jen zřídka a omezují se na kazuistická sdělení [7,8]. Beneš sr a Beneš jr v roce 1986 publikovali práci, ve které poprvé uvádí pojem „syndrom fixované míchy“ [9].
Tato práce prezentuje kazuistiku dospělé ženy s diastematomyelií a její chirurgickou terapii.
Kazuistika
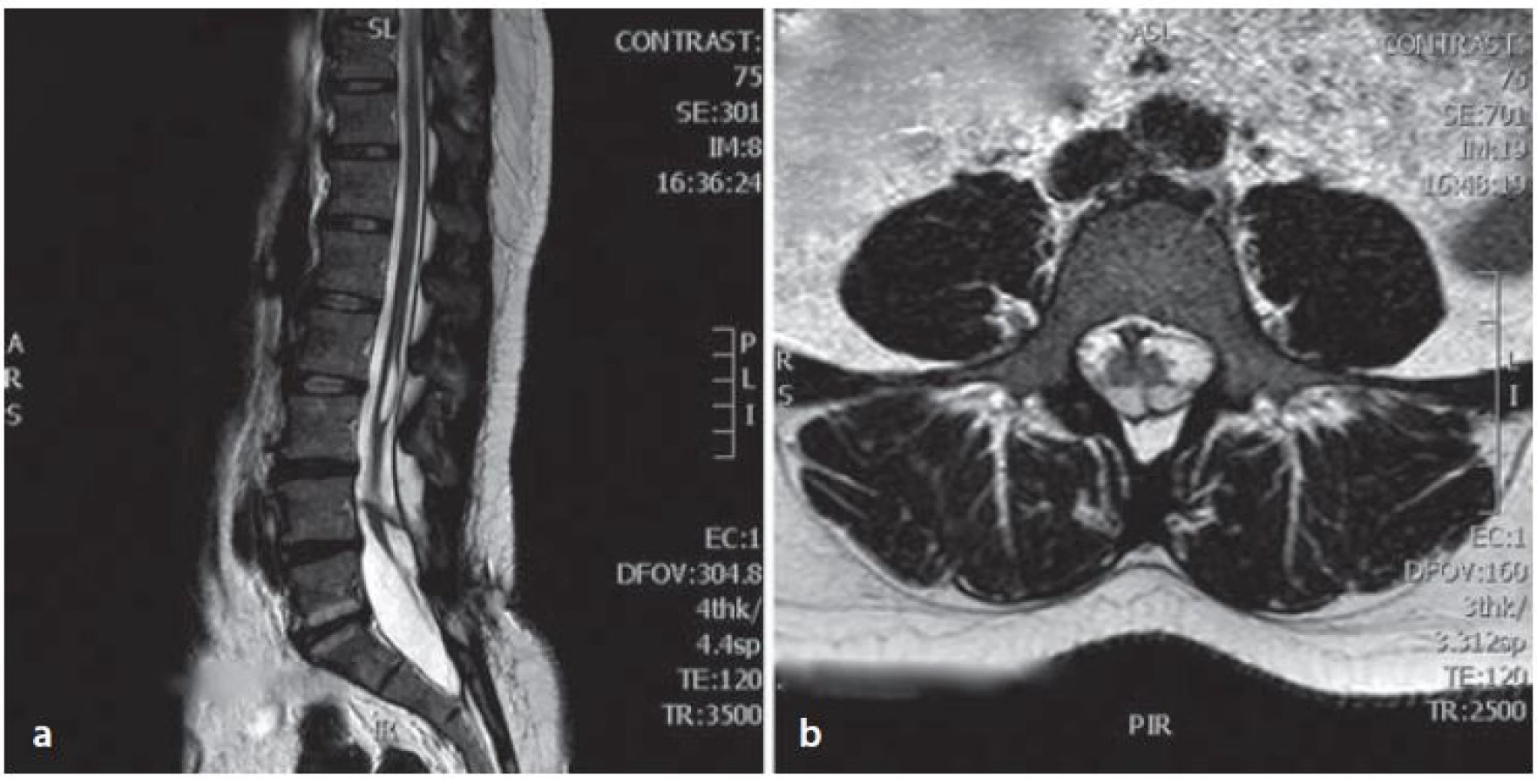
Žena ve věku 44 let s chronickými bolestmi zad byla vyšetřena pro zhoršující se obtíže trvající poslední dva měsíce. Při vyšetření udávala slabost dolních končetin a bolesti v bederní krajině s iradiací po zadní straně levé dolní končetiny. Potíže byly akcentovány vsedě a pacientka nebyla schopná se postavit. V objektivním vyšetření byla bez paréz dolních končetin, chodící, Lasegueův příznak měla pozitivní při 70 stupních bilaterálně, sníženou výbavnost patelárního reflexu a reflexu Achillovy šlachy vlevo a byla bez sfinkterových obtíží. Doplněné vyšetření magnetickou rezonancí (MR) a výpočetní tomografií (CT) bederní páteře prokázalo diastematomyelii s kostním septem v úrovni čtvrtého bederního obratle, která byla doprovázená hydrosyringomyelickou dutinou v rozsahu druhého a třetího bederního obratle. Míšní konus se nacházel v úrovni čtvrtého a pátého bederního obratle. Patrná byla také spina bifida pátého bederního obratle a diskopatie mezi pátým bederním a prvním křížovým obratlem, která se mohla podílet na bolestech v bederní krajině (obr. 1, 2). Skiagrafie a funkční skiagrafie (ve flexi a extenzi) bederní páteře neodhalily instabilitu. Předoperační elektromyografické vyšetření (EMG) prokázalo axonální lézi v myotomu prvního sakrálního kořene vlevo. Při pátrání v osobní anamnéze bylo zjištěno, že pacientka byla v dětství operována všeobecným chirurgem, nejspíše pro dermální sinus v oblasti bederní páteře bez dalšího vyšetření. Pacientka byla indikována k laminektomii, deliberaci spinální míchy a plastice dury.
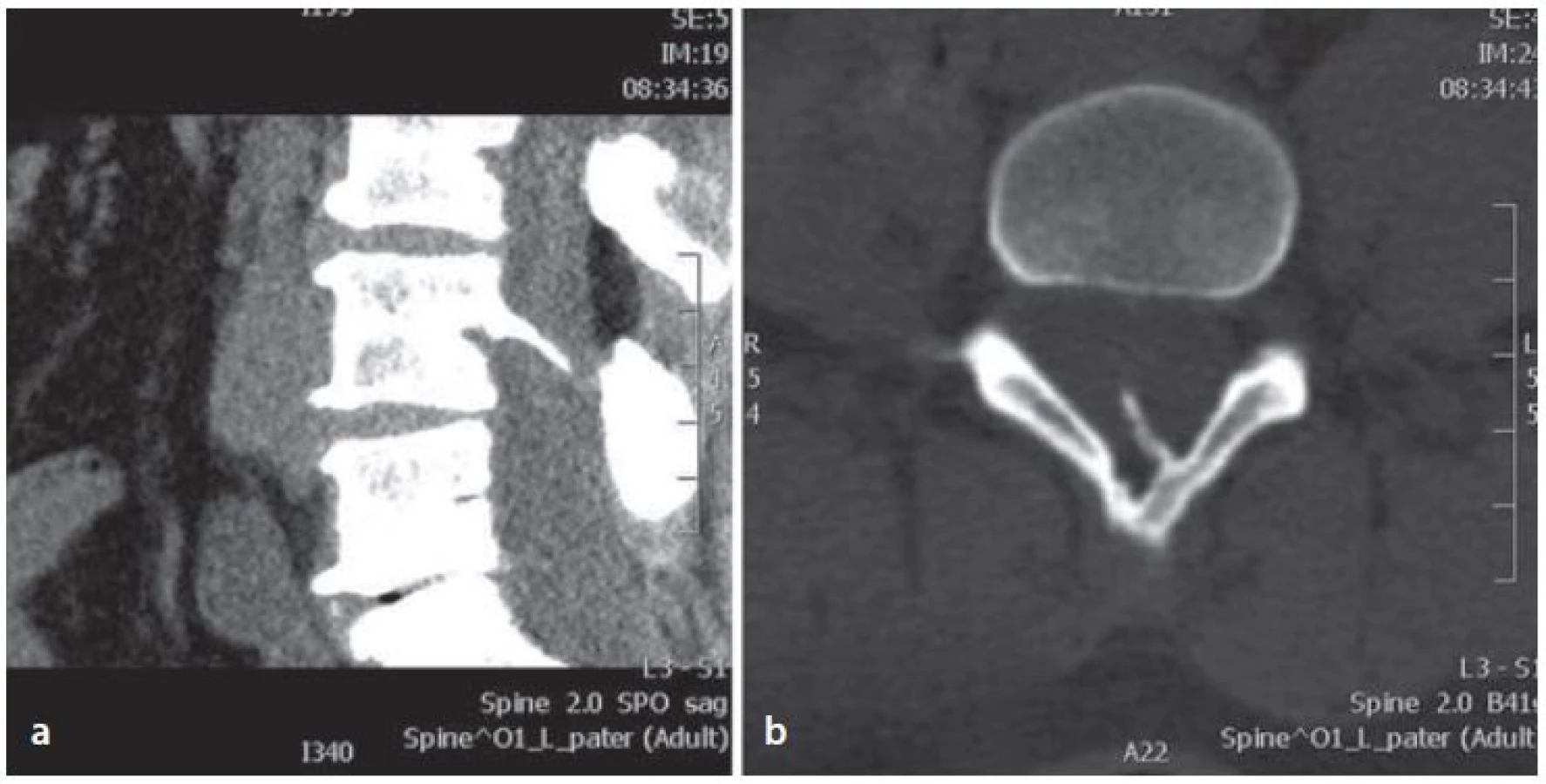
Operace byla provedena s využitím neuromonitorace. Elektrody byly umístěny na dolních končetinách v oblastech myotomu pátého bederního a prvního sakrálního kořene tak, aby bylo možné sledovat v průběhu operace evokovanou (triggered) elektromyografickou (tEMG) odpověď na stimulaci. Zákrok byl realizován v pronační poloze. Kožní incize byla provedena ve střední čáře kolmo na předchozí příčnou jizvu. Poté následovala kompletní laminektomie čtvrtého a částečná laminektomie pátého bederního obratle, při které byl nalezen zbytek komunikace z dury do předchozího kožního defektu. Kostní trn jdoucí dorzokaudálně od těla čtvrtého bederního obratle byl odseparováván od duplicitní dury a pomocí vysokootáčkové frézy částečně odstraněn (obr. 3a). Poté byla pod operačním mikroskopem provedena vřetenovitě durotomie a uvolněna mícha z adhezí (obr. 3b). Následovalo dokončení kompletního odstranění duplicitní dury a zbytku kostního trnu. Plastika dury byla provedena pomocí arteficiální dury (Neuro‑Patch®) a byl vytvořen jeden durální vak. Těsnost sutury byla pojištěna svalem a tkáňovým lepidlem (Tisseel®).

Po operaci byla pacientka přeložena na standardní oddělení, kde byla následujícího dne zahájena vertikalizace a rehabilitace. Po operaci pacientka udávala odeznění slabosti dolních končetin, výraznou úlevu od bolestí a objektivně byla bez neurologického deficitu. Třetí pooperační den byla propouštěna do domácího ošetřování.
Za čtyři měsíce po operaci bylo provedeno kontrolní MR bederní páteře (obr. 4). Na snímcích je patrné odstranění kostního septa a uvolnění míchy. Hydrosyringomyelická dutina byla po operaci zmenšena o 3 mm v podélné ose. Histologické vyšetření potvrdilo, že se jednalo o kostní septum. Pooperační vyšetření motorických evokovaných potenciálů prokázalo fyziologický nález. Při klinické kontrole pacientka udávala výraznou úlevu a byla bez bolestí.

Diskuze
Diastematomyelie je vrozená malformace, která vzniká v prvních čtyřech týdnech vývoje plodu. Její příčinou je chybná adheze ektodermu s endodermem a vytvoření akcesorního neurenterického kanálu, který je poté vyplněn mezenchymem. Oba dva typy SCM vznikají společným mechanizmem a jediný rozdíl představuje přítomnost prekurzorových buněk mening v endomezenchymálním traktu. Zda bude vada asociována s dalšími abnormalitami záleží na rozsahu ontogenetických změn v endomezenchymálním traktu. Mezi tyto vady patří otevřená myelomeningokéla, myelomeningokéla manqué, dermoidní cysta a kožní stigmata, jako jsou hypertrichóza, kapilární hemangiom a epidermální sinus. Myelomeningokéla manqué je označení pro spleť fibrózních provazců, cév a nervových kořenů vycházejících z dorzální části míchy a dosahujících dorzální části dury [1]. Mimo jiné se na syndromu ukotvení může také podílet zkrácené „filum terminale“ [2].
V námi prezentované kazuistice se jednalo o SCM typ I. Rozštěp míchy byl ve výši čtvrtého bederního obratle. Septum bylo tvořeno kostní tkání. Jak potvrzují ostatní práce, nejčastěji se jedná o kostní septum, méně často je septum chrupavčité nebo vazivové [1,2,10]. Míšní rozštěp dle četnosti je nejčastěji lokalizován v bederní páteři, poté následován výskytem v hrudní a nakonec krční páteři [1,2].
Neurologický deficit u diastematomyelie je způsoben více faktory. Jedná se zejména o nízkou anatomickou pozici míšního konu fixovanou osteokartilaginozním septem. Při normálním růstu páteře je omezen volný pohyb míchy nahoru, a tím dochází k trakci míchy. To vede k omezení krevního zásobení míchy nebo nervových kořenů. V důsledku toho následuje ischemie a nekróza nervové tkáně [11,12].
Ke stanovení diagnózy SCM se neobejdeme bez zobrazovacích vyšetření. Skiagrafické a CT vyšetření páteře umožní detailní zobrazení kostních struktur, a to zejména polohu kostního trnu, případně další kostní anomálie, jako je například spina bifida. Detailní zobrazení měkkých tkání páteře nám poskytne MR vyšetření. Umožní zobrazit rozdělení míchy, polohu míšního konu, hydrosyringomyelickou dutinu a v neposlední řadě nám umožní přesné naplánování operačního výkonu. Při rozhodování o indikaci k operační terapii je vhodné doplnit elektrofyziologické vyšetření, zejména EMG a evokované potenciály.
Cílem operační terapie je uvolnit míchu a umožnit její přirozený pohyb v durálním vaku. Operační technika se u obou typů SCM liší. Typ I je tvořen dvěma částmi rozštěpené míchy s vlastním durálním vakem a septem lokalizovaným extradurálně. Operace má za úkol odstranit extradurálně lokalizované septum a uvolnit míchu. Poté následuje durotomie s odstraněním duplicitní dury a vytvořením společného durálního vaku. V případě nálezu zkráceného filum terminale se provádí jeho přerušení. Typ II je tvořen dvěma částmi rozštěpené míchy v jednom durálním vaku a intradurálně lokalizovaným fibrózním septem. Zde se intradurálně provádí přerušení fibrózního septa a uvolnění míchy z okolních adhezí.
Cheng et al ve své práci srovnávali míru zlepšení neurologického stavu po operačním výkonu u SCM typu I a II [10]. U typu I došlo k zlepšení u 91,6 % operovaných. U typu II tomu bylo pouze v 50 %. V obdobné práci Huang et al publikovali zlepšení u 86 % pacientů operovaných pro SCM typu I [2]. U všech pacientů operovaných pro SCM typu II zůstal výsledný neurologický stav stejný jako před operací a při srovnání s konzervativně léčenou skupinou se nelišil. Huang et al konstatují, že u asymptomatických forem SCM typu I není chirurgická terapie nutná. U SCM typu II není signifikantní rozdíl v klinickém výsledku mezi chirurgickou a konzervativní terapií. Mahapatra et al prezentovali doposud největší soubor pacientů se SCM [13]. Soubor tvořilo 300 dětských pacientů. SCM typu I tvořilo 45 % a typ II byl diagnostikován v 55 %. Operační terapie byla indikována u všech pacientů včetně asymptomatických případů. Celkové zlepšení po operaci nastalo v 50 %, ve 44 % zůstali pacienti bez změny a v 6 % došlo po operaci ke zhoršení, které zůstalo trvalé pouze u 3 % pacientů. U žádného z pacientů operovaných profylakticky nedošlo ke zhoršení neurologického stavu. Dle literárních zdrojů u SCM typu I panuje shoda o vhodnosti chirurgické terapie. U SCM typu II jsou názory rozporuplné, a proto nelze jednoznačně doporučit, zda zvolit chirurgický nebo konzervativní postup.
Závěr
Diastematomyelie je vzácné vrozené onemocnění, se kterým se můžeme setkat i v dospělém věku. Dominujícím příznakem bývá bolest, která může být doprovázena slabostí končetin a sfinkterovými obtížemi. Často je tato vada kombinována s jinými anomáliemi jak spinálními, tak extraspinálními. Chirurgická terapie může významně zmírnit příznaky při správném výběru skupiny nemocných. Podle literárních údajů a na základě naší zkušenosti je chirurgická terapie u SCM typu I metodou volby.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 6. 2. 2015
Přijato do tisku: 23. 6. 2015
MUDr. Vlastimil Novák
Neurochirurgická klinika
LF UP a FN Olomouc
I. P. Pavlova 6
779 00 Olomouc
e-mail: nvlastimil@seznam.cz
Sources
1. Pang D, Dias MS, Ahab‑ Barmada M. Split cord malformation. Part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery 1992; 31(3): 451– 480.
2. Huang SL, He XJ, Wang KZ, Lan BS. Diastematomyelia: a 35‑year experience. Spine 2013; 38(6): E344– E349. doi: 10.1097/ BRS.0b013e318283f6bc.
3. Iskandar BJ, Fulmer BB, Hadley MN, Oakes WJ. Congenital tethered spinal cord syndrome in adults. Neurosurg Focus 2001; 10(1): e7.
4. Anderson FM. Occult spinal dysraphism: a series of 73 cases. Pediatrics 1975; 55(6): 826– 835.
5. Hoffman HJ, Hendrick EB, Humphreys RP. The tethered spinal cord: its protean manifestations, diagnosis and surgical correction. Childs Brain 1976; 2(3): 145– 155.
6. Pang D. Split cord malformation. Part II: clinical syndrome. Neurosurgery 1992; 31(3): 481– 500.
7. Häckel M, Beneš V. Diagnostika a léčba tethered cord syndromu – syndromu fixované míchy. Neurol Prax 2004; 5(1): 39– 41.
8. Horn F, Babala J, Studený S, Smrek M, Pevalova L, Kirnák Jet al. Diastematomyelia and the tethered spinal cord syndrome. Case report. Rozhl Chir 2001; 80(5): 242– 245.
9. Beneš V jr, Beneš V sr. Syndrom fixované míchy – tethered cord syndrome. Cesk Pediatr 1986; 41/ 78(10): 608– 611.
10. Cheng B, Li FT, Lin L. Diastematomyelia: a retrospective review of 138 patients. J Bone Joint Surg Br 2012; 94(3): 365– 372. doi: 10.1302/ 0301‑ 620X.94B3.27897.
11. Harwood‑ Nash DC, McHugh K. Diastematomyelia in 172 children: the impact of modern neuroradiology. Pediatr Neurosurg 1990– 1991; 16(4– 5): 247– 251.
12. Gan YC, Sgouros S, Walsh AR, Hockley AD. Diastematomyelia in children: treatment outcome and natural history of associated syringomyelia. Childs Nerv Syst 2007; 23(5): 515– 519.
13. Mahapatra AK. Split cord malformation – a study of 300 cases at AIIMS 1990– 2006. J Pediatr Neurosci 2011; 6 (Suppl 1): S41– S45. doi: 10.4103/ 1817‑ 1745.85708.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2015 Issue 4
Most read in this issue
- Léčba pudendální neuralgie – klinické zkušenosti po pěti letech
- Význam magnetické rezonance v diagnostice epilepsie
- Experimentální léčba poranění míchy
- Léčba foraminálního výhřezu meziobratlové ploténky u istmické spondylolistézy technikou TLIF