
Myozitida s inkluzními tělísky se slabostí šíjových svalů a pozitivním efektem imunoglobulinu – kazuistika
Inclusion Body Myositis with Neck Muscle Weakness and Positive Effect of Immunoglobulins – a Case Report
Inclusion body myositis is the most frequent inflammatory myopathy in patients aged 50 years and above. There is a chronic-progredient course of the disease without attacks and remissions. Typical clinical findings consist of proximal and distal weakness of extremity muscles and of axial muscles. Deep finger flexors of the upper extremities, quadriceps and muscles of the anterior compartment of the shin are severely affected. Dysphagia is a typical sign. Weakness of the neck muscles with a tilting head represents a rare and atypical form. We report a case of a 59-year old woman with progredient muscle weakness and atrophy with head tilting forward and dysphagia. Prednisone therapy was ineffective. After repeated intravenous administration of large immunoglobulin doses (60 g every four weeks), the posture of head a neck improved and dysphagia disappeared. One year into the treatment, immunoglobulin therapy is still highly effective.
Key words:
inclusion body myositis – dysphagia – weakness – immunoglobulins
Authors:
E. Ehler 1; A. Meleková 1; J. Latta 1; P. Mandysová 1; P. Vojtíšek 2; J. Zámečník 3
Authors‘ workplace:
Neurologická klinika FZS Univerzity Pardubice a Pardubické krajské nemocnice, a. s.
1; Interní klinika FZS Univerzity Pardubice a Pardubické krajské nemocnice, a. s.
2; Ústav patologie a molekulární medicíny 2. LF UK a FN v Motole, Praha
3
Published in:
Cesk Slov Neurol N 2013; 76/109(3): 362-366
Category:
Case Report
Overview
Myozitida s inkluzními tělísky je nejčastější zánětlivá myopatie u pacientů nad 50 let věku. Průběh choroby je chronicko-progredující bez atak a remisí. V typickém klinickém nálezu se vyskytuje kombinace proximální i distální slabosti končetin i axiálních svalů, zejména jsou postiženy hluboké flexory prstů rukou, čtyřhlavý sval a svaly přední skupiny bérce. Typickým příznakem je dysfagie. Mezi atypické formy patří slabost šíjového svalstva s přepadáním hlavy dopředu. Tento článek prezentuje kazuistiku 59leté ženy s postupným rozvojem slabostí a atrofií svalů s přepadáním hlavy dopředu a dysfagií. Terapie prednizonem neměla žádný efekt. Po opakovaném intravenózním podání imunoglobulinů (60 g každé čtyři týdny) se výrazně zlepšilo držení hlavy a trupu, vymizela dysfagie. Terapeutický efekt imunoglobulinů trvá i po roce.
Klíčová slova:
myozitida s inkluzními tělísky – dysfagie – slabost – imunoglobuliny
Úvod
Myozitida s inkluzními tělísky (IBM) je pravděpodobně nejčastější „zánětlivá“ myopatie se začátkem nad 50 let věku. Muži jsou postiženi dvakrát častěji než ženy. Diagnóza IBM bývá stanovena až po delším trvání nemoci (6–7 let) [1]. Průběh choroby je chronicko-progredující bez remisí a atak. V klinickém nálezu jsou oslabeny jak proximální svaly (šíje, trup, pletence), tak i svaly distální. Typické jsou atrofie a oslabení čtyřhlavých svalů (vedoucí k náhlým pádům), flexorů prstů rukou (nedovře pěst) i oslabení dorzální flexe nohy [2]. Vzhledem k výrazné asymetrii postižení s rozvojem paréz a atrofií může být v počátku choroby klinické podezření na amyotrofickou laterální sklerózu (ALS). U 40–60 % nemocných bývá dysfagie, která vzniká na podkladě postižení svalů hltanu a jícnu [3]. Vede k hyponutrici s úbytkem hmotnosti i k aspirační pneumonii. Mezi atypické případy IBM patří forma „skapulo-peroneální“, kamptokormie či „dropped head syndrom“, při které na podkladě slabosti šíjových svalů hlava přepadá dopředu. Pro diagnostiku je zcela klíčové vyšetření svalová biopsie (nález lymfocytárních infiltrátů, lemovaných vakuol na úrovni světelné mikroskopie a IBM inkluzí v elektronovém mikroskopu) [4]. Známky myogenní léze v biochemických parametrech (kreatinkináza, volný myoglobin) jsou relativně nízké (maximálně desetinásobek) a EMG nález není příliš charakteristický. IBM se vyznačuje trvalou progresí atrofií a oslabení svalů a léčbou je jen velmi málo ovlivnitelná. Při léčbě vysokými dávkami intravenózně podaného imunoglobulinu se v jedné ze tří studií docílil několikaměsíční efekt (6–8 měsíců) u nemocných se sporadickou formou IBM a s výraznými polykacími potížemi [5].
V péči naší neuromuskulární poradny je 59letá žena se sporadickou IBM, s výrazným dropped-head syndromem, dysfagií, u které má léčba intravenózně podanými imunoglobuliny výrazný efekt. To je důvod publikace tohoto kazuistického sdělení.
Kazuistika
Devětapadesátiletá žena je od září 2010 dispenzarizována v neuromuskulární poradně. V létě 2009 se objevila slabost dolních končetin při chůzi a téměř současně i mírná dysfagie. Postupně se přidala porucha chůze do schodů, občas padala i na rovině pro slabost stehenních svalů, vleže na zádech již nebyla schopna zvednout hlavu, hůře artikulovala a zhoršilo se polykání. Za 6 měsíců zhubla o 12 kg. S těmito potížemi a s podezřením na ALS byla poslána do naší EMG laboratoře.
Od roku 2004 léčena pro hypertenzi, od roku 2006 pro osteoporózu, od podzimu 2009 pro hypotyreózu. Pracovala jako kuchařka, je pravačka. Terapie: Euthyrox 50 ug, Vigantol, Concor COR (2,5 mg 1/2-0-0). Rodinná anamnéza nevýznamná.
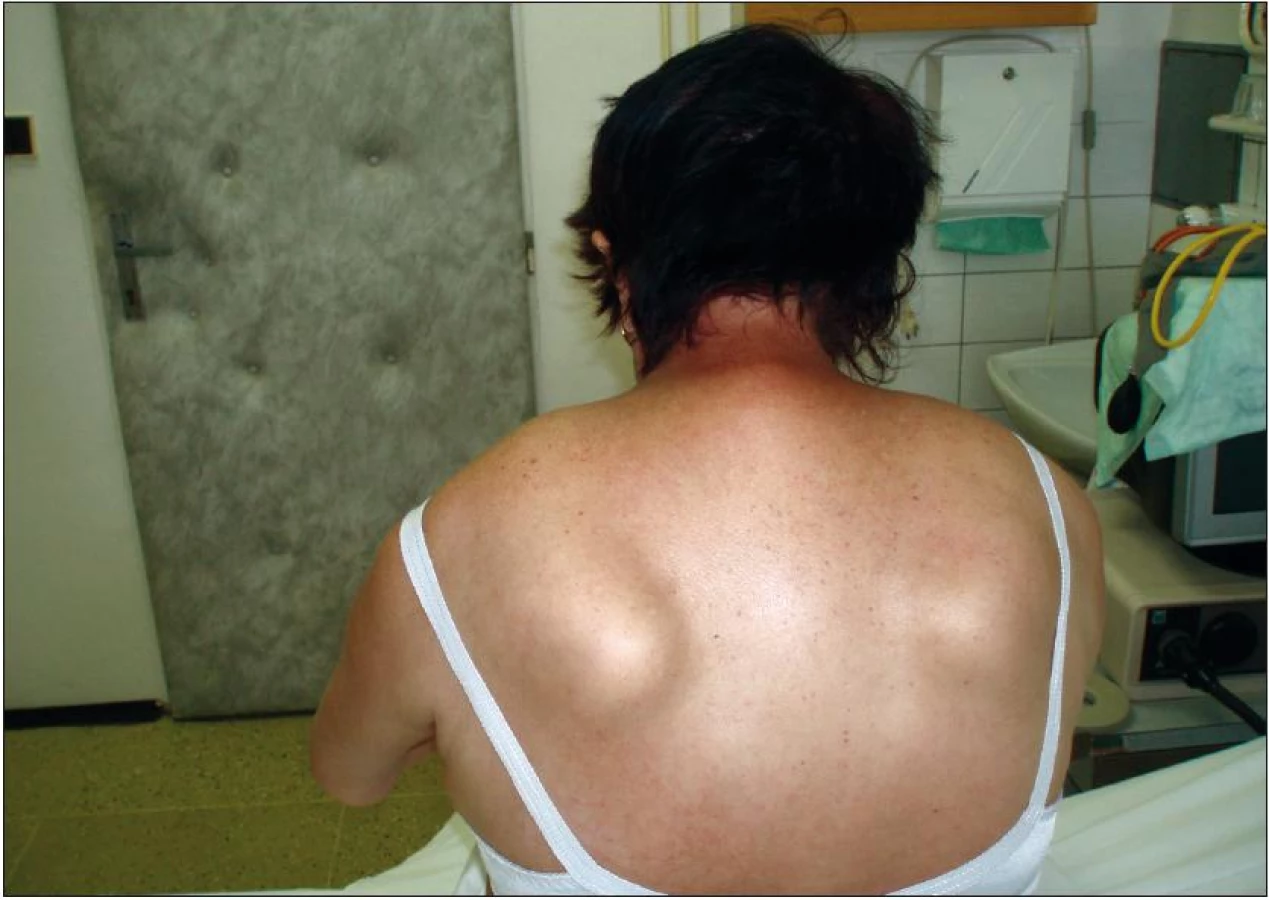
Při vyšetření je psychicky čilá, jazyk s nepravidelnými atrofiemi pomalu a omezeně plazí, zvedne špičku jazyka, zvracivý reflex živý, špatně odkašlává a obtížně polyká, plošší mimika, dysartrie, vleže nezvedne hlavu, avšak dojde přitom ke kontrakci břišních svalů, vsedě hlava přepadá dopředu a při změně polohy i dozadu, zvýšená kyfóza C-Th přechodu, HK ve výdrži rychle klesají, atrofie svalstva předloktí – zejména volární skupiny, méně svalů rukou, stisk vpravo 10 a vlevo 7 kPa, flexe prstů je různého rozsahu pro jednotlivé prsty, špetku nedotáhne pro 4. a 5. prst, DK neudrží v navozené výdrži, omezený rozsah dorzální a méně i plantární flexe nohou i prstů, RRC5–8 L2–S2 +, plantární rr. +, jemná porucha čití od kotníků distálně. Obtížně se posadí a svede jen pár nejistých krůčků o širší bázi s výraznou Th kyfózou a přepadáním hlavy dopředu (obr. 1).
V EMG byly nalezeny fibrilace, pozitivní vlny i repetitivní polyfázické výboje a v některých svalech i ojedinělé fascikulace, MUP byly převážně užší a nižší (m. tibialis anterior) a v m. vastus lateralis byly zjištěny rovněž vyšší MUP delšího trvání s polyfázií (33 %). Senzitivní neurografie na DK svědčila pro lehkou distální senzitivní polyneuropatii (SCV 33,8 a 33,3 m/s pro n. suralis a n. peroneus superficialis).
Při kardiologickém vyšetření diagnostikována lehká plicní hypertenze (PAP s výpočtem 45 mmHg).
Z laboratorních vyšetření byly zvýšeny hladiny kreatinkinázy na 8,43 (0,35–3,58 ukat/l) a volného myoglobinu 177 ug/l (25–58 ug/l). Hladiny fT3, FT4 a TSH byly v mezích normy.
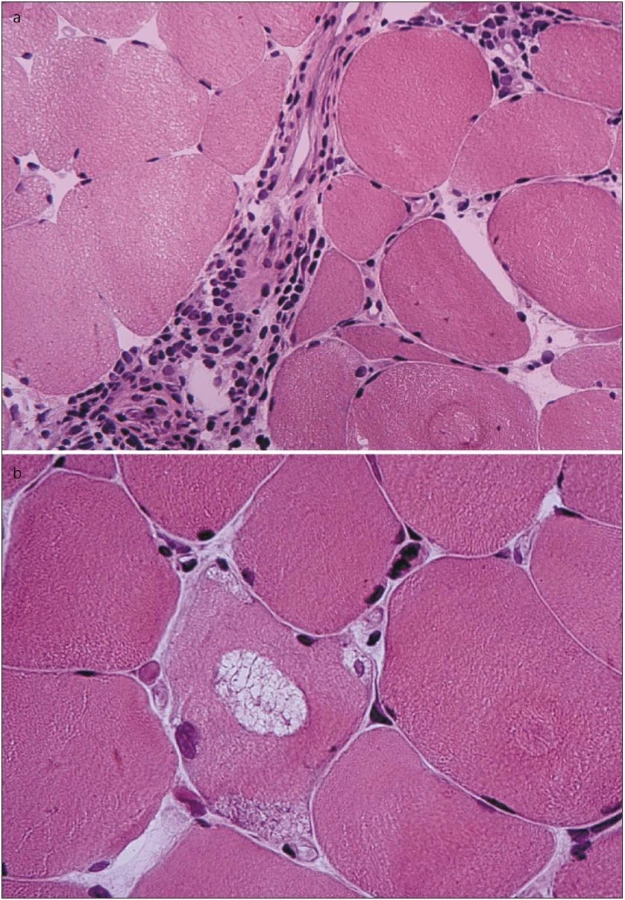
Vzhledem ke klinickému nálezu se zvýšenými svalovými enzymy v séru a EMG nálezu jsme v této fázi indikovali svalovou biopsii. Biopsií m. vastus lateralis vpravo byla prokázána poměrně pokročilá svalová atrofie s kombinací neurogenních prvků (angulární atrofická vlákna, fenomén „type grouping“) i prvků myopatického vzorce (kulatá atrofická vlákna, internalizace jader, fenomén štěpení vláken). Zastižen byl ložiskový endomysiální i perimysiální lymfocytární infiltrát sestávající převážně z CD8+ T lymfocytů. Dále byla pozorována přítomnost tzv. lemovaných vakuol s bazofilním jemně granulárním obsahem (obr. 2). Elektronmikroskopicky byly v oblastech lemovaných vakuol pozorovány membranózní víry, navíc byly v cytoplazmě detekovány shluky neuspořádaných filament a tubulofilament odpovídajícím tzv. IBM inkluzím (obr. 3).
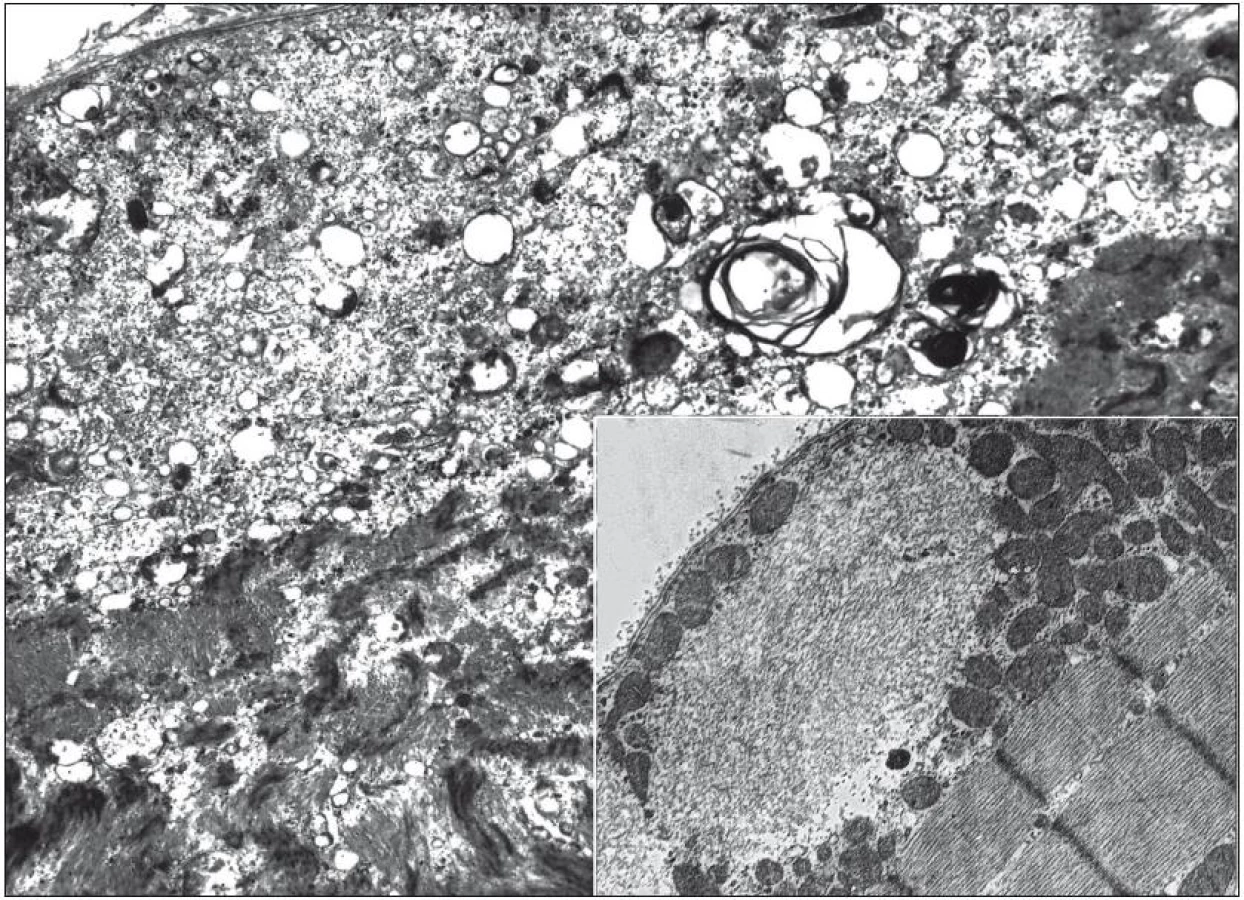
Nasadili jsme prednizon ve stoupajících dávkách. K ovlivnění potíží nemocné nedošlo. Proto byl v září 2011 aplikován intravenózní imunoglobulin (60 g) s poměrně výrazným efektem. Došlo ke zlepšení držení hlavy, významně se zmírnila dysfagie a zlepšila chůze. Od té doby je stále aplikován imunoglobulin v dávce 60 g každé čtyři týdny.
Diskuze
Prevalence IBM se liší podle zeměpisné polohy a je u různých etnik i skupin obyvatel odlišná. V Nizozemí je prevalence 4,9 případů na milión obyvatel, v Austrálii činí 9,3, v USA 7,9, avšak existují až několikanásobně vyšší (až 7krát). U Hispánců (Iberoameričanů) a u Asiatů je prevalence podstatně nižší než u bělochů [5]. U sporadické formy jsou výrazné zánětlivé změny průkazné ve svalové biopsii a vyskytují se i typické klinické projevy. U hereditární formy nepozorujeme podstatnější zánětlivé změny a klinický obraz nemá charakteristické rysy IBM. Navíc byla u recesivně dědičné hereditární formy IBM prokázána kauzální mutace v genu GNE (gen pro UDP-N-acetylglukosamin-2-epimerázu//N-acetylmannosamin kinázu) na chromozomu 9 [6]. V České republice je IBM nejspíše značně poddiagnostikována. V naší neuromuskulární poradně se jedná teprve o druhý případ.
Průběh IBM je typicky chronický, bez atak, avšak s neúnavnou progresí postižení (tab. 1). U většiny nemocných se vyskytuje svalová slabost lokalizovaná jak proximálně, tak i distálně. Typická je slabost čtyřhlavých svalů stehen s poruchou chůze po schodech a s náhlými pády. Atrofie a slabost flexorů na volární ploše předloktí vede k oslabení stisku ruky, a protože bývá elektivní oslabení hlubokého flexoru prstů, pacient nemůže dokončit flexi prstů až do dlaně. Slabost flexorů prstů (včetně palce) a ruky nápadně kontrastuje se zachovanou silou abdukce paže. Prsty jsou slabé a neohrabané (zapínání knoflíků, zipy, otevírání uzávěru láhve). Typické je oslabení dorzální flexe nohy s přepadáváním nohy, zakopáváním o špičku (není stepáž pro oslabení čtyřhlavého svalu) [7]. Pro proximální slabost DK nemocní obtížně vstávají ze židle. Až u 60 % se vyskytuje dysfagie, a to zejména na podkladě slabosti faryngeálních a ezofageálních svalů. To vede k hyponutrici i aspiraci s plicními záněty [1,8]. Mezi méně typické příznaky patří slabost paravertebrálních svalů s kyfózou či kamptokormií. Na podkladě slabosti šíjových svalů dochází k pasivnímu anteflexnímu držení hlavy – dropped head syndrom s dalším zhoršením dysfagie, orientace zrakem, dysfonie. Dropped head syndrom může být podmíněn celou řadou nemocí, a proto je u těchto nemocných velmi široká diferenciální diagnostika (tab. 2) [1,5,7]. Potíže naší nemocné se objevily v 56 letech, a to slabostí při chůzi do schodů, pak náhlými pády a rozvojem dysfagie. Postupně se rozvíjela slabost rukou a prstů, zhoršovala se dysfagie s úbytkem hmotnosti a rozvinula se těžká slabost šíjových svalů s přepadáním hlavy dopředu.

![Diferenciální diagnostika přepadání hlavy (dropped head syndrome) (volně dle [13]).](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/84126fb9709b10d47fd6a91b0d2a0523.png)
Elektromyografický nález u IBM není charakteristický a nutně vede k širší diferenciální diagnostice. Při motorické neurografii je nález normální či bývá snížena amplituda u sumační motorické odpovědi (u atrofických svalů). Až u 30 % nemocných se diagnostikuje periferní neuropatie s výraznějším postižením senzitivních vláken (typu lehké axonální poruchy). Při vyšetření EMG jehlou bývají přítomny fibrilace, pozitivní vlny i repetitivní polyfázické výboje, a to v různé svalové distribuci a různé četnosti. Potenciály motorické jednotky (MUP) bývají v převažujícím podílu užší a nižší – tedy „myogenního typu“, avšak asi u třetiny nemocných se vedle užších a nižších MUP vyskytují i vyšší a polyfázické MUP, mající „neurogenní charakter“. Tyto vyšší a polyfázické MUP jsou známkou chronicity procesu. Mohou vznikat na podkladě zánětu ve svalu s poruchou svalové membrány či dokonce i terminálního větvení a s následnou kolaterální reinervací. Mohou být i výrazem hypertrofie svalových vláken při chronicitě procesu [1,6,9]. U naší nemocné jsme při prvním vyšetření prokázali lehkou a převážně senzitivní polyneuropatii na DK a výrazné změny ve svalech – jak fibrilace a pozitivní vlny, tak i ojediněle nerytmické fascikulace. I mezi nižšími a kratšími MUP jsme nalezli nevelký podíl vysokých a polyfázických MUP. EMG nález byl také jedním z důvodů indikace svalové biopsie, kdy jsme kromě primárně myogenní příčiny byli nuceni vylučovat i případné souběžné postižení periferního motoneuronu.
U většiny nemocných s IBM byla původně diagnostikována polymyozitida (PM) [1,7]. Je proto nutné tyto dvě zánětlivé myopatie jasně odlišit. V klinickém nálezu je rozlišujícím kritériem postupná progrese asymetrického oslabení a atrofie s postižením jak proximálních, tak i distálních svalů. Pro IBM je charakteristické oslabení čtyřhlavého svalu, flexorů prstů a ruky i dorzální flexe nohy u nemocného nad 50 let. I když existují rozdíly v neurofyziologických nálezech IBM a PM, svalová biopsie je rozhodující argument. V diferenciální diagnostice přichází nezřídka do úvahy i amyotrofická laterální skleróza (ALS) a další nemoci periferního motoneuronu. Asymetrickým postižením končetin s atrofiemi a parézami včetně akra (prsty, ruka, přepadání nohy) může nemocný s IBM klinickým obrazem a trvale progredujícím průběhem připomínat nemocné s ALS. Rovněž nález příměsi vysokých a polyfázických MUP a fibrilací, pozitivních vln i repetitivních polyfázických výbojů může zčásti korelovat s ALS či motor neuron disease. V klinickém nálezu však u IBM chybí fascikulace, atrofie a slabosti svalů jsou proximální i distální. V distribuci atrofií ruky je ale značný rozdíl. U IBM jsou atrofie výraznější na předloktí, zatímco svaly ruky (thenar, hypothenar, interossei) jsou téměř přiměřené, u ALS jsou atrofie drobných svalů ruky velmi výrazné. V EMG nálezu je velký rozdíl při vyšetření histogramu MUP, kdy u IBM je podstatná převaha nízkých a krátce trvajících MUP. U ALS jsou naopak ve velké většině MUP v různém stadiu neurogenní přestavby – často vyšší, delší, polyfázické a nestabilní [1,5,9]. Naše nemocná měla při prvém EMG vyšetření nález poměrně masivní spontánní aktivity (fibrilace) a MUP byly užší s určitým podílem vyšších a polyfázických MUP. Ojediněle jsme na několika místech nalezli fascikulace (m. vastus lateralis). Proto jsme si ani my nebyli jisti, zda se jedná pouze o myogenní lézi a zda není rovněž přítomna léze periferního motoneuronu. Svalová biopsie zcela jasně prokázala charakter nemoci. Ačkoli je obraz IBM ve svalové biopsii poměrně charakteristický, je třeba i zde pečlivě korelovat nález s klinickým obrazem. IBM by mohla být pro podobnou distribuci slabosti a tempem rozvoje onemocnění snadno zaměněna např. s myofibrilární myopatií, neboť i u té bývají pozorovány filamentózní inkluze i lemované vakuoly. U myofibrilární myopatie však filamentózní depozita bývají objemnější, společně s aberantní expresí desminu a dystrofinu, což u IBM pozorováno nebývá [10].
Hereditární IBM není léčbou ovlivnitelná [1]. U sporadické formy nebývá imunosuprese – kortikoidy, azatioprin, cyklofosfamid či mykofenylát mofetil – účinná. Již počátkem 90. let byl při nekontrolovaném intravenózním podání vysokých dávek imunoglobulinů (IVIG) popisován terapeutický efekt se zlepšením svalové síly, polykání, chůze. V následujících kontrolovaných studiích v trvání 3 a 6 měsíců byl však efekt buď krátkodobý či ne již tak výrazný. V americké studii (Dalakas et al, 1997) došlo po třech měsících ke zlepšení svalové síly (složené svalové skóre), a to zejména na dolních končetinách, a zlepšilo se i polykání. V německé studii trvající 6 měsíců (Walter et al, 2000) došlo k zástavě progrese nemoci u 18 z 22 nemocných, i když se složené svalové skóre nezlepšilo. V malé sérii čtyř nemocných se po podávání IVIG po dobu 6–8 měsíců zlepšila těžké dysfagie (Cherin et al, 2002) [11,12]. K úbytku svalové síly docházelo v průměru o 4 % ročně. Naší 59leté nemocné s přepadáním hlavy, výraznou dysfagií a téměř nemožností samostatné chůze jsme aplikovali imunoglobuliny (60 g) s opakováním každé 3–4 týdny. Došlo k výraznějšímu zlepšení dysfagie, vylepšila se nutrice (přibrala 3 kg), o něco se změnilo v pozitivním směru vertikální držení hlavy i chůze. Tento příznivý efekt byl pro nemocnou podstatným přínosem.
Závěr
Prezentovali jsme kazuistiku 59leté ženy, u níž se po tři roky rozvíjela svalová slabost a atrofie svalů s distální i proximální distribucí. V diagnostickém procesu jsme hodnotili EMG vyšetření a svalovou biopsii, která prokázala IBM. Kromě charakteristických příznaků IBM měla i výrazné přepadání hlavy. Imunosupresivní léčba byla bez efektu, avšak intravenózní terapie imunoglobuliny má výrazný účinek na polykání, nutrici, držení hlavy i chůzi. Klinický efekt pravidelného podávání 60 gramů imunoglobulinů i po roce trvá.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Edvard Ehler, CSc.
Neurologická klinika
FZS Univerzity Pardubice a Pardubické krajské nemocnice, a.s.
Kyjevská 44
532 03 Pardubice
e-mail: eda.ehler@tiscali.cz
Přijato k recenzi: 4. 9. 2012
Přijato do tisku: 15. 10. 2012
Sources
1. Amato AA, Russel JA. Neuromuscular disorders. New York: McGraw Hill Medical 2008.
2. Ambler Z. Zánětlivé myopatie. Neurol Prax 2004; 5: 150–154.
3. Solorzano GE, Phillips LH 2nd. Inclusion body myositis: diagnosis, pathogenesis, and treatment options. Rheum Dis Clin N Am 2011; 37(2): 173–183.
4. Bednařík J et al. Nemoci kosterního svalstva. Praha: Triton 2001.
5. Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostics and therapeutic approaches. Lancet Neurol 2007; 6(7): 620–631.
6. Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet 2001; 29(1): 83–87.
7. Pourmand R. Immune-mediated neuromuscular diseases. Basel: Karger 2009.
8. Cox FM, Titulaer MJ, Sont JK, Wintzen AR, Verschuuren JJ, Badrising UA. A 12-year follow-up in sporadic inclusion body myositis: an end stage with major disabilities. Brain 2011; 134(11): 3167–3175.
9. Oh SJ. Clinical electromyography. Case studies. Baltimore: Williams & Wilkins 1998.
10. De Bleecker JL, Engel AG, Ertl BB. Myofibrillar myopathy with abnormal foci of desmin positivity. II. Immunocytochemical analysis reveals accumulation of multiple other proteins. J Neuropathol Exp Neurol 1996; 55(5): 563–577.
11. Bednařík J, Voháňka S, Ehler E, Ambler Z, Piťha J, Vencovský J et al. Standard pro léčbu s autoimunitními nervosvalovými onemocněními intravenózním lidským imunoglobulinem a plazmaferézou. Cesk Slov Neurol N 2010; 73/106(5): 570–589.
12. Patwa HS, Chaudry V, Katzberg H, Rae-Grant AD, So YT. Evidence-based guideline: intravenous immunoglobulin in the treatment of neuromuscular disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2012; 78(13): 1009–1015.
13. Mahjneh I, Marconi G, Paetau A, Saarinen A, Salmi T, Somer H. Axial myopathy – an unrecognised entity. J Neurol 2002; 249(6): 730–734.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 3
Most read in this issue
- Mechanizmy spasticity a její hodnocení
- Lidské prionové nemoci v České republice – 10 let zkušeností s diagnostikou
- Myozitida s inkluzními tělísky se slabostí šíjových svalů a pozitivním efektem imunoglobulinu – kazuistika
- Extrakraniálně metastazující meningeomy