
Kongenitální centrální hypoventilační syndrom (Ondinina kletba)
Congenital Central Hypoventilation Syndrome (Ondine‘s Curse)
Congenital central hypoventilation syndrome (CCHS) is a rare, lifelong disorder of the breathing centre, resulting from a mutation of the PHOX2B gene. CCHS typically manifests in newborns with alveolar hypoventilation/ apnea during sleep. Clinical severity of hypoventilation and a risk of associated conditions (Hirschsprung disease, tumors of neural crest and autonomic nervous system dysfunction) depend on the type of the PHOX2B gene mutation. Approximately 90% of individuals with the CCHS phenotype are heterozygous for the PARMs‑type mutation (Polyalanine Repeat Expansion Mutations), the remaining 10% of patients express heterozygous missense‑ , nonsense‑ or frameshift‑type mutation (non‑PARMs). Significant hypoventilation leading to severe hypercapnia and hypoxemia was observed in two of our newborns with proved CCHS during the first week after birth. Following a very short period of mechanical ventilation, we succeeded in maintaining physiological blood gases with noninvasive ventilation during sleep. With modern techniques for home ventilation and follow up at specialized centres, children with CCHS have good long‑term prognosis and low mortality.
Key words:
central hypoventilation – Ondine‘s Curse
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
T. Matějek 1; M. Šenkeříková 2; E. Ruszová 2; J. Malý 1
Authors‘ workplace:
Dětská klinika LF UK a FN Hradec Králové
1; Oddělení lékařské genetiky, FN Hradec Králové
2
Published in:
Cesk Slov Neurol N 2015; 78/111(2): 215-219
Category:
Case Report
Overview
Kongenitální centrální hypoventilační syndrom (CCHS) je vzácná celoživotní porucha dechového centra, s genetickým podkladem v mutaci genu PHOX2B. Onemocnění se obvykle manifestuje v novorozeneckém věku hypoventilací/ apnoí ve spánku. Klinická tíže hypoventilace a riziko rozvoje přidružených onemocnění (Hirschsprungova choroba, nádory neurální lišty, poruchy autonomního nervového systému) se liší dle typu mutace PHOX2B genu. U 90 % pacientů se jedná o heterozygotní mutaci typu PARMs (Polyalanin Repeat Mutations), u zbylých 10 % pacientů nacházíme heterozygotní mutaci typu missence, nonsense nebo frameshift (NON‑ PARMs). U prezentovaných novorozenců s geneticky potvrzeným CCHS byla v prvním týdnu po narození nápadná hypoventilace/ apnoe vedoucí k významné hyperkapnii a hypoxémii. Po velmi krátké potřebě umělé plicní ventilace se fyziologické krevní plyny dařilo udržovat pomocí neinvazivní spánkové ventilace. S moderními technikami pro domácí ventilaci a sledováním ve specializovaných centrech mají děti s CCHS dlouhodobě dobrou prognózu s nízkou mortalitou.
Klíčová slova:
centrální hypoventilace – Ondinina kletba
Úvod
Syndrom kongenitální centrální alveolární hypoventilace je charakterizován celoživotní hypoventilací na podkladě geneticky podmíněné poruchy centrální regulace dýchání při vyloučení infekčních (sepse, meningitida), pulmonálních (primární plicní onemocnění) a neurologických příčin (vrozené vývojové vady CNS, krvácení, tumor, trauma mozku, mozkového kmene či neuromuskulární choroby – kongenitální myopatie a myastenie, spinální svalová atrofie typ I). Starší název Ondinina kletba vychází ze starogermánské legendy, v níž Ondina byla překrásná vodní nymfa, která se vzdala daru věčného života, když porodila dítě smrtelnému muži, jehož milovala. Ten jí přísahal věrnost a oddanost s každým bdělým dechem. Když později odhalila jeho nevěru, uvrhla na něho kletbu: ,,Přísahal jsi mi věrnost a oddanost s každým bdělým dechem. Dýchej tak dlouho, jak vydržíš být vzhůru, ale až jednou usneš, pak zemřeš.“ Prvně bylo toto onemocnění popsáno v roce 1970 [1]. Do dnešního dne se poznatky o kongenitálním centrálním hypoventilačním syndromu (CCHS) významně rozrostly. V roce 2003 byl identifikován gen PHOX2B (Paired mesoderm Homeobox Protein 2B), jehož mutace jsou zodpovědné za fenotyp CCHS, a tyto mutace mají vesměs dominantní efekt (heterozygoti mají chorobu) [2]. Gen PHOX2B leží na 4. chromozonu (lokus 4p12) a je dle aktuálních medicínských znalostí jediným genem, jehož mutace prokazatelně souvisí s rozvojem CCHS. Genový produkt PHOX2B genu je významný pro maturaci neuronů autonomního nervového systému, čímž se u dětí s CCHS vysvětlují projevy dysregulace tohoto systému (srdeční arytmie), dysfunkce střevního nervového systému (Hirschprungova choroba) a deregulace proliferace a diferenciace neuronální tkáně (sklon ke vzniku nádorů). Gen PHOX2B má dvě polyalaninové oblasti v 3. exonu (standardně 9 a 20 alaninů). Pro vznik CCHS je důležitá druhá z oblastí, s fyziologickým počtem 20 alaninových tripletů (GCN).
V PHOX2B genu rozlišujeme dva základní typy mutací:
- Mutace způsobená expanzí (duplikací) polyalaninových opakování (Polyalanin Repeat expansion Mutations; PARMs). Tento typ mutace nacházíme u 90 % postižených jedinců. Patologické alely mají obvykle 24– 33 repetic. U jedinců s 24– 25 triplety není výskyt obtíží spojených s CCHS konstantní (inkompletní penetrance, event. Late Onset CCHS; LOCCHS), přítomnost 26 a více repetic je vždy spojena s rozvojem CCHS. Závažnost abnormit autonomního nervového systému stoupá s velikostí expanze. Expanze tripletů v genomu s fenotypově‑klinickými důsledky není specifickým jevem pro CCHS. Na podobném genetickém mechanizmu jsou založeny další tzv. tripletové choroby (trinucleotide repeat expansions), mezi něž řadíme Huntingtonovu chorobu, syndrom fragilního‑ X nebo různé formy spinocerebelárních ataxií.
- Dále existují mutace PHOX2B genu typu missence, nonsense a s posunem čtecího rámce (frameshift) označované jako Non‑ Polyalanin Repeat expansion Mutations; NPARMs. U NPARMs mutací může výsledný fenotyp (expresivita, penetrance) variovat dle dopadu konkrétní mutace na aktivitu PHOX2B [1,3,4].
Základní laboratorní diagnostickou metodou CCHS je mutační fragmentační analýza. Test je pozitivní při expanzi polyalaninové oblasti (PARMs), ale lze zachytit i část NPARMs. Tato metoda se využívá jako skríningová metoda a zachytí téměř 95 % mutací PHOX2B genu u jedinců s CCHS, včetně mozaikových forem. Sekvenační analýza následně upřesní počet repetic (stanoví genotyp, např. 20/ 26 – tj. expanze šesti polyalaninových tripletů) a zachytí, kromě zmíněných, také další missense, nonsense, frameshift mutace (zbytek NPARMs). Vzácně se může jednat o rozsáhlejší delece/ duplikace zahrnující celé exony i geny. V takovém případě je metodou volby např. komparativní genomická hybridizace (array CGH), protože sekvenační analýza tuto změnu nezachytí.
Kazuistiky
V této práci jsou prezentováni dva novorozenci s CCHS. V prvním případě (P1) se jednalo o nedonošenou holčičku (2 090g/ 47 cm) z dvojčetné gravidity po porodu císařským řezem v 33. gestačním týdnu při preeklampsii matky. V druhém případě (P2) šlo o donošenou holčičku z fyziologické gravidity (3 200g/ 50 cm). V prvním týdnu po narození byla u obou pacientek nápadná těžká hypoventilace/ apnoe bez známek dechové tísně. U pacientky P1 došlo při distenční dechové podpoře k retenci oxidu uhličitého, pro kterou byla nezbytná krátkodobá umělá plicní ventilace. U pacientky P2 bylo nutné záhy po narození navedení na umělou plicní ventilaci pro hypoventilaci/ apnoe s hyperkapnií a těžkými desaturacemi. Po rozdílně dlouhé potřebě umělé plicní ventilace (P1 – 12 hod, P2 – 11 dnů) se fyziologické krevní plyny dařilo udržovat pomocí neinvazivní spánkové ventilace nosní maskou v podmínkách novorozenecké intenzivní péče (vrcholový inspirační tlak – PIP 15– 18; pozitivní přetlak na konci výdechu – PEEP 4; frekvence 30– 40/ min). Obě holčičky neměly v dalším průběhu hospitalizace při bdění již žádné dechové obtíže. U obou dětí jsme zaznamenali problémy s polykáním, které odezněly do dvou měsíců od narození. U holčičky P1 byl diagnostikován Wolffův‑ Parkinsonův‑ Whiteův syndrom (WPW syndrom). Celkem prodělala tři ataky supraventrikulární tachykardie (SVT) s dobrou odpovědí na adenosin. Při dlouhodobé terapii sotalolem se ataky SVT neopakovaly. Laboratorní, zobrazovací (MR mozku, rentgen hrudníku) ani neurologické vyšetření nevedlo ke stanovení kauzální příčiny hypoventilace.
Vzhledem k vysokému klinickému podezření na CCHS bylo provedeno molekulárně‑genetické vyšetření, které u obou holčiček toto postižení prokázalo. Molekulárně‑genetická vyšetření byla provedena na Oddělení lékařské genetiky FN Hradec Králové. U pacientky P1 byla detekována NPARM mutace – duplikace osmi nukleotidů, která vede k posunu čtecího rámce a vzniku zkráceného proteinu s abnormální aminokyselinovou sekvencí od místa mutace. Tato mutace byla již v minulosti publikována jako patogenní mutace odpovědná za vznik onemocnění [1,3]. U nemocné holčičky byla zachycena v 30% mozaice v periferní krvi. V případě pacientky P2 se jednalo o jednu z nejčastějších mutací PHOX2B genu – expanzi polyalaninové oblasti na 26 alaninových tripletů. V obou případech šlo o mutaci vzniklou de novo (graf 1).

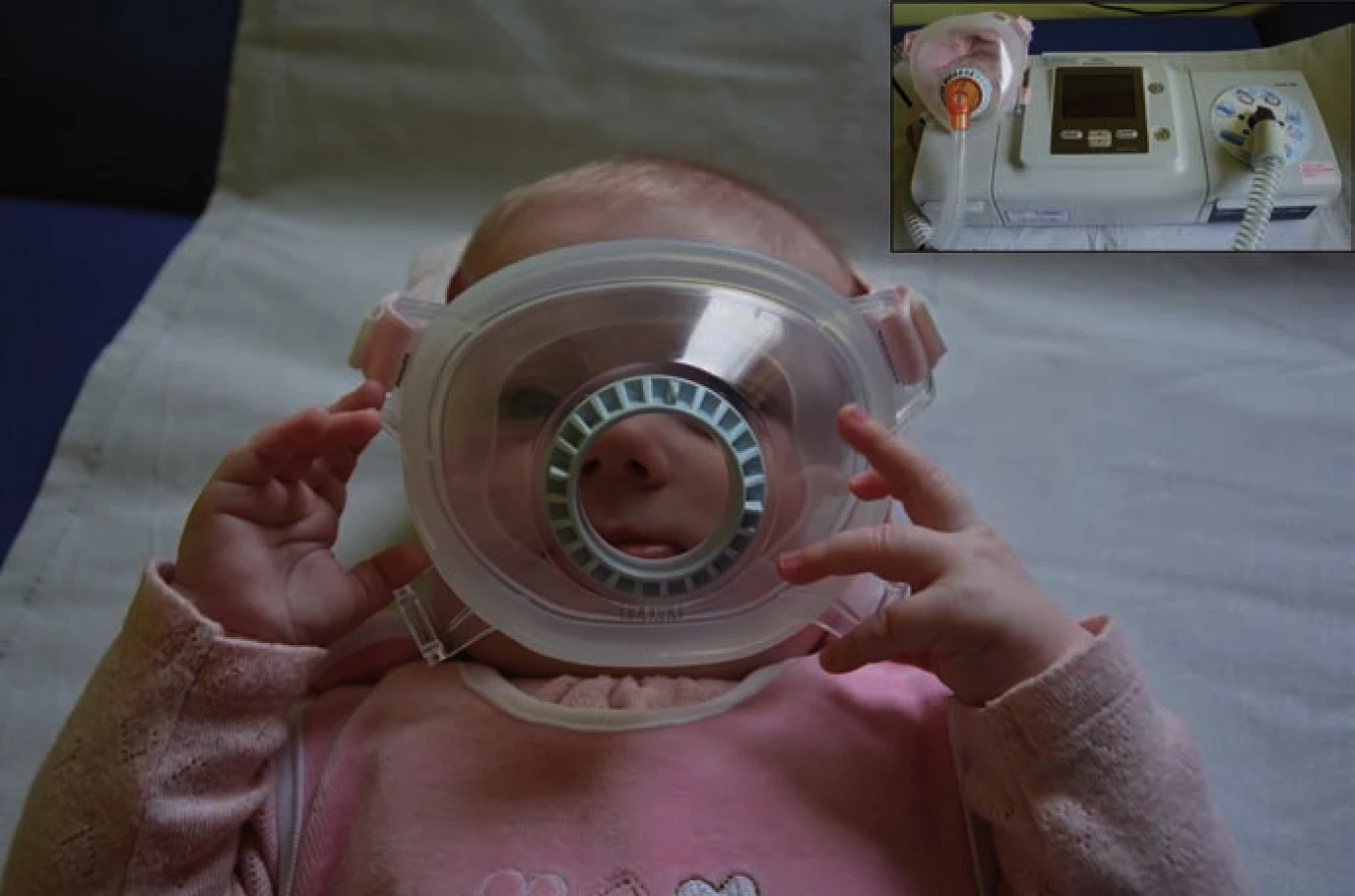
Po genetické konfirmaci se ve spolupráci s Oddělením dětské neurologie FN Ostrava, kam byly obě pacientky přeloženy, podařilo najít efektivní systém domácí spánkové neinvazivní ventilační podpory s využitím speciální celoobličejové masky pro novorozence (obr. 1). Nejlépe vyhovujícím ventilačním přístrojem se ukázal být Philips BiPAP A40 (režim Bi‑ level Positive Airway Pressure). Po seznámení rodičů s obsluhou přístroje a ověření jeho bezpečnosti byly holčičky propuštěny do domácí péče.
Diskuze
Kongenitální centrální hypoventilační syndrom (Ondinina kletba) je velmi vzácné onemocnění centrálního nervového systému, při němž zcela chybí nebo je porušeno automatické řízení dýchání. CCHS má neznámou incidenci a obvykle se manifestuje v novorozeneckém věku hypoventilací/ apnoí ve spánku, nejzávažněji postižení jedinci mají obtíže i při bdění. Možný je i pozdější nástup onemocnění (kojenci, starší děti), včetně příznaků souvisejících s rozvojem komplikací chronické hypoxie a hyperkapnie (cor pulmonale, křeče, psychomotorická retardace) [5]. Klinická tíže hypoventilace se liší dle typu mutace PHOX2B genu. NPARMs jsou obecně spojeny s vyšším rizikem kontinuální ventilační podpory (67 % u NPARMs vs. 35 % u PARMs) [1]. Nástup onemocnění byl u obou holčiček zcela typický. Určitou zvláštností je tranzientní hypoventilace/ apnoe i při bdění v prvních dnech života (zejm. u pacientky P2), což může souviset s určitou nezralostí centrálního nervového systému. V dalším průběhu se hypoventilace manifestovala standardně pouze ve spánku.
Mezi další projevy popisované při CCHS patří Hirschsprungova choroba (až u 20 % ze všech CCHS), nádory neurální lišty a poruchy autonomního nervového systému (sinusové bradykardie/ asystolie, teplotní nestabilita, poruchy polykání) [5]. Hirschsprungova choroba se s mnohem vyšší prevalencí vyskytuje u typu NPARMs (80 %) oproti PARMs (do 20 %). Nádory neurální lišty se objevují u 5 % ze všech případů CCHS (zejm. neuroblastom, ganglioneurom, ganglioneuroblastom). Velmi vzácně (cca v 1 %) se objevují u PARMs (ganglioneurom a ganglioneuroblastom u genotypu 20/ 28 až 20/ 33), mnohem vyšší riziko (41 %) mají pacienti s NPARMs (neuroblastom). Ataky náhlých asystolií jsou život ohrožující komplikace. Gronli et al [6] identifikovali korelaci mezi typem PARMs a délkou intervalu R‑ R při holterovském monitorování. Děti s genotypem 20/ 25 neměly žádné asystolie delší než 3 vteřiny, nicméně genotyp 20/ 26 je spojen s 19% rizikem těchto asystolií a genotyp 20/ 27 má toto riziko až 83 %. Naše pacientky neměly problémy s odchodem stolice ani tendence k bradyarytmiím. Diagnostikovaný WPW syndrom u pacientky P1 či jiné tachyarytmie nebyly v nám známé literatuře popsány. Dle našeho soudu WPW syndrom se základním onemocněním nesouvisí. Pro pacientku P2 s genotypem 20/ 26 existuje přibližně 20% riziko náhlé asystolie s rizikem syndromu náhlého úmrtí kojence (SIDS) a je možné, že v budoucnu bude vyžadovat zavedení pacemakeru. Výrazným problémem byly v obou případech obtíže s polykáním, které odezněly až na konci druhého měsíce.
Molekulárně‑genetické vyšetření má nezastupitelné místo v diagnostice CCHS. Nízký počet diagnosticky dotažených případů je nejen důsledkem vzácnosti onemocnění, částečně ale také dosud špatnou dostupností molekulární genetiky. Velmi podstatným přínosem je zavedení molekulárně‑genetické diagnostiky CCHS na Oddělení lékařské genetiky ve FN Hradec Králové. V případě podezření na CCHS je možné kontaktovat Oddělení lékařské genetiky FN HK (tel.: 495 833 348, Mgr. Ema Ruszová, Ph.D).
Většina případů CCHS vzniká jako de novo mutace v PHOX2B genu. Vzácněji je onemocnění zděděno od některého z rodičů. U části zdravých rodičů (5– 10 %) postižených dětí byla prokázána somatická mutace. Riziko opakování nemoci u dalších sourozenců dítěte může v takovém případě dosáhnout až 50 %. Riziko výskytu nemoci u sourozenců pacientů nelze nikdy zcela vyloučit ani při negativitě testovaných rodičů pro malou možnost germinální mozaiky. V případě jasné kauzální mutace je indikováno prekoncepční poradenství s možností zvážení preimplantační a prenatální diagnostiky k vyloučení opakování nemoci v rodině.
Cílem léčby CCHS je zajištění adekvátní ventilace a oxygenace ve spánku a u nejtěžších forem onemocnění i při bdění. Všichni pacienti s tímto onemocněním vyžadují určitou formu spánkové ventilační podpory po celý život. V čase se závislost na ventilační podpoře snižuje, což je dáno zkracující se délkou spánku. Dle dostupné literatury převažuje v prvních letech života invazivní ventilace přes tracheostomii (více než 60 %) s převedením na neivazivní ventilaci maskou v předškolním věku [7]. V posledních letech se objevují zprávy i o časnějším převedení na neinvazivní ventilaci [8]. Ventilační mód pro neinvazivní ventilaci se označuje jako BiPAP (Bi‑ level Positive Airway Pressure), což znamená, že přístroj nejen že je schopen udržovat pozitivní přetlak na konci výdechu (PEEP), ale umí současně generovat nastavitelný vrcholový inspirační tlak (PIP) k dosažení potřebného dechového objemu. Další ovladatelný parametr je dechová frekvence, jejíž nastavení by mělo přibližně kopírovat fyziologické potřeby pacienta dle jeho stáří (novorozenec – 40 dechů/ min, kojenec – 30 dechů/ min, předškolák – 20 dechů/ min, starší dítě – 15 dechů/ min, dospělý 10 dechů/ min) [9]. Hlavním faktorem určujícím nastavení ventilace jsou fyziologické krevní plyny. V případě selhání BiPAPu je nutná invazivní ventilace přes tracheostomii. Naše pacientky byly pomocí BiPAPu léčeny od prvního (P1) respektive 11. (P2) dne života, tudíž ještě před stanovením diagnózy. Domácí neinvazivní ventilace pak byla možná od druhého (P2) respektive čtvrtého (P1) měsíce života. Použití neinvazivní ventilace namísto tracheostomie přináší řadu pozitiv jako odstranění rizik invazivní ventilace, zlepšení rozvoje řeči a psychosociálního postavení. Přesto má i některé nevýhody – zvýšenou četnost infekcí horních cest dýchacích, gastroezofageální reflux, deformity obličeje a poruchy prořezávání chrupu. Těmto problémům lze čelit střídáním různých typů nostril a sledování obličeje pacienta s využitím fotografií [8].
Vzhledem k rizikům dlouhodobých kom-plikací stran možného selhání ventilace (cor pulmonale, psychomotorická retardace) a přidružených onemocnění je nutné pacienty s CCHS pravidelně sledovat (tab. 1). U všech dětí do tří let věku s CCHS bez ohledu na typ mutace je vhodné v půlročních intervalech provádět komplexní fyziologické testování za hospitalizace (zejm. krevní plyny – bdění/ spánek, krevní obraz, retikulocyty, ultrazvuk srdce, 72hod EKG holter, neurokognitivní vyšetření a pátrat po příznacích Hirschsprungovy choroby). U starších dětí postačí roční intervaly. U pacientů s mutací NPARMs je nutné k časné detekci neuroblastomu provést ultrazvukové vyšetření břicha a stanovit katecholaminy v moči každé tři měsíce první dva roky, dále každých šest měsíců do sedmi let. A u pacientů s mutací PARMs 20/ 28– 20/ 33 je vhodné provádět jedenkrát ročně cílené zobrazovací vyšetření k odhalení nádorů neurální lišty (ganglioneuromu a ganglioneuroblastomu) [1].
![Doporučení sledování pacientů s CCHS dle zjištěného genotypu [1].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/1b24e27181407a09d1896c7b049979f9.png)
Závěr
Na diagnózu CCHS je nutné pomýšlet zvláště u všech novorozenců/ kojenců s hypoventilací/ apnoí při absenci primárního plicního onemocnění, neuromuskulárního onemocnění či organické léze dechových center mozkového kmene. Pro definitivní potvrzení diagnózy CCHS je nutný průkaz mutace PHOX2B genu, nově dostupné i v České republice (Oddělení lékařské genetiky FN HK). S moderními technikami pro domácí ventilaci mají děti s CCHS dlouhodobě dobrou prognózu s nízkou mortalitou a malým rizikem psychomotorického opoždění. Neinvazivní ventilace pomocí BiPAPu je praktická a spolehlivá alternativa k tracheostomii a může zlepšit kvalitu života dítěte a jeho rodiny.
Podpořeno MZ ČR RVO (FNHK, 00179906, Z1/ 8142).
Děkujeme MU Dr. Vilému Novákovi z Oddělení dětské neurologie FN Ostrava za spolupráci s výběrem ventilačního přístroje a navedení obou dětí na systém neinvazivní domácí spánkové ventilace.
Přijato k recenzi: 24. 2. 2014
Přijato do tisku: 23. 12. 2014
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Tomáš Matějek
Dětská klinika
LF UK a FN Hradec Králové
Sokolská 581
500 05 Hradec Králové
e-mail: xmatt01@seznam.cz
Sources
1. Weese‑Mayer DE, Berry‑ Kravis EM, Ceccherini I, Keens TG,Loghmanee DA, Trang H. An official ATS clinical policy statement: Congenital central hypoventilation syndrome. Am J Respir Crit Care Med 2010; 181(6): 624– 644. doi: 10.1164/ rccm.200807‑ 1069ST.
2. Amiel J, Laudier B, Attie‑ Bitach T, Trang H, de Pontual L,Gener B et al. Polyalanine expansion and frameshift mutations of the paired‑like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 2003; 33(4): 459– 461.
3. Berry‑ Kravis EM, Zhou L, Rand CM, Weese‑Mayer DE. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med 2006; 174(10): 1139– 1144.
4. Weese‑Mayer DE, Marazita ML, Berry‑ Kravis EM, Patwari PP. Congenital Central Hypoventilation Syndrome. GeneReviews™ [online]. Available from URL: http:/ / www.ncbi.nlm.nih.gov/ books/ NBK1427.
5. Healy F, Marcus CL. Congenital central hypoventilation syndrome in children. Paediatr Respir Rev 2011; 12(4): 253– 263. doi: 10.1016/ j.prrv.2010.10.001.
6. Gronli JO, Santucci BA, Leurgans SE, Berry‑ Kravis EM, Weese‑Mayer DE. Congenital central hypoventilation syndrome: PHOX2B genotype determines risk for sudden death. Pediatr Pulmonol 2008; 43(1): 77– 86.
7. Vanderlaan M, Holbrook CR, Wang M, Tuell A, Gozal D.Epidemiologic survey of 196 patients with congenital central hypoventilation syndrome. Pediatr Pulmonol 2004; 37(3): 217– 229.
8. Kam K, Bjornson C, Mitchell I. Congenital central hypoventilation syndrome; safety of early transition to non‑invasive ventilation. Pediatr Pulmonol 2013; 49(4): 410– 413. doi: 10.1002/ ppul.22848.
9. Nováková Z. Fyziologické zvláštnosti dětského věku. Prakt Lékaren 2012; 8(6): 279– 282.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2015 Issue 2
Most read in this issue
- Agresivní hemangiom obratle
- Neuromyelitis optica
- Kongenitální centrální hypoventilační syndrom (Ondinina kletba)
- Radiologické hodnocení lumbální spinální stenózy a jeho klinická korelace